

Abstract
Type I interferon (IFN) is critical for resistance of mice to infection with vesicular stomatitis virus (VSV). Wild type (wt) VSV infection did not induce type I IFN production in vitro or in the central nervous system (CNS) of mice; however IFN-β was detected in lungs, spleen, and serum within 24 h. The M protein mutant VSV, T1026R1 (also referred to as M51R), induced type I IFN production in vitro and in the CNS, with poor expression in spleens. In addition, VSV T1026R1 was not pathogenic to mice after intranasal infection, illustrating the importance of IFN in controlling VSV replication in the CNS. Experiments with chemical sympathectomy, sRAGE, and neutralizing antibody to VSV were performed to investigate the mechanism(s) utilized for induction of peripheral IFN; neither sRAGE infusion nor chemical sympathectomy had an effect on peripheral IFN production. In contrast, administration of neutralizing antibody (Ab) readily blocked the response. Infectious VSV was transiently present in lungs and spleens at 24 h post infection. The results are consistent with VSV traffic from the olfactory neuroepithelium to peripheral lymphoid organs hematogenously or via lymphatic circulation. These results suggest that VSV replicates to high titers in the brains of mice because of the lack of IFN production in the CNS after intranasal VSV infection. In contrast, replication of VSV in peripheral organs is controlled by the production of large amounts of IFN.
Keywords: dendritic cells, iron production, viral encephalitis
Introduction
The rhabdovirus vesicular stomatitis virus (VSV) causes severe central nervous system (CNS) pathology when administered to mice intranasally (i.n.) (Huneycutt et al, 1994; Sabin and Olitsky, 1938). Immunocompetent mice exhibit high morbidity and mortality at low doses of virus, succumbing to infection between 6 and 11 days post infection (p.i.). In contrast, inoculation of immunocompetent mice with high doses of VSV by the intramuscular, subcutaneous, or intraperitoneal routes generally leads to limited viral replication and no apparent disease (Reiss and Aoki, 1994). Similarly, intravenous (i.v.) inoculation of mice with high doses of VSV leads to limited viral replication in the periphery, but can cause CNS pathology if virus gains access to the brain. One of the major factors controlling VSV replication in peripheral organs is interferon (IFN); type I IFN receptor (IFNAR1)-deficient mice succumb rapidly to i.v. infection by VSV (100% mortality within 3 to 6 days p.i.), with high viral titers in peripheral organs (Muller et al, 1994). Similarly, STAT1-deficient mice succumb very rapidly to i.v. infection with VSV, with 100% mortality occurring within 2 days p.i. and high viral titers observed in the liver (Durbin et al, 1996).
The ability of VSV to replicate in the mouse CNS to high titers while replication is controlled in the periphery suggests that either IFN is not produced in the CNS or neurons are not sensitive to IFN-mediated inhibition of VSV. Neurons are exquisitely sensitive to the antiviral effects of type I IFN, as both cultured lines and primary mouse neurons restrict VSV replication after pretreatment with IFN-β (Trottier et al, 2005). Such results suggest a lack of IFN induction in the mouse brain may contribute to the high pathogenicity of VSV in neural tissues.
The VSV M protein has been shown to play an important role in suppressing IFN induction by VSV infection of L929, HeLa, and Baby Hampster Kidney (BHK) cells in vitro (Ahmed et al, 2003; Stojdl et al, 2003). VSV T1026R1 has been reported as an IFN-inducing mutant of VSV due to a mutation in the M protein (methionine to arginine in amino acid 51; M51R) (Francoeur et al, 1980, 1987). Subsequent studies using the cloned M gene from wild-type (wt) and T1026R1 virus have shown that wt M inhibits transcription of cellular genes including IFN-β, whereas M from T1026R1 does not (Ferran and Lucas-Lenard, 1997). wt M has also been shown to be able to block nucleocytoplasmic transport, in contrast to the M51R mutant (Petersen et al, 2000), and to be associated with induction of apoptosis (Gaddy and Lyles, 2005).
Host induction of type I IFNs during innate immune responses to antigens, viral, fungal, or bacterial challenge, to single-stranded RNA (ssRNA), double-stranded (dsRNA), and CpG in DNA has been associated with the triggering of a family of receptors termed Toll-like receptors (TLRs) for their sequence similarity to Toll, which was first identified in the fruit fly (reviewed in Boehme and Compton, 2004; Conzelmann, 2005; Kawai and Akira, 2005; Yang et al, 2005). Although many cell types are able to express some TLRs, research has indicated that the plasmacytoid dendritic cell (pDC) maintains a central role in the TLR-triggered IFN response (reviewed in Malmgaard, 2005). With viral challenges, type 1 IFN not only regulates host responses, but also can suppress viral replication. ssRNA viruses, like VSV, have been shown to elicit pDC activation through TLR7 and TLR8 (Diebold et al, 2004; Heil et al, 2004; Lund et al, 2004; Malmgaard, 2005), which signal through the MyD88 pathway (reviewed in Crozat and Beutler, 2004). A role for TLR 4 in VSV activation of pDCs has been suggested (Georgel et al, 2007; Jiang et al, 2005), as has the requirement for MyD88 signaling in VSV infection of the neuroepithelium (Zhou et al, 2007). In the case of VSV, activation of IFN production by pDCs has been demonstrated in vitro and in vivo following peripheral inoculation (Barchet et al, 2002; Lund et al, 2004).
The sympathetic nervous system (SNS) (Blalock, 1989; Madden, 2003; Weihe et al, 1991) and the hypothalamic-pituitary-adrenal axis (reviewed by Bailey et al, 2003; McEwen et al, 1997; Silverman et al, 2005) regulate many peripheral immune functions in health and disease. Thus, VSV infection of the CNS might activate host responses through the SNS. A common way to evaluate the contribution of the SNS to host immune responses is by chemical sympathectomy with 6-hydroxydopamine (6-OHDA). In excitotoxicity or cerebrovascular ischemic events, traumatic insults to the brain, the host is alerted by a number of pathways, including the release of S100B by astrocytes (Herrmann and Ehrenreich, 2003). The receptor for S100B (and also for many other proinflammatory molecules including High Mobility Group B-1 (HMGB-1). (Chen et al, 2004) is RAGE (Hofmann et al, 1999), originally characterized as the receptor for advanced glycation endproducts in hyperglycemia (Neeper et al, 1992). Schmidt's group has elucidated many proinflammatory pathways and diseases in which RAGE contributes by the use of the soluble receptor, sRAGE (Lalla et al, 2000; Liliensiek et al, 2004; Yan et al, 2003).
In this report, we analyze the ability of VSV to induce IFN synthesis in the periphery and CNS. Infection of NB41A3 neuroblastoma cells by VSV did not result in IFN production, whereas infection by Newcastle disease virus (NDV) did, suggesting that neuronal cells were capable of producing type I IFN, but not in response to wt VSV infection. No IFN mRNA could be detected in the brains of VSV-infected mice as late as 6 days p.i. In contrast, i.n. infection by VSV induces high levels of type I IFN mRNA in spleens and lungs, and protein detected by enzyme-linked immunosorbent assay (ELISA) and bioassay in the serum of mice 24 h p.i. To determine the mechanism of IFN induction in the spleen, we examined two well-characterized pathways of inflammatory signaling between the CNS and the immune system, the SNS and RAGE; neither was found to contribute to the production of IFN by the spleen. Infectious VSV was detectable in lungs and spleens of VSV-infected mice at 24 h p.i., but little was found in serum. When we used a virus that was mutated in the M protein (T1026R1), IFN mRNA was induced in vitro and also in the CNS; T1026R1 virus infection of mice showed low pathogenicity, suggesting that the viral M protein is critical for evasion of host responses. IFN production required hematogenous or lymphatic transfer of infectious virus from the nasal neuroepithelium, because it could be prevented by passive immunization with neutralizing antibody (Ab). The results suggest that the M protein of VSV blocks IFN synthesis in neurons and other infected CNS cells, whereas peripheral IFN production is not antagonized by the viral M protein. The failure to elicit IFN in the CNS may be attributable to the lack of expression of TLR7 by neurons, and contributes to the disease pathogenesis (Trottier et al, in preparation).
Results
VSV does not induce type I IFN in infected neuroblastoma cells
NB41A3 cells, a neuroblastoma cell line, have been shown to have many properties of neurons, including the ability to form neurotubules and neurofilaments and in the storage and release of catecholamines (Breakfield et al, 1975; Schubert et al, 1969); they constitutively express neuronal nitric oxide synthase (NOS-1), IFN-γ eceptor (IFN-γ R), interleukin-12 receptor (IL-12R), and the N-methyl-d-aspartate (NMDA) receptor (Bi and Reiss, 1995; Ireland and Reiss, 2004; Komatsu et al, 1996). The effects of type I and II IFN on these cells have also been studied; the inhibitory effects of these cytokines on VSV replication are similar in these cells and in primary murine neurons (Chesler et al, 2003; Trottier et al, 2005).
To determine if NB41A3 cells were able to synthesize type I IFN in response to virus infection, NB41A3 cells were infected with either VSV or NDV at various multiplicities of infection (m.o.i.). At 12 and 24 h p.i., cell supernatants were harvested and assayed for type I IFN by bioassay. IFN bioassays take advantage of the stability of type I IFN to both ultraviolet (UV) irradiation and to acid pH. Test samples were treated with HCl to pH 2 and then neutralized, or (in the case of small volumes) with UV light. Treated samples were then applied to an indicator cell line, in this case NB41A3 cells. If samples contained IFN, the cells were able to enter an antiviral state, and thus resist subsequent challenge with virus. Thus results of the IFN bioassay were expressed as plaque-forming units (PFU)/ml or log PFU/ml. In the absence of IFN, the sample would yield a high viral titer, whereas if IFN were present in the serum or cell culture supernatant tested, the indicator cell line would be protected from infection, and a low viral titer would be apparent. Supernatants from NDV-infected NB41A3 cells infected at a m.o.i. = 10 had antiviral activity in IFN bioassays when diluted up to 20-fold (Figure 1). At lower moi, IFN production was at or below the level of detection of the assay (∼1 unit/ml; not shown) (Figure 1). In contrast, VSV infection of NB41A3 cells did not result in detectable IFN production at 12 and 24 h p.i. (Figure 1). VSV infection of NB41A3 cells resulted in death of all cells within 12 to 16 h (not shown), indicating that further incubation of these cells would not increase the yield of IFN production.
Figure 1.
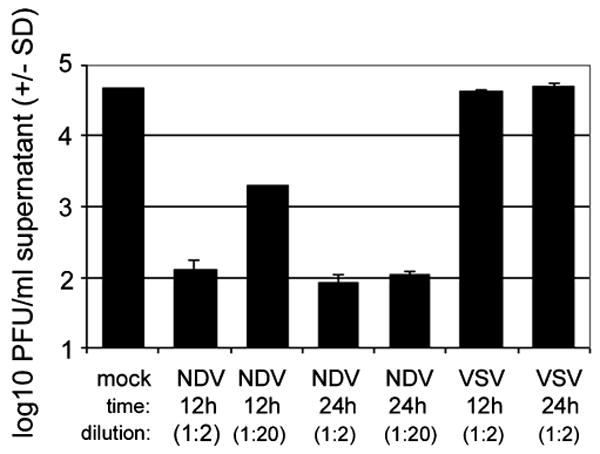
NDV but not VSV induces type I IFN production from neuroblastoma cells in vitro. Approximately 2 × 105 NB41A3 cells were infected with NDV or VSV (m.o.i. = 10) or mock-infected with medium for 12 or 24 h. Supernatants were assayed for the presence of type I IFN bioactivity by IFN bioassay. The presence of IFN in supernatants was indicated by inhibition of VSV replication, shown as a reduction of viral titers in supernatants. Dilution represents fold dilution (from 1 ml) of supernatant added to NB41A3 cells.
VSV infection of neuroblastoma cells activates IRF-3 signaling pathway
To determine whether neuroblastoma cells sense VSV infection, the ability of VSV to induce IFN regulatory factor-3 (IRF-3) nuclear localization was tested. NB41A3 cells were infected for 4 h with wt VSV and assayed by immunofluorescence. Infection with VSV induced nuclear localization of IRF-3 (Figure 2), indicating that VSV infection was recognized by neuroblastoma cells, leading to phosphorylation and nuclear translocation of IRF-3. Both defective-interfering VSV particles (DI-VSV) and UV-inactivated VSV (UV-VSV) also promoted IRF-3 nuclear localization after infection of NB41A3 cells (not shown). We interpret these experiments to be compatible with an early cellular event during VSV infection leading to activation of IRF-3 signaling; however, IFN-β production is blocked after nuclear translocation of IRF-3 in neuronal cells.
Figure 2.
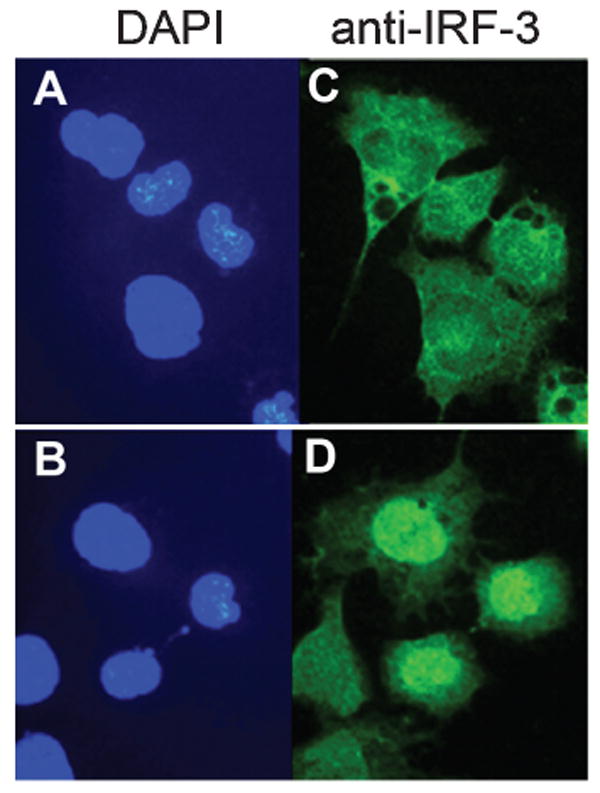
VSV infection of neuroblastoma cells results in nuclear localization of IRF-3. Approximately 2 × 105 NB41A3 cells were mock-infected (A, C) or infected with VSV (B, D) at an m.o.i. of 10. After 4 h, cells were fixed and nuclear localization of IRF-3 was determined using immunofluorescence (C, D). Cells were stained with DAPI (A, B) to visualize nuclei.
VSV infection does not induce IFN mRNA synthesis in mouse brains
VSV infection of mice after i.n. inoculation results in rapid spread to the brain and resulting encephalitis. To determine whether i.n. VSV infection induced a type I IFN response in mouse brains, male BALB/c mice were infected intranasally with VSV. Brains from VSV-infected mice were harvested on days 1, 3, and 6 p.i. and analyzed for the presence of type I IFN mRNA. No IFN-β mRNA was detected in VSV-infected mouse brains (Figure 3A), even at times when mice were exhibiting signs of morbidity (weight loss, lack of grooming, reduced activity, and hind-limb paralysis in some mice) due to CNS infection (day 3) and when mice first began to succumb to disease (day 6). Similar results were observed with IFN-α mRNA (not shown). The results are consistent with a failure of CNS parenchymal tissue to synthesize IFN mRNA.
Figure 3.
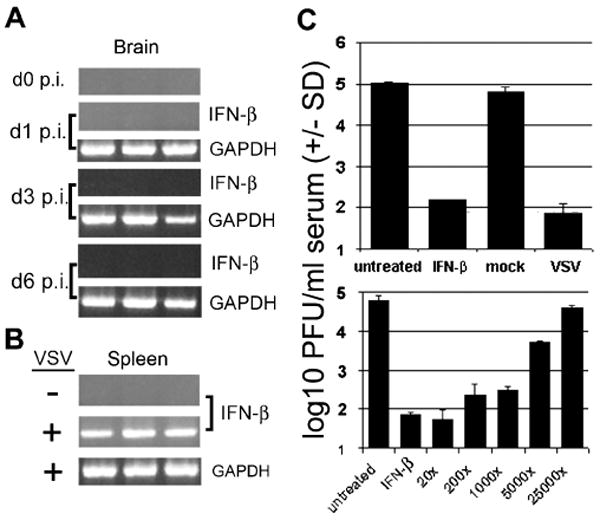
Intranasal infection of mice with VSV induces no detectable IFN mRNA expression in the brain but high levels of type I IFN mRNA in the spleen and IFN bioactivity in serum. Mice were mock-infected (receiving PBS) or infected with 1 × 104 PFU VSV i.n. and sacrificed at various times p.i. Total RNA was isolated from brains and subjected to RT-PCR to detect IFN-β mRNA at days 1, 3, or 6 p.i. (A). RT-PCR to detect IFN-β mRNA was also performed on total RNA from mock- or VSV-infected mouse spleens taken at 24 h p.i. (B). Mouse sera collected from mock- and VSV-infected mice was subjected to IFN bioassay to detect the presence of type I IFN (C, top panel). Serial dilutions of serum were analyzed to semiquantify IFN levels in VSV-infected mouse sera (C, bottom panel). IFN controls in bioassays were used at a concentration of 100 units/ml. GAPDH served as housekeeping gene control for RT-PCR reactions.
Intranasal VSV infection induces splenic and serum IFN
To analyze the peripheral IFN response to VSV infection, mice were infected i.n. with VSV and spleens and serum harvested at 24 h p.i. reverse transcriptase–polymerase chain reaction (RT-PCR) assays performed on spleen total RNA revealed the presence of IFN-β mRNA at 24 h p.i. (Figure 3B). Similar results were observed for IFN-α mRNA (not shown). Analysis of mouse serum by bioassay revealed that IFN was present in blood at 24 h p.i. (Figure 3C, top panel); quantification of IFN in serum showed that IFN bioactivity corresponded to a concentration of approximately 200 to 1000 units per ml of serum (Figure 3C, bottom panel) (Trottier et al, 2005). In contrast, mock-infected animals showed no detectable expression of IFN mRNA in the spleen and no IFN bioactivity in serum.
Mutant VSV replicates and induces IFN in neuronal cells in vitro and in vivo
VSV T1026R1 has been reported as an IFN-inducing mutant of VSV due to the M51R mutation in the M protein (Francoeur et al, 1980, 1987). To test the ability of this virus to replicate in neuronal cells, NB41A3 cells were infected with wt VSV or VSV T1026R1 at m.o.i. = 0.1. At 8, 24, 32, and 36 h p.i., supernatants were harvested and subjected to plaque assay to determine viral titers. VSV T1026R1 replicated to high titers in NB41A3 cells, similar to that observed for wild type VSV (Figure 4A). Cells infected with wt virus appeared apoptotic by 14 to 18 h p.i., whereas cells infected with T1026R1 virus showed significant cytopathic effect at 24 h p.i. and were dead by 28 h p.i. (data not shown). These results indicate that VSV T1026R1 can replicate to high titers in neuronal cells in vitro.
Figure 4.
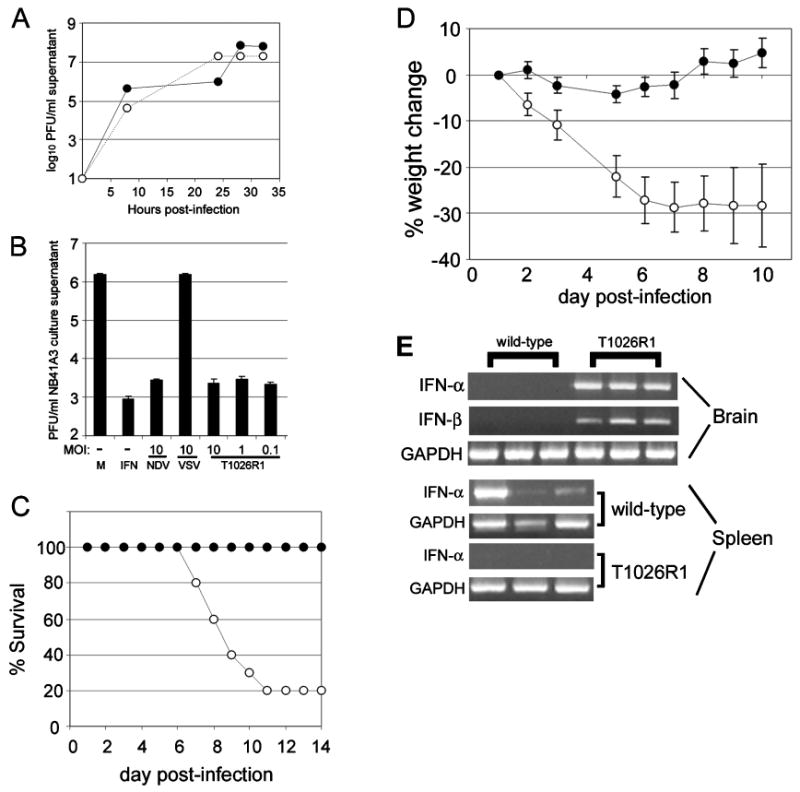
Pathogenicity of mutant VSV inversely correlates with ability to induce type I IFN. (A) NB41A3 cells were infected with wild type VSV (open circles) or VSV T1026R1 (filled circles) and supernatants were analyzed for viral titer by plaque assay at various times post-infection. (B) Approximately 2 × 105 NB41A3 cells were also mock-infected (M), or infected with NDV, wild-type VSV, or VSV T1026R1 at various m.o.i. for 24 h. Supernatants were subjected to IFN bioassay to determine presence of type I IFN. For in vivo studies, mice (n = 10 per group) were infected intranasally with VSV-Indiana (100 PFU) or VSV T1026R1 (4 * 106 PFU) and monitored for weight loss (D) and mortality (C). (E) VSV T1026R1 was apathogenic in mice infected i.n. Some animals were sacrificed at 24 h p.i. and tissues were harvested. RT-PCR was performed on total RNA from brains (top panel) and spleens (bottom panel) to detect IFN-β and -α mRNA in wt VSV and VSV T1026R1 infected mice. Data shown are from three individual mice, representative of two experiments.
To determine the ability of mutant VSV to induce IFN production by neuronal cells, NB41A3 cells were infected with VSV, NDV, or VSV T1026R1 for 24 h at various m.o.i. and cell supernatants were assayed for the presence of IFN (Figure 4B). NDV induced type I IFN production from NB41A3 cells at an m.o.i. = 10, while wt VSV did not induce detectable IFN bioactivity. Mutant VSV T1026R1 induced type I IFN bioactivity, even at m.o.i. as low as 0.1 (Figure 4B). These results indicated that mutant VSV T1026R1 was able to induce type I IFN production by neuronal cells and are consistent with wt VSV M protein blockade of cellular transcription and nuclear transport, as has been described for non-neuronal host cells (Ahmed et al, 2003; Ferran and Lucas-Lenard, 1997; Petersen et al, 2000; Stojdl et al, 2003).
To investigate the pathogenicity of and IFN induction by VSV T1026R1 in vivo, groups of 10 male BALB/c mice were infected i.n. with wt or T1026R1 VSV. wt VSV induced severe morbidity (Figure 4D) and 80% mortality (Figure 4C) upon intranasal infection of mice with 100 PFU. In contrast, VSV T1026R1 induced virtually no morbidity and no mortality after inoculation with 4 × 106 PFU (Figures 4D and 3C). Intranasal VSV T1026R1 infection induced both IFN-α and IFN-β mRNA production in the brain at 24 h p.i., in contrast to wt VSV (Figure 4E, top panel). These results suggest that IFN induction in the brain prevents VSV induced encephalitis. Interestingly, no detectable IFN mRNA was detected in spleens of VSVT1026R1 infected mice, despite the high dose given (Figure 4E, bottom panel). ELISA analysis of IFN-β in serum indicated that T1026R1 infection induced 170.8 ± 5.71 pg/ml IFN-β, whereas wt VSV elicited 1283 ± 21.6 pg/ml at 24 h p.i., or a 7.5-fold difference. Thus, the M protein mutation permitted IFN production in neurons in vitro and in vivo.
How does the periphery know that there is an intranasal infection?
In order to determine how the spleen “learned” that the nasal neuroepithelium and, after 6 to12 h, the olfactory bulb were infected, we investigated the possibility that the SNS was involved. Every lymphoid organ is innervated by sympathetic neurons (Blalock, 1989; Madden, 2003; Weihe et al, 1991). Mice were treated with 6-OHDA to establish a chemical sympathectomy and then challenged with VSV i.n. Serum was harvested at 24 h p.i. and presence of type I IFN was determined by bioassay. There was no difference between control and 6-OHDA–treated mice in the level of type I IFN present in serum (Figure 5A), or in the level of type I IFN mRNA detected in spleens from these mice (data not shown), indicating that the spleen was not alerted by the SNS.
Figure 5.
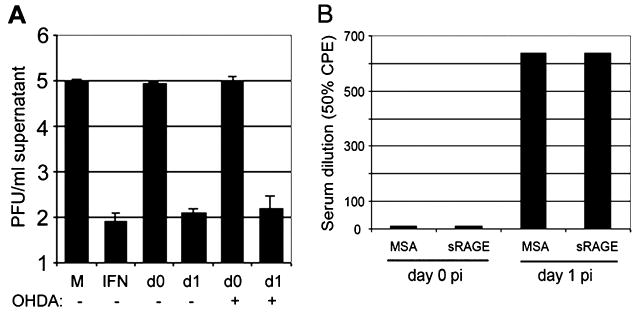
Chemical sympathectomy and infusion of sRAGE. (A) Three male BALB/c mice were treated daily with 100 mg/kg/day 6-OHDA and two mice were injected with PBS for 10 days. One control mouse and 2 sympathectomized mice were infected i.n. with 1 × 104 PFU VSV, the remaining mice were mock-infected with PBS. Spleens were assayed for mRNA specific for IFN-βsing IFN bioassay. “M,” referring to mock-treated NB41A3 cells and “IFN,” referring to IFN-β (100 U/ml)-treated NB41A3 cells, are IFN bioassay controls. “d0” refers to mock-infected mouse serum treated NB41A3 cells, and “d1” refers to VSV-infected mouse serum (taken at 24 h p.i.)-treated NB41A3 cells. There was no influence of sympathetic innervation on the IFN-β response. (B) sRAGE, the soluble form of the receptor for S100B, was infused into mice to determine if this proinflammatory pathway contributed to the IFN-β response. Mice were injected with 100 μg mouse serum albumin (MSA) in saline or 100 μg sRAGE immediately prior to i.n. infection with 1 × 104 PFU VSV. At 24 h p.i. blood was obtained from individuals by cardiac puncture, and then serum was analyzed for type I IFN by bioassay. There was no influence of RAGE on the IFN-β response.
If the infection in the nasal neuroepithelium and olfactory bulb were perceived of as traumatic by astrocytes, it was possible that S100B would be released (Herrmann and Ehrenreich, 2003). Previous studies in our laboratory had indicated that glial fibrillary acidic protein (GFAP) is readily expressed by astrocytes in the olfactory bulb as early as when viral antigens are first detected in the CNS (Barna et al, 1996b), which would be consistent with astrocytic responsiveness to the viral infection. To assess whether S100B was released and could activate splenic macrophages or plasmacytoid dendritic cells through the receptor, the soluble receptor, sRAGE, was obtained from Ann-Marie Schmidt (Columbia University) and administered to mice which were infected with VSV i.n. As shown in Figure 5B, there was no difference between controls (albumin-treated) and sRAGE-treated mice in the production of splenic IFN-βRNA.
Intranasal VSV-infected mice produce IFN mRNA in the lung
Another possibility for how i.n. infection of mice leads to peripheral IFN is via replication of VSV in the lungs. With delivery of large volumes of inoculum, and high titers of virus, VSV can be inhaled into the lung; however, we use 5 μl, a volume essentially restricted to the nasal turbinates. Alternatively, infection may lead to enhanced secretion of fluid by Bowman's glands in the neuroepithelial tissue, which could result in virus being carried into the lower respiratory tract. RT-PCR analysis of lung total RNA revealed the presence of type I IFN mRNA in lungs of infected animals at day 1 p.i. (Figure 6A), suggesting that VSV gains access to the lungs after intranasal administration.
Figure 6.
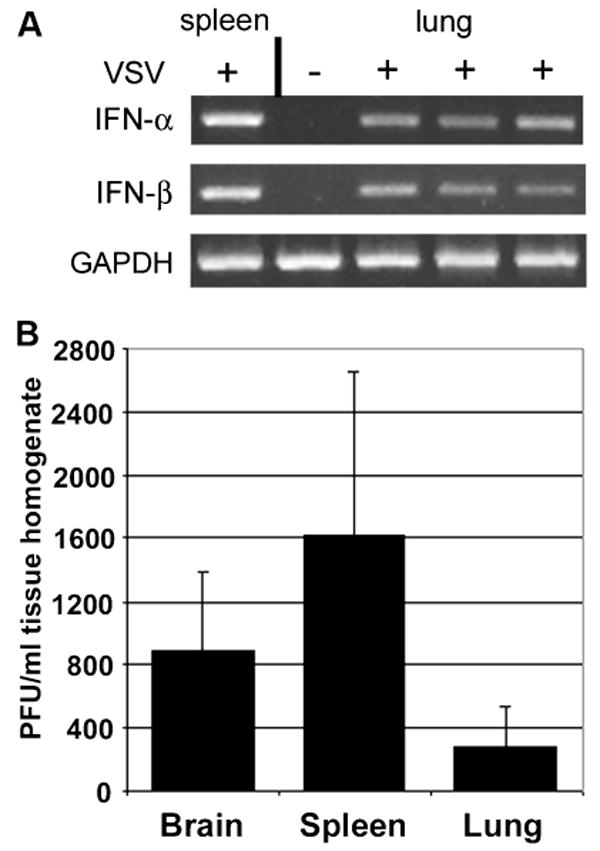
Intrnasal infection of mice with wt VSV induces type I IFN expression in mouse lungs and results in replication of VSV in mouse brains, spleens and lungs. Mice were infected i.n. with wt VSV (1 × 104 PFU) or mock-infected and sacrificed 24 h p.i. RT-PCR was used to detect IFN-α and IFN-β mRNA expression in the lungs of 3 infected and 1 mock-infected mice (A). GAPDH was used as housekeeping gene control. Homogenates from mouse brains, spleens and lungs taken at day 1 p.i. were subjected to plaque assays to detect the presence of VSV in these tissues (B; n = 3 per group).
VSV replicates in lung and spleen at 24 h p.i.
To determine if VSV is inducing peripheral IFN synthesis by infecting peripheral tissues, male BALB/c mice were infected i.n. with 1 × 104 PFU VSV and brains, lungs, spleens, and serum were harvested. Tissues homogenates were subjected to plaque assays to determine the presence of infectious VSV. As expected, infectious VSV was detectable in the brain at 24 h p.i. both by plaque assay and by RT-PCR for VSV M RNA (Figures 6B, 7A). VSV was also found in the lungs, albeit at lower levels (Figure 6B), indicating that i.n. infection leads to infection of the respiratory tract. Surprisingly, at 24 h post i.n. application, VSV was found in mouse spleens, at titers similar to that observed in the brain (Figure 6B). Virus was rapidly cleared from the spleen (data not shown). These results suggest that viral replication in peripheral organs leads to induction of IFN. In keeping with this finding, neither UV-VSV nor DI-VSV induced IFN production in the spleen or serum upon i.n. administration (data not shown).
Figure 7.
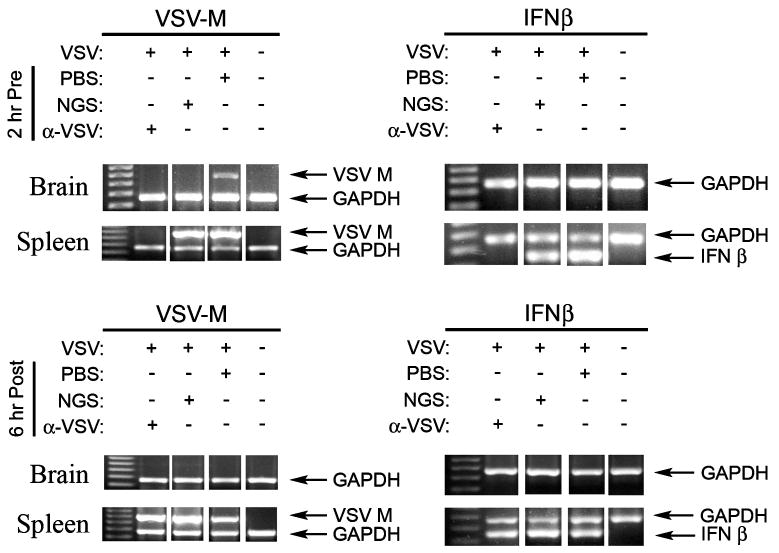
Administration of neutralizing antibody to VSV prevents splenic IFN-β mRNA production. Anti-VSV polyclonal Ab was administered i.p. either 2 h prior to infection (top panels) or 6 h after i.n. application of 1 × 104 PFU virus (bottom panels) to groups of three BALB/c mice. Controls included i.n. mock-infection with PBS and i.p. injection of normal goat serum. mRNA was harvested at 24 h. p.i. from brains or spleens and RT-PCR was performed for amplification of VSV M protein, IFN-β or GAPDH message. Where viral gene expression was observed, IFN-β mRNA was produced. Anti-VSV Ab neutralized virus and prevented splenic mRNA production if present before infection.
How does VSV get from the neuroepithelium to the spleen?
Although it is possible that VSV is passively carried by fluid in the nasal turbinates to the lung, the mechanism of transport of infectious virus from the nasal neuroepithlium to the spleen was investigated. One possibility we evaluated was the hematogenous route or via lymphatic drainage. If VSV were cell associated, it would be inaccessible to neutralizing Ab. To determine if free (that is, cell-free) virus were transmitted in either plasma or lymphatic fluid, groups of BALB/c mice were injected intraperitoneally (i.p.) with neutralizing sheep anti-VSV Ab or normal sheep serum either 2 h before the i.n. inoculation of VSV or 6 h thereafter. Spleens were harvested at 24 h p.i. and assessed for expression of VSV M and IFN-β mRNA. As shown in Figure 7, prior administration of anti-VSV Ab prevented infection of the spleen and prevented the expression of IFN-β mRNA at 24 h p.i. However, delay of 6 h p.i. prior to treatment with Ab was sufficient to permit a measurable host IFN response. VSV mRNA was not found in all brain mRNA samples tested, which we attribute to the large amount of RNA harvested from whole brains, compared to the low level of viral infection, localized to the olfactory bulb, at 24 h p.i. In more sensitive assays, such as viral replication, infectious virus could be detected by plaque assay in all samples (data not shown). We interpret these data to be consistent with circulation of VSV by blood or lymph from the neuroepithelium almost immediately after initial inoculation of the virus.
Discussion
Previous studies have revealed the critical importance of the type I IFN pathway in control of VSV replication in mice. IFNAR1 −/− mice and STAT1 −/− mice are highly susceptible to lethal VSV infection, showing high viral titers in organs not normally infected productively in wt mice (Durbin et al, 1996; Muller et al, 1994). Similarly, dsRNA–dependent protein kinase (PKR) −/− mice were shown to be highly susceptible to lethal infection with VSV administered i.n. (Durbin et al, 2002; Stojdl et al, 2000). Thus, pathways involving type I IFN are crucial for control of VSV in peripheral organs. In contrast, wt mice are highly susceptible to viral encephalitis following i.n. inoculation, which allows VSV efficient access to the brain through olfactory receptor neurons (Plakhov et al, 1995b). In this study we tested the hypothesis that IFN rapidly controls peripheral infections but fails to suppress VSV infection of the CNS, and investigated the mechanisms that contribute to this innate immune failure.
Of note, Stojdl et al (2000) showed low mortality in wild-type BALB/c mice infected i.n. with VSV. These results conflict with the severe mortality observed in wt VSV infected mice in our study (see Figure 4). The discrepancy in the susceptibility of BALB/c mice to i.n. VSV infection can be attributed in part to the age of the mice used: 5 to 7 weeks of age in this work and 8 to 10 weeks of age in Durbin et al (2002). We have found that mice quickly lose susceptibility to VSV i.n. infection at ages greater than 8 weeks (unpublished results) (Sabin and Olitsky, 1938). In addition, Durbin et al used female mice (2002), which are less susceptible than male mice to VSV-induced encephalitis (Barna et al, 1996a).
We have shown previously that VSV replication in neurons is strongly inhibited by type I IFN in vitro (Trottier et al, 2005), suggesting that if IFN was produced in the CNS in response to VSV infection, neurons could be protected from VSV infection. In this study we investigated that possibility, showing that type I IFN was detected in mouse spleens, lungs, and serum of i.n. infected mice at 24 h p.i., but no IFN mRNA was found in brain tissue homogenates as late as day 6 p.i (Figure 3). Similarly, VSV infection of neuronal cells in vitro did not induce type I IFN synthesis (Figure 1), an observation that has been made with VSV infection of a number of cell lines of different tissue origin in past studies (Marcus et al, 1998). Importantly, VSV infection of neuronal cells induced the nuclear translocation of IRF-3, indicating that these cells sensed VSV infection and activated IFN signaling pathways. The wt VSV M protein blocks nucleocytoplasmic transport, thereby inhibiting IFN production from infected cells (Ahmed et al, 2003; Stojdl et al, 2003). In contrast, neuronal cells were able to produce type I IFN in response to infection by NDV (Figure 1) and the M protein mutant VSV T1026R1 infection in vitro (Figure 5B), indicating that neurons can respond to and produce type I IFN in response to viral infection, and when VSV M is inactivated, produce IFN in response to VSV infection. The mutant VSV T1026R1 also replicated to high levels in neuronal cells indicating that reduced replicative capacity of T1026R1 was not the reason these cells were able to produce IFN (Figure 4A). Together, the results suggest that wt VSV is able to replicate to high levels in the CNS of mice because of a lack of local induction of type I IFN. The lack of IFN induction is likely caused by the ability of VSV to block IFN production by infected cells. This point is emphasized by IFN-inducing mutant VSV T1026R1, which induces strong IFN mRNA expression in the CNS at 24 h p.i., and causes no apparent disease in mice (Figure 4C and D). Thus, it appears that blockade of IFN production by infected cells is critical for VSV replication in the CNS.
Astrocytes respond to i.n. VSV infection as early as 24 h by the production of GFAP and NOS-3 (Barna et al, 1996b). Similarly, Campbell and colleagues have observed that the liver rapidly responds to CNS injury, triggering leukocyte recruitment and chemokine production (Campbell et al, 2005). To determine if there were a contribution of either S100B release by astrocytes reacting to infection of adjacent neurons, or the SNS, to the induction of the peripheral IFN in response to i.n. VSV infection, we administered the soluble receptor for S100B, sRAGE, and performed a chemical sympathectomy, respectively. Neither treatment abrogated the production of IFN in the spleen or serum (Figure 5A and B).
Examination of lung and spleen for infectious VSV or VSV RNA confirmed that virus had rapidly entered these organs and replicated (Figures 6 and 7). To determine whether the virus traveled to the spleen in a cell-associated or free form, mice were injected with neuralizing Ab to VSV and the presence of VSV RNA and IFN-β mRNA was determined at 24 h p.i. Prior administration of Ab prevented infection from spreading and abrogated the IFN response of the spleen, but if Ab treatments were delayed 6 h, a modest response was observed (Figure 7). These experiments indicated that virus rapidly spreads free in plasma or lymphatic fluid to the spleen. VSV replication in peripheral organs after i.n. infection has been reported (Publicover et al, 2006; Ramsburg et al, 2005); however, those studies inoculated mice with much higher titers and in a larger volume than in our work, which can have dramatic effects on the ability of viruses to replicate in the lung (Yetter et al, 1980). Despite the small dose of virus given in our work (as low as 100 PFU in 5 μl), VSV was still able to productively infect peripheral tissues.
The relatively high VSV titer in mouse spleens at 24 h p.i. suggests that transient viral replication in the spleen leads to IFN induction. Indeed, i.n. application of UV-inactivated VSV does not induce peripheral IFN production indicating a need for viral replication. Thus, it appears that VSV gains access to either blood or lymphatic circulation and then travels to the lung and spleen. Analysis of mouse blood by plaque assay has shown no evidence of infectious VSV after our inoculum with low dose (data not shown); however, VSV in serum has been described after mice were infected with higher titers (Publicover et al, 2005). Administration of neutralizing Ab to VSV 2 h before, but not 6 h after, i.n. infection abrogated the IFN response in the spleen at 24 h p.i; this was also associated with inhibition of infection of the spleen when Ab was present before i.n. application of virus (Figure 6).
Although the mutant virus T1026R1 replicated in the brains of mice at 24 h post i.n. infection, no virus was detected in the lung or spleen. In spite of this, 4 of 4 mice infected with T1026R1 virus produced detectable amounts of IFN-β in blood. The ELISA titers of IFN-β were substantially lower, however, than those obtained in mice administered wt VSV. In contrast to mice infected with wt VSV, IFN-β was found in the brains of T1026R1-infected mice. It is likely that the rapid production of IFN by this mutant virus profoundly limited its replication and spread in vivo, by eliciting a protective antiviral state in adjoining cells.
The lack of IFN production in the CNS after VSV infection does not appear to be a universal phenomenon with viruses. Type I IFN is strongly induced, early, in brains of MHV-JHM infected mice, and IFN production was sustained for at least 7 days (Rempel et al, 2004). Similarly, the GDVII strain of Theiler's murine encephalomyelitis virus (TMEV) induces type I IFN in the CNS, and this IFN is produced by neurons (Delhaye et al, 2006). Interestingly, both of these viruses cause severe, acute encephalitis similar to that observed with VSV. In contrast, studies with lymphocytic choriomeningitis virus (LCMV) and the BeAn strain of TMEV have shown a lack IFN production after early viral replication in the brain (Sandberg et al, 1994; Trottier et al, 2004). Similar to our findings, LCMV induced peripheral, but not CNS, production of IFN (Sandberg et al, 1994).
One question in this viral encephalitis model is why a type I IFN response is not rapidly induced in the brains of wt VSV-infected mice whereas it was in the spleen. There are abundant pDCs in the spleen and lymph nodes, which are absent in the resting CNS, and pDCs monitor for pathogen exposure and produce IFN. It is clear that wt VSV blocks IFN synthesis in infected cells via the biological effects of M protein (Ahmed et al, 2003; Ferran and Lucas-Lenard, 1997; Francoeur et al, 1980, 1987; Petersen et al, 2000; Stojdl et al, 2003). Any VSV-infected neuronal or extraneuronal cell may have IFN gene expression limited by the activity of VSV M protein. In contrast, pDCs are poised to recognize Pathogen associated molecular pattern (PAMPs), through receptors including TLRs, and rapidly produce and secrete IFNs, despite the evasive pathways developed by pathogens. The cell type and critical molecules involved in this system are currently under investigation.
Although many peripheral cell types infiltrate the CNS during the course of VSV encephalitis, starting with neutrophils as early as 24 to 36 h p.i. (Ireland and Reiss, 2006), followed by NK cells at 3 days p.i. and both CD4 and CD8 cells as well as macrophages around day 7 p.i. (Christian et al, 1996), we have found no evidence for the presence of plasmacytoid dendritic cells during the first 14 days after VSV infection (Palian et al, unpublished). Indeed, pDCs are reportedly absent from the brain parenchyma, but have been found in brain during chronic inflammation (Ambrosini et al, 2005; Bailey et al, 2007; Matyszak and Perry, 1997; Newman et al, 2005; Rosicarelli et al, 2005; Serafini et al, 2006; Zozulya et al, 2007). IFN-producing cells do not enter the CNS at early times after infection by wt VSV, and peripheral IFN does not cross the blood-brain barrier efficiently (Dafny and Yang, 2005). Thus it appears the lack of pDCs in the brain and the inability of infected neurons to produce IFN provides VSV with an ideal site for replication.
Materials and methods
Cells, animals, and viruses
Mouse neuroblastoma (NB41A3) cells were purchased from ATCC and maintained in F12K medium with 15% horse serum, 2.5% fetal bovine serum, 1% penicillin/streptomycin. Chinese hamster ovarian (CHO) cells were a gift of Alice Huang and were maintained in Cellgro Free medium with 10% fetal bovine serum, 1 mM l-glutamine, and 1% penicillin/streptomycin.
BALB/c and C57BL/6 mice were obtained from Taconic Laboratories (Germantown, NY). All mice were maintained in specific pathogen-free facilities at New York University in accordance with University Animal Welfare Committee (UAWC) guidelines.
VSV (Indiana strain, San Juan serotype; originally obtained from Alice S. Huang) and Newcastle disease virus (NDV strain B1; generously provided by Peter Palese, Mt Sinai School of Medicine) were used for virus infections. UV-VSV and DI particles were prepared as previously described (Bi et al, 1995; Browning et al, 1991; Plakhov et al, 1995a, 1995b). VSV T1026R1 was isolated from the HR strain was generously provided by Laurent Poliquin (University of Quebec); the virus has been shown to have a mutation in the M protein and to induce IFN in several cell types including mouse L cells (Francoeur et al, 1980, 1987).
Immunofluorescence studies
NB41A3 cells were grown on 4-well chamber slides and infected with either VSV or NDV at a multiplicity of infection (m.o.i.) = 10. At various times p.i., cells were washed and treated with 200 μl 10% buffered formalin in phosphate-buffered saline (PBS) for 5 min at 4°C, washed again, and then permeablized with 1% Triton X-100 in PBS at 4°C for 10 min. After washing, slides were incubated overnight at 4°C with 5% goat serum, 1% bovine serum albumin (BSA) in PBS, then incubated for 2 h at 20°C (room temperature [rt]) with rabbit anti-IRF-3 (Zymed, San Francisco, CA) at a concentration of 0.75 μg/ml in 1% BSA-PBS. Cells were washed with PBS and incubated with goat anti-rabbit Alexafluor 488–conjugated secondary antibody (Molecular Probes) for 1 h at rt. Slides were washed before mounting in Permount containing Pro-Long antifade reagent (Molecular Probes) and 4′-6′-Diamidino-2-phenylindole (DAPI) (0.5 mg/ml).
Experimental infection and treatment of mice
Male BALB/c mice (6 to 7 weeks of age) were anesthetized with isofluorane and infected i.n. with various doses of VSV ranging from 30 to 1 × 104 PFU in PBS with a dose of 5 μl per nostril; mock-infected mice were inoculated with the same volume of PBS, i.n. Mice were treated with UV-VSV at a dose 5 × 106 PFU. At various times p.i., mice were anesthetized with Avertin. Blood was obtained from animals by cardiac puncture into Eppendorf tubes and stored at 4°C overnight for clotting, and subsequent collection of serum. Spleens, lungs, and brains were aseptically removed, divided in half and then immediately homogenized using Potter-Elvijem tubes either in PBS (for plaque assays) or in Trizol (Invitrogen) for isolation of total RNA.
Mutant VSV T1026R1 was also used for intranasal infection at a dose of 4 × 106 PFU, and mice were processed as above.
Chemical sympathectomy was performed by i.p. injection of groups of five male BALB/c mice with 100 mg/kg/day 6-hydroxydopamine (6-OHDA, Sigma) or media for 10 days prior to i.n. infection with VSV as described above.
To assess the possible role of S100B release, groups of five male BALB/c mice were injected i.p. with either 100 μg mouse serum albumin (Sigma) diluted in saline or 100 μg sRAGE (generously provided by Ann-Marie Schmidt, College of Physicians and Surgeons, Columbia University) and immediately infected i.n. with 100 PFU VSV or mock-infected with PBS. Blood and organs were sampled as described above at 24 h p.i.
Normal serum (Sigma) or sheep neutralizing polyclonal anti-VSV antibody (0.5 ml) were administered i.p. to groups of 10 mice either 2 h prior to or 6 h post i.n. infection with 30 PFU VSV. Tissues were harvested at 24 h p.i. and processed as above.
| Sense 5′ → 3′ | Antisense 5′ → 3′ | Product sizes | |
|
| |||
| IFN-α4 | ATGGCTAGRCTCTGTGCTTTCCT | AGGGCTCTCCAGAYTTCTGCTCTG | 511bp |
| IFN-β | CATCAACTATAAGCAGCTCCA | TTCAAGTGGAGAGCAGTTGAG | 353bp |
| GAPDH | ATTCAACGGCACAGTCAAGG | TGGATGCAGGGATGATGTTC | 467bp |
RT-PCR
Total RNA was isolated from mouse tissues using Trizol Reagent (Invitrogen). Total RNA was treated with DNase I (DNA-free kit, Ambion) to remove chromosomal DNA contamination. One microgram of total RNA was reverse transcribed using Omniscript RT kit (Qiagen) in a volume of 20 μl. Two μl cDNA was used in PCR reactions using the GeneAmp Gold RNA-PCR kit (Applied Biosystems), with PCR conditions being 95°C 10 s followed by 35 cycles of 94°C 30 s, 55°C 30 s, 72°C 1 min. PCR products were visualized by agarose gel electrophoresis. Primers for IFN-β and IFN-α were as reported previously (Marie et al, 1998). Primers and the size of the products follow.
IFN bioassays
IFN bioassays were used to estimate the amount of type I IFN in mouse serum or cell culture supernatants. Mouse serum was irradiated for 3 min in a 50-μl volume on ice using a Stratalinker (Stratagene). Tissue culture supernatants from NB41A3 cells (1 ml) were adjusted to pH 2 with 0.25 N HCl and incubated at 4°C for 24 h, after which the pH was adjusted to 7 with 0.25 N NaOH. Irradiated serum (50 μl) or acid-treated supernatants (500 μl) were added to NB41A3 monolayers and allowed to incubate 18 to 24 h to allow cells to achieve an antiviral state. After incubation, monolayers were challenged with VSV at m.o.i. = 0.1 for 30 min in 200 μl F12K medium, after which virus was removed and fresh media added. After 7 h, cell supernatants were harvested and serially diluted before adsorption on CHO cell monolayers. Following overnight incubation in 0.9% agar-containing medium, the monolayers were fixed and stained with methylene blue and plaques were counted. The presence of IFN in either serum or culture supernatant resulted in a 500- to 1000-fold decrease in plaques in the treated monolayers. Alternatively, to semiquantify the amount of IFN in serum samples, serial dilutions were added to NB41A3 cells and allowed to incubate for 18 h. Cells were challenged with VSV at m.o.i. = 0.01. After 24 h, cells were fixed with 10% buffered formalin and stained with methylene blue. Preserved monolayers stained blue after 24-h infection indicated the presence of IFN. Mouse IFN-β (R&D Systems; 100 U/ml) was used as positive control.
IFN ELISA assays
ELISA assays to determine concentration of murine IFN-β in plasma samples was performed according to manufacturer's directions (PBL Medical Laboratories). Mouse IFN-β (R&D Systems) was used as positive control. Normal mouse serum was used as a negative control.
Acknowledgments
The authors are very appreciative of Peter Palese and Neva Morales (Mt Sinai School of Medicine) for providing NDV, Laurent Poliquin (University of Quebec) for providing T1026R1 virus, and Ann Marie Schmidt (College of Physicians & Surgeons, Columbia University) for sRAGE. Valuable conversations with Akiko Iwasaki (Yale University), V. Hugh Perry (University of Southampton), and Susan Morgello (Mt. Sinai School of Medicine) during the course of this work provided insights and are gratefully acknowledged. Technical support was provided by Marta Crowe and Ernest Yakob. Paul M. D'Agostino assisted in preparation of some figures. This work was supported by postdoctoral fellowship NS11073 to M.D.T. and a Research Challenge Fund award N5385 from New York University, and DC003536 and NS039746 to C.S.R.
Footnotes
Publisher's Disclaimer: Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf
This article maybe used for research, teaching and private study purposes. Any substantial or systematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply or distribution in any form to anyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contents will be complete or accurate or up to date. The accuracy of any instructions, formulae and drug doses should be independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directly or indirectly in connection with or arising out of the use of this material.
References
- Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol. 2003;77:4646–4657. doi: 10.1128/JVI.77.8.4646-4657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini E, Remoli ME, Giacomini E, Rosicarelli B, Serafini B, Lande R, Aloisi F, Coccia EM. Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. J Neuropathol Exp Neurol. 2005;64:706–715. doi: 10.1097/01.jnen.0000173893.01929.fc. [DOI] [PubMed] [Google Scholar]
- Bailey M, Engler H, Hunzeker J, Sheridan JF. The hypothalamic-pituitary-adrenal axis and viral infection. Viral Immunol. 2003;16:141–157. doi: 10.1089/088282403322017884. [DOI] [PubMed] [Google Scholar]
- Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ TH-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- Barchet W, Cella M, Odermatt B, Asselin-Paturel C, Colonna M, Kalinke U. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J Exp Med. 2002;195:507–516. doi: 10.1084/jem.20011666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna M, Komatsu T, Bi Z, Reiss CS. Sex differences in susceptibility to viral infection of the central nervous system. J Neuroimmunol. 1996a;67:31–39. doi: 10.1016/0165-5728(96)00022-7. [DOI] [PubMed] [Google Scholar]
- Barna M, Komatsu T, Reiss CS. Activation of type III nitric oxide synthase in astrocytes following a neurotropic viral infection. Virology. 1996b;223:331–343. doi: 10.1006/viro.1996.0484. [DOI] [PubMed] [Google Scholar]
- Bi Z, Barna M, Komatsu T, Reiss CS. Vesicular stomatitis virus infection of the central nervous system activates both innate and acquired immunity. J Virol. 1995;69:6466–6472. doi: 10.1128/jvi.69.10.6466-6472.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi Z, Reiss CS. Inhibition of vesicular stomatitis virus infection by nitric oxide. J Virol. 1995;69:2208–2213. doi: 10.1128/jvi.69.4.2208-2213.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock JE. A molecular basis for bidirectional communication between the immune and neuroendocrine systems. Physiol Rev. 1989;69:1–32. doi: 10.1152/physrev.1989.69.1.1. [DOI] [PubMed] [Google Scholar]
- Boehme KW, Compton T. Innate sensing of viruses by toll-like receptors. J Virol. 2004;78:7867–7873. doi: 10.1128/JVI.78.15.7867-7873.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breakfield XO, Neale EA, Neale JH, Jacobowitz DM. Localized catecholamine storage associated with granules in murine neuroblastoma cells. Brain Res. 1975;92:237–256. doi: 10.1016/0006-8993(75)90273-5. [DOI] [PubMed] [Google Scholar]
- Browning MJ, Huneycutt BS, Huang AS, Reiss CS. Replication-defective viruses modulate immune responses. J Immunol. 1991;147:2685–2691. [PubMed] [Google Scholar]
- Campbell SJ, Perry VH, Pitossi FJ, Butchart AG, Chertoff M, Waters S, Dempster R, Anthony DC. Central nervous system injury triggers hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. Am J Pathol. 2005;166:1487–1497. doi: 10.1016/S0002-9440(10)62365-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Ward MF, Sama AE, Wang H. Extracellular HMGB1 as a proinflammatory cytokine. J Interferon Cytokine Res. 2004;24:329–333. doi: 10.1089/107999004323142187. [DOI] [PubMed] [Google Scholar]
- Chesler DA, Munoz-Jordan JL, Donelan N, Garcia-Sastre A, Reiss CS. PKR is not required for interferon-gamma inhibition of VSV replication in neurons. Viral Immunol. 2003;16:87–96. doi: 10.1089/088282403763635474. [DOI] [PubMed] [Google Scholar]
- Christian AY, Barna M, Bi Z, Reiss CS. Host immune response to vesicular stomatitis virus infection of the central nervous system in C57BL/6 mice. Viral Immunol. 1996;9:195–205. doi: 10.1089/vim.1996.9.195. [DOI] [PubMed] [Google Scholar]
- Conzelmann KK. Transcriptional activation of alpha/beta interferon genes: interference by nonsegmented negative-strand RNA viruses. J Virol. 2005;79:5241–5248. doi: 10.1128/JVI.79.9.5241-5248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozat K, Beutler B. TLR7: a new sensor of viral infection. Proc Natl Acad Sci USA. 2004;101:6835–6836. doi: 10.1073/pnas.0401347101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafny N, Yang PB. Interferon and the central nervous system. Eur J Pharmacol. 2005;523:1–15. doi: 10.1016/j.ejphar.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, Michiels T. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci USA. 2006;103:7835–7840. doi: 10.1073/pnas.0602460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- Durbin RK, Mertz SE, Koromilas AE, Durbin JE. PKR protection against intranasal vesicular stomatitis virus infection is mouse strain dependent. Viral Immunol. 2002;15:41–51. doi: 10.1089/088282402317340224. [DOI] [PubMed] [Google Scholar]
- Ferran MC, Lucas-Lenard JM. The vesicular stomatitis virus matrix protein inhibits transcription from the human beta interferon promoter. J Virol. 1997;71:371–377. doi: 10.1128/jvi.71.1.371-377.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francoeur AM, Lam T, Stanners CP. PIF, a highly sensitive plaque assay for the induction of interferon. Virology. 1980;105:526–536. doi: 10.1016/0042-6822(80)90053-7. [DOI] [PubMed] [Google Scholar]
- Francoeur AM, Poliquin L, Stanners CP. The isolation of interferon-inducing mutants of vesicular stomatitis virus with altered viral P function for the inhibition of total protein synthesis. Virology. 1987;160:236–245. doi: 10.1016/0042-6822(87)90065-1. [DOI] [PubMed] [Google Scholar]
- Gaddy DF, Lyles DS. Vesicular stomatitis viruses expressing wild-type or mutant M proteins activate apoptosis through distinct pathways. J Virol. 2005;79:4170–4179. doi: 10.1128/JVI.79.7.4170-4179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgel P, Jiang Z, Kunz S, Janssen E, Mols J, Hoebe K, Bahram S, Oldstone MBA, Beutler B. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology. 362:304–313. doi: 10.1016/j.virol.2006.12.032. [DOI] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Ehrenreich H. Brain derived proteins as markers of acute stroke: their relation to pathophysiology, outcome prediction and neuroprotective drug monitoring. Restor Neurol Neurosci. 2003;21:177–190. [PubMed] [Google Scholar]
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- Huneycutt BS, Plakhov IV, Shusterman Z, Bartido SM, Huang A, Reiss CS, Aoki C. Distribution of vesicular stomatitis virus proteins in the brains of BALB/c mice following intranasal inoculation: an immunohistochemical analysis. Brain Res. 1994;635:81–95. doi: 10.1016/0006-8993(94)91426-5. [DOI] [PubMed] [Google Scholar]
- Ireland DD, Reiss CS. Expression of IL-12 receptor by neurons. Viral Immunol. 2004;17:411–422. doi: 10.1089/vim.2004.17.411. [DOI] [PubMed] [Google Scholar]
- Ireland DD, Reiss CS. Gene expression contributing to recruitment of circulating cells in response to vesicular stomatitis virus infection of the CNS. Viral Immunol. 2006;19:536–545. doi: 10.1089/vim.2006.19.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, Beutler B. CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptor downstream signaling. Arthritis Res Ther. 2005;7:12–19. doi: 10.1186/ar1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T, Bi Z, Reiss CS. Interferon-gamma induced type I nitric oxide synthase activity inhibits viral replication in neurons. J Neuroimmunol. 1996;68:101–108. doi: 10.1016/0165-5728(96)00083-5. [DOI] [PubMed] [Google Scholar]
- Lalla E, Lamster IB, Feit M, Huang L, Spessot A, Qu W, Kislinger T, Lu Y, Stern DM, Schmidt AM. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J Clin Invest. 2000;105:1117–1124. doi: 10.1172/JCI8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Grone HJ, Kurschus FC, Schmidt AM, Yan SD, Martin E, Schleicher E, Stern DM, Hammerling GG, Nawroth PP, Arnold B. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004;113:1641–1650. doi: 10.1172/JCI18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden KS. Catecholamines, sympathetic innervation, and immunity. Brain Behav Immun. 2003;17 1:S5–S10. doi: 10.1016/s0889-1591(02)00059-4. [DOI] [PubMed] [Google Scholar]
- Malmgaard L. Dendritic cells, toll-like receptors, and T-cell responses: lessons from viral infections in vivo. Viral Immunol. 2005;18:584–594. doi: 10.1089/vim.2005.18.584. [DOI] [PubMed] [Google Scholar]
- Marcus PI, Rodriguez LL, Sekellick MJ. Interferon induction as a quasispecies marker of vesicular stomatitis virus populations. J Virol. 1998;72:542–549. doi: 10.1128/jvi.72.1.542-549.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyszak MK, Perry VH. Dendritic cells in inflammatory responses in the CNS. Adv Exp Med Biol. 1997;417:295–299. doi: 10.1007/978-1-4757-9966-8_48. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Biron CA, Brunson KW, Bullock K, Chambers WH, Dhabhar FS, Goldfarb RH, Kitson RP, Miller AH, Spencer RL, Weiss JM. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Brain Res Rev. 1997;23:79–133. doi: 10.1016/s0165-0173(96)00012-4. [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- Newman TA, Galea I, van RN, Perry VH. Blood-derived dendritic cells in an acute brain injury. J Neuroimmunol. 2005;166:167–172. doi: 10.1016/j.jneuroim.2005.04.026. [DOI] [PubMed] [Google Scholar]
- Petersen JM, Her LS, Varvel V, Lund E, Dahlberg JE. The matrix protein of vesicular stomatitis virus inhibits nucleocytoplasmic transport when it is in the nucleus and associated with nuclear pore complexes. Mol Cell Biol. 2000;20:8590–8601. doi: 10.1128/mcb.20.22.8590-8601.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plakhov IV, Aoki C, Reiss CS, Huang AS. Pathogenesis of murine encephalitis limited by defective interfering particles. An immunohistochemical study. J NeuroVirol. 1995a;1:207–218. doi: 10.3109/13550289509113967. [DOI] [PubMed] [Google Scholar]
- Plakhov IV, Arlund EE, Aoki C, Reiss CS. The earliest events in vesicular stomatitis virus infection of the murine olfactory neuroepithelium and entry of the central nervous system. Virology. 1995b;209:257–262. doi: 10.1006/viro.1995.1252. [DOI] [PubMed] [Google Scholar]
- Publicover J, Ramsburg E, Robek M, Rose JK. Rapid pathogenesis induced by a vesicular stomatitis virus matrix protein mutant: viral pathogenesis is linked to induction of tumor necrosis factor alpha. J Virol. 2006;80:7028–7036. doi: 10.1128/JVI.00478-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Publicover J, Ramsburg E, Rose JK. A single-cycle vaccine vector based on vesicular stomatitis virus can induce immune responses comparable to those generated by a replication-competent vector. J Virol. 2005;79:13231–13238. doi: 10.1128/JVI.79.21.13231-13238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsburg E, Publicover J, Buonocore L, Poholek A, Robek M, Palin A, Rose JK. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced t-cell responses and is highly attenuated for replication in animals. J Virol. 2005;79:15043–15053. doi: 10.1128/JVI.79.24.15043-15053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel JD, Murray SJ, Meisner J, Buchmeier MJ. Differential regulation of innate and adaptive immune responses in viral encephalitis. Virology. 2004;318:381–392. doi: 10.1016/j.virol.2003.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosicarelli B, Serafini B, Sbriccoli M, Lu M, Cardone F, Pocchiari M, Aloisi F. Migration of dendritic cells into the brain in a mouse model of prion disease. J Neuroimmunol. 2005;165:114–120. doi: 10.1016/j.jneuroim.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Sabin AB, Olitsky PK. Influence of host factors on neuroinvasiveness of vesicular stomatitis virus: III. Effect of age and pathway of infection on the character and localization of lesions in the central nervous system. J Exp Med. 1938;67:201–228. doi: 10.1084/jem.67.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg K, Eloranta ML, Campbell IL. Expression of alpha/beta interferons (IFN-alpha/beta) and their relationship to IFN-alpha/beta-induced genes in lymphocytic choriomeningitis. J Virol. 1994;68:7358–7366. doi: 10.1128/jvi.68.11.7358-7366.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Humphreys S, Baroni C, Cohn M. In vitro differentiation of a mouse neuroblastoma. Proc Natl Acad Sci USA. 1969;64:316–323. doi: 10.1073/pnas.64.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, Aloisi F. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol. 2006;65:124–141. doi: 10.1097/01.jnen.0000199572.96472.1c. [DOI] [PubMed] [Google Scholar]
- Silverman MN, Pearce BD, Biron CA, Miller AH. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005;18:41–78. doi: 10.1089/vim.2005.18.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl DF, Abraham N, Knowles S, Marius R, Brasey A, Lichty BD, Brown EG, Sonenberg N, Bell JC. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J Virol. 2000;74:9580–9585. doi: 10.1128/jvi.74.20.9580-9585.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- Trottier M, Schlitt BP, Kung AY, Lipton HL. Transition from acute to persistent Theiler's virus infection requires active viral replication that drives proinflammatory cytokine expression and chronic demyelinating disease. J Virol. 2004;78:12480–12488. doi: 10.1128/JVI.78.22.12480-12488.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trottier MDJ, Palian BM, Shoshkes Reiss C. VSV replication in neurons is inhibited by type I IFN at multiple stages of infection. Virology. 2005;333:215–225. doi: 10.1016/j.virol.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Weihe E, Nohr D, Michel S, Muller S, Zentel HJ, Fink T, Krekel J. Molecular anatomy of the neuroimmune connection. Int J Neurosci. 1991;59:1–23. doi: 10.3109/00207459108985446. [DOI] [PubMed] [Google Scholar]
- Yan SS, Wu ZY, Zhang HP, Furtado G, Chen X, Yan SF, Schmidt AM, Brown C, Stern A, LaFaille J, Chess L, Stern DM, Jiang H. Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat Med. 2003;9:287–293. doi: 10.1038/nm831. [DOI] [PubMed] [Google Scholar]
- Yang K, Puel A, Zhang S, Eidenschenk C, Ku CL, Casrouge A, Picard C, von BH, Senechal B, Plancoulaine S, Al-Hajjar S, Al-Ghonaium A, Marodi L, Davidson D, Speert D, Roifman C, Garty BZ, Ozinsky A, Barrat FJ, Coffman RL, Miller RL, Li X, Lebon P, Rodriguez-Gallego C, Chapel H, Geissmann F, Jouanguy E, Casanova JL. Human TLR-7-, -8-, and -9-mediated induction of IFN-alpha/beta and -lambda Is IRAK-4 dependent and redundant for protective immunity to viruses. Immunity. 2005;23:465–478. doi: 10.1016/j.immuni.2005.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yetter RA, Lehrer S, Ramphal R, Small PA., Jr Outcome of influenza infection: effect of site of initial infection and heterotypic immunity. Infect Immun. 1980;29:654–662. doi: 10.1128/iai.29.2.654-662.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Kurt-Jones EA, Fitzgerald KA, Wang JP, Cerny AM, Chan M, Finberg RW. Role of MyD88 in route-dependent susceptibility to vesicular stomatitis virus infection. J Immunol. 2007;178:5173–5181. doi: 10.4049/jimmunol.178.8.5173. [DOI] [PubMed] [Google Scholar]
- Zozulya AL, Reinke E, Baiu DC, Karman J, Sandor M, Fabry Z. Dendritic cell transmigration through brain microvessel endothelium is regulated by MIP-1alpha chemokine and matrix metalloproteinases. J Immunol. 2007;178:520–529. doi: 10.4049/jimmunol.178.1.520. [DOI] [PMC free article] [PubMed] [Google Scholar]