

Abstract
Genetically, schizophrenia is a complex disease whose pathogenesis is likely governed by a number of different risk factors. While substantial efforts have been made to identify the underlying susceptibility alleles over the past 2 decades, they have been of only limited success. Each year, the field is enriched with nearly 150 additional genetic association studies, each of which either proposes or refutes the existence of certain schizophrenia genes. To facilitate the evaluation and interpretation of these findings, we have recently created a database for genetic association studies in schizophrenia (“SzGene”; available at http://www.szgene.org). In addition to systematically screening the scientific literature for eligible studies, SzGene also reports the results of allele-based meta-analyses for polymorphisms with sufficient genotype data. Currently, these meta-analyses highlight not only over 20 different potential schizophrenia genes, many of which represent the “usual suspects” (eg, various dopamine receptors and neuregulin 1), but also several that were never meta-analyzed previously. All the highlighted loci contain at least one variant showing modest (summary odds ratios approximately 1.20 [range 1.06–1.45]) but nominally significant risk effects. This review discusses some of the strengths and limitations of the SzGene database, which could become a useful bioinformatics tool within the schizophrenia research community.
Keywords: epidemiologic methods, genetic associations, meta-analysis, schizophrenia
Introduction
Although the heritability—the proportion by which phenotypic variation is determined by genetic variation—of schizophrenia is high (approximately 80%1), efforts to identify the underlying putative schizophrenia genes have met with only limited success. This is at least in part due to problems that generally aggravate epidemiological research in many psychiatric diseases, eg, the considerable degree of phenotypic variability and diagnostic uncertainty, the lack of extended pedigrees with typical Mendelian inheritance, and the absence of disease-specific neuropathological features or biomarkers.2 The identification of susceptibility genes is further complicated by gene-gene interactions that are difficult to predict and model and a likely substantial but difficult to detect environmental component. Notwithstanding these challenges, several chromosomal regions thought to harbor schizophrenia genes have been identified via whole-genome linkage analyses, a few even overlapping across different samples.3,4 Furthermore, over 1300 “candidate gene” studies—ie, studies that focus on certain genes based on some prior hypothesis regarding their potential involvement in the disease process—have been published over the past 2 decades claiming or refuting genetic association between putative schizophrenia genes and affection status and/or certain endophenotypes.5 Currently, more than 150 schizophrenia genetic association articles are published each year, at increasing pace. Despite these efforts, no single gene or genetic variant has yet been established as bona fide schizophrenia gene, at least not with the confidence attributed to other complex disease genes, such as APOE in Alzheimer disease6 or CFH in macular degeneration.7 For geneticists as well as clinicians, the growing number of (mostly conflicting) genetic findings has become increasingly difficult to follow, evaluate, and interpret.
Genome-Wide Association Screening
An alternative to the traditional candidate gene approach is afforded by recent advances in large-scale genotyping technologies that now enable researchers to perform comprehensive and largely hypothesis-free genome-wide association (GWA) analyses. As of today, 5 groups have reported the results using this approach in schizophrenia8–12 (table 1). The first study by Mah et al8 tested roughly 25 000 polymorphisms in or very near genetic coding regions and described significant association between the risk for schizophrenia and a polymorphism in PLXNA2. The following studies were much more comprehensive, testing between 400 000 and 500 000 simple nucleotide polymorphisms (SNPs) each, though 2 of them were based on pooled genotyping. This led to the reporting of 5 additional putative schizophrenia loci (CSF2RA and IL3RA,9 CCDC60 and RBP1,10 and RELN11). Only the report by Sullivan et al12 concluded that even after testing nearly 500 000 markers they did not find “evidence for the involvement of any genomic region with schizophrenia detectable with moderate sample size” in their analyses.
Table 1.
Overview of All Published GWA Studies in SZ (Current on June 1, 2008)
| GWA Study | Design | Population | Platform | Number of SNPs | Data Available? | Number of SZ Casesa (Total) | Number of Controlsa (Total) | “Featured” Genes |
| Mah et al8 | Case-control and family based | United States, Australia, and other | Customized cSNPs | 25 494 | No | 320 (1082) | 325 (1123) | PLXNA2 |
| Lencz et al9 | Case-control | United States | Affymetrix (500K) | 439 511 | No | 178 (249) | 144 (175) | CSF2RA, IL3RA |
| Shifman et al11 | Case-control | Israel, United States, Europe | Affymetrix (500k) | 510 552 | No | 660 (3015) | 2771 (7183) | RELN |
| Kirov et al10 | Family based | Bulgaria | Illumina (550K) | 43 680 | No | 574 (na) | 1753 (na) | CCDC60, RBP1 |
| Sullivan et al12 | Case-control | United States | Affymetrix (500K) and Perlegen (custom) | 492 900 | Yesb | 738 (na) | 733 (na) | None |
Note: Modified after content on the SzGene Web site (http://www.szgene.org). Studies are listed in order of publication date. “Featured Genes” are those genes/loci that were declared as “associated” in the original publication, note that criteria for declaring association may vary across studies. The studies by Mah et al,8 Shifman et al,11 and Kirov et al10 used pooled genotypes in their initial GWA analyses. SZ, schizophrenia.
Numbers of “schizophrenia Cases” and “Controls” refer to sample sizes used in initial GWA screening, whereas “Total” refers to initial sample plus any follow-up samples (where applicable); please consult SzGene Web site for more details on these studies.
Original publication states that “individual phenotype and genotype data [has been] made available to the scientific community”; application from SzGene curatorial team for access to these data is currently pending.
While these and several of the forthcoming GWA studies have the potential to significantly advance our understanding of the genetics and pathogenetic mechanisms of schizophrenia, it needs to be emphasized that in many ways GWA screens are actually not so different from conventional candidate gene association analyses. Both search for significantly different allele or genotype distributions or transmissions in subjects affected by the disease/phenotype as compared with presumably healthy individuals. The 2 approaches differ mostly on a quantitative level: instead of testing a few tens of markers (or less), GWA studies simultaneously screen a few hundreds of thousands of markers (or more). The major qualitative difference between GWA and candidate gene analysis is that the former investigates the whole genome in a more or less unbiased fashion, whereas the latter only investigates a limited number of specific loci proven or thought to be involved in disease predisposition or progression (eg, in schizophrenia many of these loci are involved in the release and regulation of certain neurotransmitters such as dopamine or serotonin). On the downside and owing to their sheer number of polymorphisms tested, GWA analyses actually substantially compound the problem that has plagued genetic studies of complex phenotypes in the past, ie, to determine which of the many reported putative risk alleles are “real” as opposed to which merely reflect statistical artifacts. The first essential step in differentiating these 2 alternatives is to provide independent replication of the association,13 just as for any result emerging from “old-fashioned” candidate gene analyses.
Systematic Field Synopsis and Meta-analyses: The SzGene Database
In an attempt to facilitate the interpretation of association findings regardless of the technology used for initial detection, we have recently created a publicly available database, “SzGene” (http://www.szgene.org), which systematically collects, summarizes, and meta-analyzes all genetic association studies published in the field of schizophrenia, including GWA studies5 (table 2; figure 1). After thorough (and still ongoing) searches of the available scientific literature, key variables are extracted from the original publications and summarized on the SzGene Web site (see below). Furthermore, if published genotype data are available from at least 4 independent case-control studies, they are subjected to random-effects meta-analyses on study-specific allelic odds ratios (ORs). Currently, SzGene includes over 1300 individual studies and showcases the results of over 150 meta-analyses. In these, more than 20 genes show nominally significant risk effects (table 2). The average allelic summary ORs are generally very modest, ie, approximately 1.2 (range: 1.06–1.34) for “risk” alleles and approximately 0.8 (range: 0.69–0.94) for “protective” alleles, compared with an OR of approximately 3–4 for a single copy of the APOE ϵ4 allele in Alzheimer disease.6 These modest effect sizes are in good agreement with those found in other large-scale studies on the genetics of complex diseases6,14,15 and have important (and well-known) implications for the design of future genetic association studies in schizophrenia because sample sizes will need to be vastly increased in order to detect or exclude ORs of 1.5 or below with sufficient confidence. For instance, to detect an allelic OR of 1.25 with 80% power at a P value of .05, sample sizes between approximately 1400 and 6000 combined cases and controls are needed for disease allele frequencies ranging from 0.50 to 0.05, respectively (based on calculations using the tools described in Purcell et al16 and Lange et al17). Sample sizes need to be increased approximately 5-fold to detect such modest effects with the same power at P values below 5 × 10−8, ie, one proposed threshold for declaring genome-wide significance13,18.
Table 2.
SzGene “Top Results” (Current on June 1, 2008)
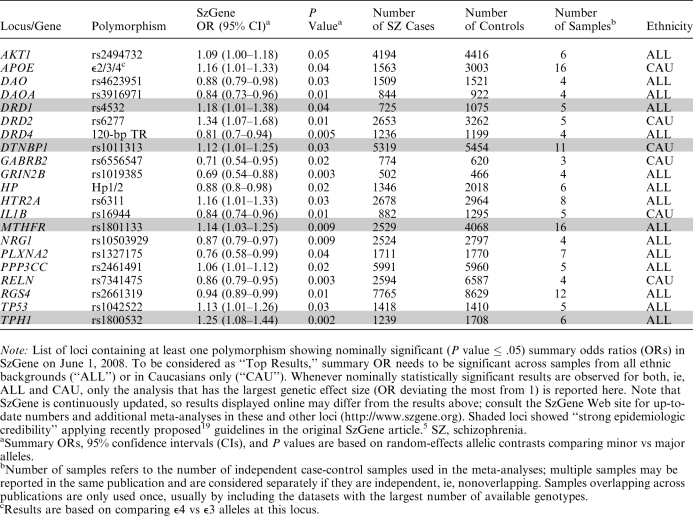 |
Fig. 1.

Simplified Flowchart of Methodology Related to Data Identification, Processing, and Analysis Used in SzGene. †Averaged on total number of publications included in SzGene for 2005–2007. ‡Current on June 1, 2008; see table 2 for details on these loci (For more details on all methods and statistical analyses see Allen et al5).
The remainder of this review will cover in more detail the methods underlying the SzGene approach, its strengths and limitations, and also how GWA studies are being included.
SzGene: Background and First Results
For the purpose of an extensive assessment of the existing genetic literature at the time, the database content was “frozen” on April 30, 2007. At that time, 1179 individual publications reporting on 3608 genetic variants (or: polymorphisms) in 516 different genes were included, after screening approximately 15 000 titles and abstracts (note that in the 12 months following the freeze approximately 150 additional articles were published and have been added to the database). From these numbers, it becomes clear that the underlying “engine” of SzGene is the ongoing search for publications eligible for inclusion. Studies are considered eligible if (a) they represent genetic association studies, (b) they are published in a peer-reviewed journal, and (c) they are published in English. Clearly, these criteria are arbitrary and nonexhaustive and therefore may lead to bias in the resulting meta-analyses (eg, because data presented at scientific meetings or published in a language other than English are ignored). However, to the degree that it can be detected, we found no evidence that this strategy, which could lead to the exclusion of a disproportionate amount of “negative” data, resulted in any significant bias, at least not in the majority of meta-analyses with a nominally significant outcome. For more details on the methods related to the literature searches, data management, and statistical procedures, please consult the original SzGene article5 and the database's Web site (http://www.szgene.org).
Of the 3608 included polymorphisms, 118 variants in 52 genes had sufficient data to warrant meta-analysis (ie, genotype data available from at least 4 independent case-control samples) on April 30, 2007. On average, these meta-analyses were based on approximately 3600 combined cases and controls, originating from 6 independent datasets. While 24 of the meta-analyzed variants in 16 genes showed nominally significant (P ≤ .05) summary ORs, the vast majority of polymorphisms yielded no significant association with schizophrenia risk. Interestingly, the average sample size of the “negative” meta-analyses was not significantly different from that of genes with “positive” outcomes. However, in only about half of all meta-analyses was the combined sample size sufficient to detect an allelic OR of approximately 1.25 with 80% power (see above), which could have affected both positive and negative results. Overall, our systematic approach applied to the schizophrenia genetics literature nearly doubled the number of meta-analyses published in the field at that time, including the detection of significant effects in 7 genes (DAO, DRD1, DTNBP1, GABRB2, HP, PLXNA2, and TP53) that were not meta-analyzed prior to April 30, 2007.
In addition to the actual meta-analyses, SzGene also systematically examines potential sources of bias for all nominally significant results, using recently proposed guidelines19 by the Human Genome Epidemiology Network (HuGENet). The criteria formulated in these guidelines take into consideration a whole range of possible biases (such as small sample size, heterogeneity across study-specific ORs, small-study bias, and first-study bias, etc) with the aim to appraise its “epidemiologic credibility” using a 3-tiered grading scheme (grade A signifies “strong credibility,” B “moderate,” and C “weak”). Application of these guidelines to the data included in the April 30, 2007, datafreeze suggested that variants in 4 genes obtained an overall “A grade,” implying that their meta-analysis results showed a strong degree of epidemiological credibility (ie, DRD1, DTNBP1, MTHFR, TPH1).5 Thus, at least based on the HuGENet criteria, these genes currently appear as the best contenders to harbor genuine susceptibility alleles within the whole domain of genetic epidemiology in schizophrenia (see below for important limitations of this interpretation).
Inclusion of GWA Studies
The sheer scale of GWA studies makes their systematic inclusion in SzGene a daunting and computationally demanding task. We have devised a 3-stage protocol that captures the most relevant genetic information from GWA studies without the need to display each data point or result online. Stage I focuses on the inclusion and display of genes and polymorphisms highlighted (or “featured”) by the authors of a GWA study (table 1). Usually, these loci are emphasized in the original publication because they show some degree of genetic association after completion of all analyses, eg, correction for multiple comparisons and/or replication in multiple independent datasets. Stage I data represent the core findings of each GWA study, and their inclusion is relatively straightforward because the genotype distributions of these genes/markers are usually readily available in the original publication. Stage II makes use of “nonfeatured” genotype distributions (provided the complete GWA data are publicly available), ie, of polymorphisms not believed to be associated with schizophrenia in the original publications. Practically, this entails identifying all markers not covered in Stage I for overlap with polymorphisms already included in SzGene and recalculation of the meta-analyses. Note that the failure to identify previously proposed candidate gene effects within the setting of a GWA screen does not necessarily preclude such effects from existing. Rather, this scenario could reflect insufficient power due to small sample size. For instance, the combined (cases and controls) sample sizes used for GWA screening across the 5 currently published studies in schizophrenia ranged from 322 to 3431 (table 1). Thus, none of these studies came even close to the minimum sample sizes needed (approximately 7000 combined cases and controls, see above) to detect ORs of approximately 1.25 with 80% power at P values ≤5 × 10−8. Stage III entails systematic meta-analyses for all variants overlapping across independent samples, provided that at least 4 complete GWA datasets have been made publicly available. Only variants showing genome-wide significant summary ORs will be displayed on the SzGene Web site. The threshold for declaring statistical significance in this context will be more stringent than for meta-analyses of individual candidate polymorphisms, due to the large number of tests performed. Procedures for implementing this stage and the definition of appropriate threshold criteria are currently being developed by our group and by others.20
At the day of this writing, only Stage I of the above protocol has been implemented in SzGene, due to the lack of publicly available genotype data (an application for access to data from Sullivan et al12 is pending). Of the 21 genes currently showing nominally significant association in SzGene, 2 (PLXNA2 and RELN) were implicated by GWA analysis (table 1). Note, however, that the putative association between certain schizophrenia endophenotypes and variants in RELN was originally described in a conventional candidate gene study21 and merely “confirmed” by the subsequent GWA screen.
Strengths and Limitations of the SzGene Approach
The strengths of SzGene are obvious: assuming that the literature searches, inclusion criteria, data management, and data analysis procedures are working flawlessly and actually provide a correct and exhaustive account of the available literature, SzGene is the single most comprehensive and sophisticated resource for the status of genetics research in schizophrenia available to date (but see below). In our original datafreeze,5 we could show that literature searches in SzGene outperformed those of several other literature/genetics databases and that the results of our meta-analyses were in very good agreement with those published previously in nearly 60 individual articles. Published meta-analyses, however, have one important disadvantage: by nature of their design, they run the risk of becoming outdated quickly, possibly as soon as new data from 1 or 2 additional studies are published. Provided that sufficient funding remains available, SzGene does not have this caveat. Any meta-analysis in the database can be updated literally within hours after the publication of new data. Another strength of SzGene is that it is not limited to meta-analyses on certain genes or networks of genes (eg, those that are in the same pathway or gene family) but considers all published loci simultaneously, making the comparison of results across studies, genes, pathways, chromosomal regions, etc, extremely easy. Furthermore, all loci containing at least one polymorphism nominally significant by meta-analysis are separately highlighted on the database's home page in a section called “Top Results”. Thus, consulting this section of the SzGene Web site will provide the user with a complete—and essentially real time—snapshot of the “most promising” schizophrenia candidate genes, based on the systematic evaluation of literally hundreds of individual studies and thousands of data points. As such, the “Top Results” list could help prioritize future genetic association studies (eg, for further independent replication or fine mapping) and guide functional genomics and molecular studies investigating the potential pathogenetic mechanisms underlying the putative genetic associations.
While SzGene undoubtedly represents a leap forward in managing and displaying the data gathered within the domain of schizophrenia genetics research, its overall approach, too, comes with some strings attached. First and foremost, despite our comprehensive and systematic searches of the scientific literature, we cannot exclude the possibility that some schizophrenia association studies were overlooked or entered erroneously. This can be partly alleviated with the help of database users who are explicitly encouraged to alert the curatorial team of any errors or omissions, which will be fixed as soon as possible. Other limitations include our restriction to allele contrasts in the meta-analyses (which allows no inference of the true underlying mode of inheritance and is usually less powerful than genotype-based tests), the nonconsideration of haplotype-based genotype data or imputed single-locus genotypes (possibly missing important associations), the exclusive focus on “main effects” (and the inherent inability to account for gene-gene and gene-environment interactions), and the lack of adjustment for certain covariates such as age or gender (which is impossible to do systematically without access to study-level raw data). Finally, protection from bias is particularly difficult to ensure or assess because latent bias is always possible and no test can have very high sensitivity and specificity for all types of possible biases. There are good reasons why the aforementioned problems cannot be addressed in the context of the current SzGene methodology, but covering these issues in greater detail would go beyond the scope of this review. The interested reader is encouraged to consult our orginal publication5 for a more in-depth discussion of these and related topics.
Despite space constraints, this section cannot conclude without briefly highlighting the single most important caveat of the SzGene approach, ie, that the number of “true” associations is almost certainly going to be smaller than the number of nominally significant findings listed at any time on the SzGene Web site.22,23 This has a number of causes, including multiple testing, linkage disequilibrium among associated variants, undetected publication or other reporting biases, as well as study-level technical artifacts that may have gone unnoticed or may be impossible to detect. Furthermore, most of the “positive” meta-analysis outcomes currently do not reach very high levels of statistical significance (see table 2), none even approaching the threshold needed to declare genome-wide significance, eg, a P value ≤ 5 × 10−8. While this, too, could be due to a number of factors including small effect size and insufficient power even after combining all the available data, it is important to emphasize that the possibility of a false-positive finding always exists, even for the highest ranked “Top Results.” Eventually, genuine risk effects can only be “proven” by the accumulation of sufficient unbiased and high-quality genotype data in favor of the presumed association in combination with functional genomics and biological evidence suggesting a direct biochemical involvement of the associated variant.13 Of course, such evidence can be difficult to come by. For instance, despite the umabigous role of the APOE ϵ4 allele in increasing the risk for Alzheimer disease, the precise mechanism underlying this association remains only poorly understood.24 Notwithstanding these limitations, there is good reason to believe that the variants and loci highlighted in the “Top Results” section of SzGene and related databases currently represent our best bets as to which of the hundreds of putative candidate genes might genuinely contribute to disease susceptibility and pathogenesis. As such, they probably warrant follow-up with high priority.
Conclusions
Despite intensifying efforts to unravel the genetic underpinnings of schizophrenia, successes to date have been modest at best. This situation is expected to change dramatically with the advent of novel, genome-wide analysis tools that are becoming increasingly popular25,26. For schizophrenia and other psychiatric disorders it remains to be seen whether GWA studies will live up to these expectations. In the interim, systematic bioinformatics approaches encompassing the results from both candidate gene and GWA analyses will help researchers to keep track of the myriad of genetic association findings to come. One such approach, the SzGene database, is now available, highlighting a number of promising schizophrenia loci by means of systematic meta-analysis.
As should be clear from the above discussion, SzGene explicitly does not aim to deliver the last piece in the puzzle that genetic epidemiology research is trying to solve. Rather, it attempts to provide a tool that can help researchers of many disciplines decide which piece of the puzzle to try next. In the best case scenario, it will also help to sharpen the overall picture of the genetic forces driving schizophrenia predisposition and pathogenesis. Eventually, only the concerted efforts of genetics, genomics, proteomics and clinical disciplines will give rise to new diagnostic and therapeutic targets that, hopefully in the not too distant future, will benefit the millions of patients afflicted with this debilitating disorder.
Funding
National Alliance on Research in Schizophrenia and Depression (to L.B.).
Acknowledgments
We are grateful to the Schizophrenia Research Forum for hosting SzGene on their Web site.
References
- 1.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60:1187–1192. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy JL, Farrer LA, Andreasen NC, Mayeux R, St George-Hyslop P. The genetics of adult-onset neuropsychiatric disease: complexities and conundra? Science. 2003;302:822–826. doi: 10.1126/science.1092132. [DOI] [PubMed] [Google Scholar]
- 3.Badner JA, Gershon ES. Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol Psychiatry. 2002;7:405–411. doi: 10.1038/sj.mp.4001012. [DOI] [PubMed] [Google Scholar]
- 4.Lewis CM, Levinson DF, Wise LH, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am J Hum Genet. 2003;73:34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40(7):827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- 6.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 7.Thakkinstian A, Han P, McEvoy M, et al. Systematic review and meta-analysis of the association between complement factor H Y402H polymorphisms and age-related macular degeneration. Hum Mol Genet. 2006;15:2784–2790. doi: 10.1093/hmg/ddl220. [DOI] [PubMed] [Google Scholar]
- 8.Mah S, Nelson MR, Delisi LE, et al. Identification of the semaphorin receptor PLXNA2 as a candidate for susceptibility to schizophrenia. Mol Psychiatry. 2006;11:471–478. doi: 10.1038/sj.mp.4001785. [DOI] [PubMed] [Google Scholar]
- 9.Lencz T, Morgan TV, Athanasiou M, et al. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry. 2007;12:572–580. doi: 10.1038/sj.mp.4001983. [DOI] [PubMed] [Google Scholar]
- 10.Kirov G, Zaharieva I, Georgieva L, et al. A genome-wide association study in 574 schizophrenia trios using DNA pooling. Mol Psychiatry. doi: 10.1038/mp.2008.33. March 11, 2008; doi:10.1038/mp.2008.33. [DOI] [PubMed] [Google Scholar]
- 11.Shifman S, Johannesson M, Bronstein M, et al. Genome-wide association identifies a common variant in the reelin gene that increases the risk of schizophrenia only in women. PLoS Genet. 2008;4:e28. doi: 10.1371/journal.pgen.0040028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sullivan PF, Lin D, Tzeng JY, et al. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol Psychiatry. 2008;13:570–584. doi: 10.1038/mp.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 14.Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003;33:177–182. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- 15.Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. 2001;29:306–309. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- 16.Purcell S, Cherny SS, Sham PC. Genetic power calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 17.Lange C, DeMeo D, Silverman EK, Weiss ST, Laird NM. PBAT: tools for family-based association studies. Am J Hum Genet. 2004;74:367–369. doi: 10.1086/381563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoggart CJ, Clark TG, De Iorio M, Whittaker JC, Balding DJ. Genome-wide significance for dense SNP and resequencing data. Genet Epidemiol. 2008;32:179–185. doi: 10.1002/gepi.20292. [DOI] [PubMed] [Google Scholar]
- 19.Ioannidis JP, Boffetta P, Little J, et al. Assessment of cumulative evidence on genetic associations: interim guidelines. Int J Epidemiol. 2008;37:120–132. doi: 10.1093/ije/dym159. [DOI] [PubMed] [Google Scholar]
- 20.Evangelou E, Maraganore DM, Ioannidis JP. Meta-analysis in genome-wide association datasets: strategies and application in Parkinson disease. PLoS ONE. 2007;2:e196. doi: 10.1371/journal.pone.0000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wedenoja J, Loukola A, Tuulio-Henriksson A, et al. Replication of linkage on chromosome 7q22 and association of the regional Reelin gene with working memory in schizophrenia families. Mol Psychiatry. 2008;13(7):673–684. doi: 10.1038/sj.mp.4002047. [DOI] [PubMed] [Google Scholar]
- 22.Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst. 2004;96:434–442. doi: 10.1093/jnci/djh075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ioannidis JP. Why most published research findings are false. PLoS Med. 2005;2:e124. doi: 10.1371/journal.pmed.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Craddock N, O'Donovan MC, Owen MJ. Genome-wide association studies in psychiatry: lessons from early studies of non-psychiatric and psychiatric phenotypes. Mol Psychiatry. 2008;13(7):649–653. doi: 10.1038/mp.2008.45. [DOI] [PubMed] [Google Scholar]
- 26.Sullivan PF. Schizophrenia genetics: the search for a hard lead. Curr Opin Psychiatry. 2008;21:157–160. doi: 10.1097/YCO.0b013e3282f4efde. [DOI] [PMC free article] [PubMed] [Google Scholar]