

Abstract
Chronic hepatitis B virus (HBV) infection is the result of an inadequate immune response towards the virus. Myeloid dendritic cells (mDC) of patients with chronic HBV are impaired in their maturation and function, resulting in more tolerogenic rather than immunogenic responses, which may contribute to viral persistence. The mechanism responsible for altered mDC function remains unclear. The HBV-infected patients display large amounts of HBV particles and viral proteins in their circulation, especially the surface antigen HBsAg, which allows multiple interactions between the virus, its viral proteins and DC. To assess whether HBV directly influences mDC function, the effects of HBV and HBsAg on human mDC maturation and function were investigated in vitro. As already described for internalization of HBV by DC, the present study shows that peripheral blood-derived mDC of healthy controls also actively take up HBsAg in a time-dependent manner. Cytokine-induced maturation in the presence of HBV or HBsAg resulted in a significantly more tolerogenic mDC phenotype as demonstrated by a diminished up-regulation of costimulatory molecules and a decreased T-cell stimulatory capacity, as assessed by T-cell proliferation and interferon-γ production. In addition, the presence of HBV significantly reduced interleukin-12 production by mDC. These results show that both HBV particles and purified HBsAg have an immune modulatory capacity and may directly contribute to the dysfunction of mDC in patients with chronic HBV. The direct immune regulatory effect of HBV and circulating HBsAg particles on the function of DC can be considered as part of the mechanism by which HBV escapes immunity.
Keywords: hepatitis B surface antigen, myeloid dendritic cells, tolerance, viral hepatitis
Introduction
Hepatitis B virus (HBV) infects the liver as its primary target, resulting in the majority of cases in self-limiting acute hepatitis, which confers protective HBV-specific T-cell and B-cell responses.1,2 Nevertheless, more than 350 million people are chronically infected with HBV, as the result of a complex interaction between the replicating virus and an inadequate immune response. The immune system evolved to recognize and eliminate foreign antigens. However, if the immune system for any reason has become tolerant of HBV, the HBV-specific T-cell and B-cell responses are generally undetectable.3,4 The exact mechanism by which HBV escapes immunity is still not known.
Dendritic cells (DC) play an important role in antiviral immunity and have the unique capacity to activate naïve T cells and stimulate B and natural killer cells.5,6 Both circulating and tissue-resident immature DC sample the environment for the presence of foreign antigens and upon activation, DC migrate to lymphoid tissues to initiate immune responses. Depending on their maturation status, represented by the expression level of costimulatory and human leucocyte antigen (HLA) molecules and the capacity to produce proinflammatory cytokines, DC can induce either immunity or tolerance.7,8 Immature and semi-mature DC are associated with tolerogenic responses, so in the context of HBV a defect in the maturation process of DC may lead to tolerogenic T-cell responses and HBV persistence. We and others have previously shown that a specific subset of DC, the myeloid dendritic cells (mDC), of patients with chronic HBV are indeed impaired in their capacity to mature compared to mDC of healthy controls, as shown by a decreased capacity to up-regulate costimulatory molecules, produce proinflammatory cytokines and stimulate T cells.9,10 Monocyte-derived DC are also reported to be functionally impaired by the presence of HBV11,12 although deficits remained minor in other studies.13 Whether HBV directly interferes with DC function is not known. In theory, the different steps of binding, uptake and subsequent replication of HBV could all compromise DC function. Contradictory results have been reported about the possible presence and active replication of HBV in in vitro-generated monocyte-derived DC of patients with chronic HBV.11,14 Concerning circulating mDC, viral DNA could be detected in a subset of patients with chronic HBV but no evidence was found for viral replication in these cells.15 These observations have been confirmed and expanded by a study of Untergasser et al.16 that demonstrates that monocyte-derived DC are capable of internalizing HBV, but do not allow HBV replication.
Although HBV seems to be incapable of replicating in DC, the binding and uptake of viral particles by these cells may introduce some functional changes. The liver and peripheral blood of HBV-infected individuals can reach levels of 109–1010 infectious particles per ml, which allows multiple interactions between the virus and DC. In addition, the liver and peripheral blood of patients with HBV contain large amounts of viral proteins, especially the HBV surface antigens.17,18 The HBV envelope consists of small (S), medium (M) and large (L) surface antigens, generally referred to as HBsAg. All three surface antigens contain a common S domain, both M and L proteins contain a preS2 domain and L exclusively contains a preS1 domain. S is the main component of both HBV virions and HBsAg subviral particles, while M and L are highly enriched on HBV virions. HBsAg is secreted from infected hepatocytes as subviral particles, which can accumulate up to concentrations of 100 μg/ml in peripheral blood. The high amounts of HBsAg in the circulation of patients can theoretically contribute to the hampered immune response. Whether HBsAg interacts with DC is not known. Therefore, the aim of the present study was to assess the nature of interaction between HBsAg or HBV and mDC, and the possible role of this interaction for the persistence of HBV infection.
Materials and methods
HBV surface antigens
The following viral antigens were kindly provided by M. van Roosmalen (bioMérieux, Boxtel, the Netherlands) and are listed in Fig. 1: pepsin-treated HBsAg particles immunopurified from sera of patients infected with HBV of serological subtypes ad and ay (patient serum-derived HBsAg; amino acids 1–226)19 and recombinant (r)HBsAg (amino acids 1–281, subtypes ad and ay) purified from transfected Chinese Hamster Ovary (CHO) cells. Cell lysates of non-transfected CHO cells served as a negative control. Representative Coomassie-stained protein gels and Western blots of patient serum-derived and rHBsAg, provided by M. van Roosmalen, show the native p24 and the glycosylated gp27 forms of the S protein and the two glycosylated forms of the M protein, the latter only present in rHBsAg. The higher molecular weight bands represent dimer and trimer forms of HBsAg (Fig. 1b). HBsAg was used at a final concentration of 1 μg/ml.
Figure 1.
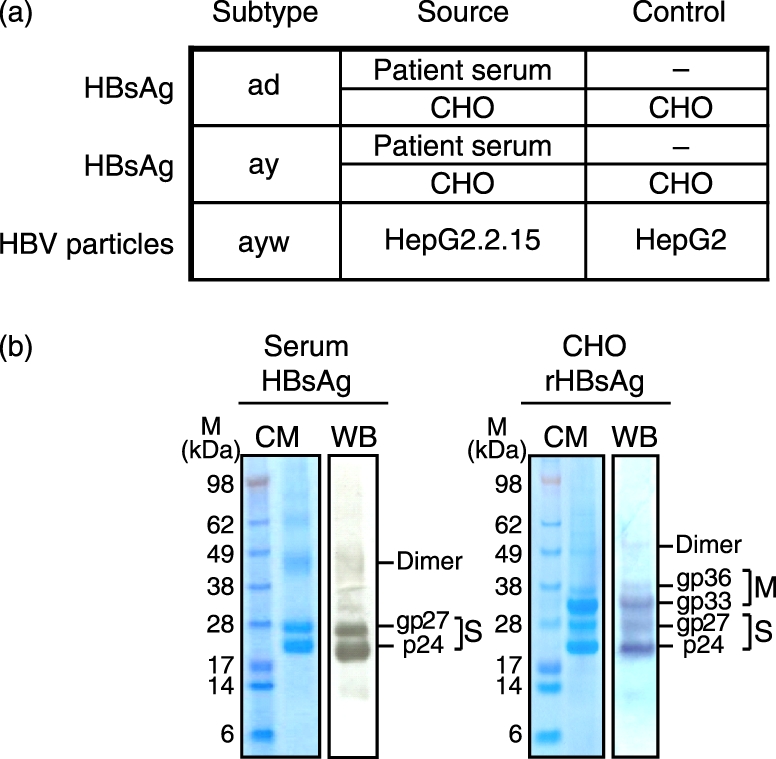
Characteristics of hepatitis B virus surface antigen (HBsAg) and hepatitis B virus (HBV). (a) Source and subtypes of HBsAg and HBV and available controls. (b) Representative Coomassie stained protein gel (CM) and Western blot (WB) of HBsAg purified from pooled patient sera and recombinant Chinese hamster ovary-derived HBsAg.
HepG2.2.15-derived HBV
HepG2.2.15 cells20,21 were grown until confluence in Williams’ E medium (Gibco, Paisley, UK) supplemented with 5% fetal calf serum (FCS; Hyclone, Logan, UT) and HBV particles were concentrated from the medium as described before.16 As a negative control, the same procedure was followed with supernatant from untransfected HepG2 cells. Unless otherwise stated, HBV particles were used at a multiplicity of infection of 100.
Isolation of mDC from peripheral blood
Peripheral blood mononuclear cells were isolated from healthy control blood using Ficoll–Isopaque gradient centrifugation. In total, blood was obtained from 24 different healthy donors [15 male and 9 female; median age (range) 30 (23–54) years]. All healthy controls gave written informed consent before blood donation and the institutional ethical committee gave declaration of no objection for this study. Myeloid DC (BDCA1+) were isolated from peripheral blood mononuclear cells by negative depletion with anti-CD19-conjugated microbeads, followed by positive selection using phycoerythrin (PE)-conjugated anti-BDCA1 and PE-conjugated microbeads using the mini-MACS system (Miltenyi Biotec, Bergisch Gladbach, Germany) as described previously.9 The purity and viability of the isolated mDC were determined by flow cytometry by staining for BDCA1 and CD20 (to ensure the absence of contaminating BDCA1+ CD20+ B lymphocytes) and trypan blue and 7AAD staining [Becton, Dickinson and Company (BD), Franklin Lakes, NJ], respectively. Isolated mDC were cultured in DC medium consisting of RPMI-1640 (Bio Whittaker, Verviers, Belgium) containing 10% FCS (Hyclone), penicillin (50 IU/ml), streptomycin (50 μg/ml) and granulocyte–macrophage colony-stimulating factor (GM-CSF, 500 U/ml; Leucomax, Novartis Pharma, Arnhem, the Netherlands).
HBsAg binding and uptake by mDC
Isolated mDC were incubated for 2 or 18 hr with rHBsAg in the presence of GM-CSF (500 U/ml; Leucomax) and antigen uptake was stopped by a cold wash in phosphate-buffered saline (PBS) containing 1% FCS and 0·02% NaN3. Intracellular staining was performed according to the manufacturer’s protocol (IntraPrep; Beckman Coulter, Fullerton, CA), HBsAg was stained with fluorescein isothiocyante (FITC)–conjugated anti-HBsAg (recognizing subtypes ad and ay, Acris Antibodies GmbH, Hiddenhausen, Germany) and detected by flow cytometry (FACScalibur; BD). Negative controls were incubated without antigen or with antigen at 4°.
Cytospin slides with 15 000 mDC were fixed in 4% paraformaldehyde in PBS and permeabilized with 0·1% saponin (Merck, Darmstadt, Germany). After blocking with 0·1% saponin, 10% normal mouse serum (CLB, Amsterdam, the Netherlands) and 10% normal human plasma (Sanquin, Amsterdam, the Netherlands), slides were stained for 5 hr with allophycocyanin (APC)-conjugated anti-HLA-DR (clone L243; BD) and anti-HBsAg-FITC (Acris Antibodies GmbH) at 4°. After washing in PBS, slides were sealed with Vectashield/DAPI (Vector Laboratories Inc., Burlingame, CA). Signals were captured with a Zeiss Laser Scanning Microscope 510 Meta JNI (Carl Zeiss B.V., Sliedrecht, the Netherlands).
Expression of cell surface molecules on mDC
Isolated mDC (30 × 103/200 μl) were matured in the presence of HBV, HBsAg or the appropriate controls. After 24 hr, cells were harvested and stained with combinations of anti-BDCA1-PE (Miltenyi Biotec), anti-CD80-FITC (clone MAB104; Immunotech, Marseilles, France), anti-CD86-APC (clone 2331; BD), anti-HLA-DR-PerCP (clone 243; BD) and anti-CD40-APC (clone 5C3; BD). Cells were analysed by flow cytometry with corresponding isotype-matched control antibodies to determine background staining.
Cytokine production by mDC
Isolated mDC (40 × 103cells/200 μl) were stimulated in culture medium containing synthetic double-stranded poly[I : C] RNA (20 μg/ml; Sigma-Aldrich, St Louis, MO) and recombinant human interferon-γ (IFN-γ; 1000 U/ml; Strathmann Biotech, Hannover, Germany) in the presence of HBV, HBsAg or the appropriate controls. Supernatants were harvested after 24 hr. Levels of tumour necrosis factor-α (TNF-α) and interleukin-10 (IL-10) were determined by enzyme-linked immunosorbent assay (ELISA) using a kit from Biosource International (Nivelles, Belgium) and IL-12p70 was determined using an ELISA kit from Diaclone (Besançon, France).
T-cell stimulatory capacity of mDC
T-cell stimulatory capacity of mDC was determined in an allogeneic mixed lymphocyte reaction (MLR). Isolated mDC were matured in the presence of HBsAg, HBV or the appropriate controls in culture medium containing IL-1β and TNF-α in a 96-well culture plate (10 × 103 or 5 × 103/200 μl). After 24 hr, culture medium was removed and untouched isolated CD3+ T cells (Miltenyi Biotec) from a buffy coat were added (1·5 × 105/200 μl) and cultured for 6 days. As a positive control, T cells were stimulated with phytohaemagglutinin (5 μg/ml; Murex, Paris, France) or anti-CD3 (100 ng/ml, clone UCHT1; eBioscience, San Diego, CA) and anti-CD28 (1 μg/ml, clone CD28·2; eBioscience). Supernatant was harvested at day 5 to determine IFN-γ production by ELISA (U-CyTech, Utrecht, the Netherlands). During the last 16 hr of a 6-day culture period, 0·5 μCi [3H]thymidine (Amersham, Little Chalfont, UK) was present per well to determine T-cell proliferation. All measurements were performed in triplicate. In a subset of experiments, the percentage of regulatory T cells was determined at day 5 by Foxp3 staining according to the manufacturer’s protocol. Briefly, T-cell surface markers were stained with a combination of anti-CD25-PE (clone M-A251; BD) and peridinin chlorophyll protein (PerCP)-conjugated anti-CD4 (clone SK3; BD). The cells were fixed and permeabilized with fixation/permeabilization and permeabilization buffer (eBioscience) and anti-Foxp3-APC (clone PCH101; eBioscience) was present during permeabilization. Cells were analysed by flow cytometry and CD4+ CD25high Foxp3+ cells were defined as regulatory T cells.
Statistical analysis
All experimental conditions were pair analysed against their controls. Data on costimulatory molecules and cytokine expression were analysed using the non-parametric Wilcoxon Signed Ranks Test with two-tailed P-values. Comparison of the data from MLR and IFN-γ production was performed after logarithmic transformation of raw data. A mixed linear model with subject specific random effects was used with the Satterthwaite approximation method for correction of degrees of freedom. This method allows inclusion of all individual values obtained from triplicate measurements. Data from cytokine expression and MLR are normalized and given as percentages of the appropriate control. Data from expression of costimulatory molecules are expressed as mean ± SEM. A P-value of < 0·05 was considered statistically significant.
Results
HBsAg is internalized by mDC
Although HBV is not capable of replicating within mDC, HBV particles and HBV DNA are detected on the surface or within DC.11–13,16 To investigate whether HBsAg is also taken up by mDC, isolated mDC (Fig. 2a) were cultured in the presence of rHBsAg. Intracellular fluorescence-activated cell sorting showed an increase of HBsAg uptake over time, increasing from 4 ± 1% after 2 hr, to 27 ± 2% after 18 hr. Control mDC incubated at 4° showed no increase in HBsAg positivity, indicating that HBsAg internalization is an active process (Fig. 2b,c). To confirm the uptake of HBsAg, confocal microscopy was performed on cytospin slides of mDC incubated with rHBsAg or CHO control lysates. HBsAg was detected within the cytoplasm of mDC (Fig. 2d) and the percentage of HBsAg-positive mDC increased to 12 ± 1% after overnight culture (data not shown). No staining was detected in cells incubated with the CHO control lysate.
Figure 2.

Hepatitis B virus surface antigen (HBsAg) is actively internalized by myeloid dendritic cells (mDC). (a) Phenotypic analysis of isolated mDC. Purity and viability were checked by anti-CD20-fluorescein isothiocyanate and 7AAD staining respectively. (b, c) Myeloid DC were incubated in the presence of granulocyte–macrophage colony-stimulating factor with or without recombinant HBsAg for 2–18 hr at either 4° or 37°. After incubation, mDC were washed and HBsAg uptake was determined by flow cytometry. Data demonstrate a representative experiment (b) and the quantification of six independent experiments with mDC from different donors (mean ± SEM) (c). (d) Cytospins of mDC cultured in the presence of HBsAg were stained for human leucocyte antigen DR and HBsAg as described in the Materials and methods. Cell nuclei were visualized with DAPI. Two representative mDC were selected by confocal microscopy, one HBsAg-positive and one HBsAg-negative.
HBsAg inhibits the up-regulation of costimulatory molecules on mDC
Freshly isolated mDC from peripheral blood express only low levels of the costimulatory molecules CD40, CD80 and CD86. The TNF-α/IL-1β-induced maturation resulted in a strongly increased expression of these costimulatory molecules (Fig. 3a–c) that is required for proper T-cell activation. Since mDC are capable of internalizing HBsAg, the effect of HBsAg on mDC maturation was studied. rHBsAg derived from CHO cells significantly inhibited up-regulation of CD86 and also the up-regulation of CD40 and CD80 tended to be inhibited. Next to the recombinant proteins, also patient serum-derived HBsAg was examined for its effect on mDC maturation. Both subtypes ad and ay of serum-derived HBsAg significantly inhibited the up-regulation of CD40 and CD86 (P< 0·05). Like the subviral protein HBsAg, purified HBV particles, which are also known to bind and be taken up by mDC, significantly inhibited the up-regulation of CD40 and CD80 during TNF-α/IL-1β-induced maturation compared to control conditions (Fig. 3d–f). Neither HBsAg, nor HBV affected cell viability (data not shown).
Figure 3.

Hepatitis B virus surface antigen (HBsAg) and hepatitis B virus (HBV) inhibit up-regulation of costimulatory molecules during myeloid dendritic cell (mDC) maturation. The mDC were activated with tumour necrosis factor-α and interleukin-1β for 24 hr, in the presence of 1 μg/ml HBsAg derived from patient serum or recombinant HBsAg from Chinese hamster ovary cells (subtype ad and ay), HBV particles (multiplicity of infection of 100) or the appropriate controls. The expression levels of costimulatory molecules CD40, CD80 and CD86 were determined by flow cytometry. (a–c) Mean fluorescence intensities of untreated mDC before (grey line) and after maturation (black line) of a representative experiment. (d–f) Quantification of the mean fluorescence intensities of CD40, CD80 and CD86 expressed on mDC matured in the presence of HBsAg and HBV, or the appropriate controls (n= 8, mean ± SEM, *P< 0·05).
HBV but not HBsAg inhibits the production of IL-12 after in vitro stimulation
In addition to costimulatory molecules as important players in T-cell activation, cytokines produced by DC also influence their T-cell stimulatory capacity and their ability to polarize T cells. To study the effect of HBsAg and HBV on the cytokine expression profile of mDC, levels of IL-10, TNF-α and IL-12p70 were determined after 24 hr maturation in the presence or absence of HBsAg or HBV. Nonstimulated mDC did not show detectable levels of cytokine production (data not shown), whereas activated mDC clearly produced IL-10, TNF-α and IL-12 within 24 hr. No difference in expression levels of these cytokines was observed in the presence of serum-derived or recombinant HBsAg (Fig. 4a–c). Also HBV particles did not influence the production of IL-10 and TNF-α (Fig. 4a,b). Interestingly, the presence of HBV particles induced a significant fourfold reduction in the production of IL-12 (P= 0·028, Fig. 4c,d).
Figure 4.

Hepatitis B virus (HBV) but not hepatitis B virus surface antigen (HBsAg) reduced interleukin-12 (IL-12) production by myeloid dendritic cells (mDC). The mDC were stimulated with poly(I : C) and interferon-γ in the presence of 1 μg/ml HBsAg derived from patient serum or recombinant HBsAg from Chinese hamster ovary cells (subtype ad and ay), HBV particles (multiplicity of infection of 100) or the appropriate controls. After 24 hr, supernatants were harvested and examined for the production of IL-10 (a), tumour necrosis factor-α (TNF-α) (b) or IL-12 (c, d). (a–c) Normalized data derived from six independent experiments with different donors are shown as mean ± SEM percentage compared to appropriate control. (d) Mean IL-12 production of stimulated mDC of six donors in the absence or presence of HBV particles. *P< 0·05.
Both HBV and HBsAg inhibit the T-cell stimulatory capacity of mDC
Since both the expression of costimulatory molecules and the production of cytokines like IL-12 play a major role in T-cell activation by DC, the effects of HBsAg and HBV on the T-cell stimulatory capacity of mDC were determined in an allogeneic MLR. Matured non-treated mDC induced T-cell proliferation, which varied between different donors (range 4215–40 977 counts/min; ratio mDC : T = 1 : 15). Maturation of mDC in the presence of serum-derived HBsAg subtype ad reduced the T-cell proliferation approximately 1·7-fold (Fig. 5a,b). Likewise, the T-cell proliferation was inhibited in the presence of HBsAg subtype ay. Comparable to serum-derived HBsAg, recombinant HBsAg significantly reduced the T-cell stimulatory capacity compared to control conditions (mean reduction: HBsAg ad 35 ± 5%P= 0·012, HBsAg ay 35 ± 3%P= 0·034, rHBsAg ad 22 ± 7%P= 0·007 and rHBsAg ay 33 ± 9%P= 0·0006; Fig. 5b). This effect was dose-dependently regulated and already observed with an HBsAg concentration of 0·1 μg/ml. The HBsAg-induced inhibition reached a plateau at a concentration of 1 μg/ml.
Figure 5.

Hepatitis B virus surface antigen (HBsAg) and hepatitis B virus (HBV) inhibit antigen presentation capacity of myeloid dendritic cells (mDC). The mDC were activated for 24 hr with tumour necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) in the presence of 1 μg/ml HBsAg or HBV particles (multiplicity of infection of 100) or the appropriate controls and cultured for 6 days with allogeneic T cells. (a) Representative experiment of the effect of serum derived-HBsAg on the T-cell stimulatory capacity of mDC, measured by [3H]thymidine incorporation in proliferating T cells (ratio mDC : T cells = 1 : 15). (b) Normalized data derived from six independent experiments with mDC isolated from different donors are shown as mean ± SEM percentage compared to appropriate control. (c) Representative experiment of interferon-γ (IFN-γ) production after 5 days of coculture of HBsAg-treated mDC and allogeneic T cells (ratio mDC : T cells = 1 : 15). (d) Normalized IFN-γ production in a subset of the same experiments as in (b) was determined and shown as mean ± SEM percentage compared to appropriate control (n= 4). (e, f) Representative experiment of the effect of HBV particles on the T-cell stimulatory capacity of mDC measured by T-cell proliferation (e) and IFN-γ production (f). *P< 0·05.
Next to T-cell proliferation, IFN-γ produced during these MLRs was measured in the supernatants after 5 days of culture. Non-treated mDC induced IFN-γ production, which ranged between 3·7 and 71 ng/ml. The HBsAg-treated mDC reduced induction of IFN-γ production by T cells to the same extent as observed for proliferation (Fig. 5c,d). HBsAg inhibited neither phytohaemagglutinin nor anti-CD3/anti-CD28 induced T-cell proliferation, indicating HBsAg has no direct effect on T-cell proliferation (data not shown). Hepatitis B virus particles reduced the T-cell stimulatory capacity of mDC to a similar extent to HBsAg as demonstrated by the significantly reduced T-cell proliferation (mean reduction 34 ± 6%, P< 0·0001 Fig. 5e). Furthermore, HBV-treated mDC reduced the induction of IFN-γ production by T cells by approximately twofold (Fig. 5f). Both HBV and HBsAg inhibit mDC maturation and function, thereby giving rise to more tolerogenic mDC.
HBV-treated and HBsAg-treated mDC do not induce regulatory T cells
One of the possible mechanisms by which HBsAg-treated and HBV-treated mDC can reduce T-cell proliferation is the induction of regulatory T cells, which can actively suppress T-cell responses. The induction of this T-cell subset during MLRs was analysed by the expression of the regulatory T-cell-specific transcription factor Foxp3.22 After 5 days of culture with non-treated mDC, approximately 2.5% of CD4+ T cells could be defined as regulatory T cells (CD4+ CD25high Foxp3+, Fig. 6). No difference in regulatory T-cell numbers was observed when T cells were stimulated with HBsAg-treated or HBV-treated mDC. Interestingly, both HBsAg-treated and HBV-treated mDC showed less T-cell activation as indicated by the lower percentage of CD4+ CD25high Foxp3− T-cell subset (Fig. 6).
Figure 6.

Myeloid dendritic cells (mDC) treated with hepatitis B virus (HBV) or HBV surface antigen (HBsAg) do not induce regulatory T cells. The mDC were activated for 24 hr with tumour necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) in the presence of 1 μg/ml HBsAg or HBV particles (multiplicity of infection of 100) or the appropriate controls and cultured for 5 days with allogeneic T cells. The percentage of regulatory T cells within the CD4+ cell fraction was determined by intracellular fluorescence-activated cell sorter staining. The numbers in the upper right quadrants indicate the percentage of regulatory T cells (CD4+ CD25high Foxp3+). Representative dot blots of triplicate measurements of two independent experiments are shown.
Discussion
The liver and peripheral blood of HBV-infected individuals can reach levels of 109–1010 infectious HBV particles per ml and 100 μg/ml circulating HBsAg, which allows multiple interactions between the virus and DC. The present study demonstrates that myeloid DC actively internalize HBsAg and that the presence of either purified HBsAg or intact viral particles during mDC maturation gives rise to mDC with a significantly reduced immunogenic phenotype and function as demonstrated by the reduced expression of costimulatory molecules and decreased T-cell stimulatory capacity.
Because of their pivotal role in antiviral immunity, viruses, such as HBV, might have evolved to interfere with the function of DC to escape the host immune response. It has been demonstrated that DC of patients with chronic HBV display a less immunogenic function compared to DC of healthy controls.9–11,23 This DC impairment was recently challenged by a study of Tavakoli et al.24 However, in that study only five to eight patients with chronic HBV were included in the functional assays and the experiments were performed with cryopreserved instead of freshly isolated material, which may have a strong impact on the function. Both HBV particles and HBsAg reduced the immunogenicity of mDC in vitro, which indicates a possible immune escape mechanism of HBV by the virus itself and/or by the production of subviral particles, i.e. HBsAg. The similar tolerogenic effects of HBsAg and HBV could easily be explained by the fact that the HBV envelope consists mainly of HBsAg. No major difference was observed between native patient serum-derived HBsAg and CHO rHBsAg although the latter contains not only S but also M forms of HBsAg. The HBsAg subtypes ad and ay are very similar proteins according to charge and protein folding25,26 and indeed showed comparable results in all assays. These data suggest that HBsAg, either as subviral particles or as part of the viral envelope, is at least partially responsible for the impaired mDC function observed in chronic HBV patients. This is in line with data from an HBsAg/HLA-A2 transgenic mouse model showing that high serum levels of HBsAg were able to induce T-cell tolerance.27 Whether HBsAg can also be detected on or within mDC of patients with chronic HBV in vivo needs to be addressed in future studies.
Recombinant yeast-derived HBsAg has been shown to interact with monocytes through the lipopolysaccharide (LPS) binding protein and the LPS receptor CD14, resulting in diminished LPS-induced monocyte activation. Using differentiated THP-1 cells as a model for monocytes, HBsAg was shown to reduce the LPS-induced TNF-α production through interference with the activation of ERK-1,2 and JNK-1,2 kinases.28,29 The lack of effect of HBsAg on TNF-α production by mDC in the present study might be explained by differences between cell types and stimuli.
The direct effect of HBsAg on monocytes might be the explanation for the impaired function of DC generated from monocytes of patients with chronic HBV.11 In addition, the presence of HBV during the generation of monocyte-derived DC resulted in monocyte-derived DC with a reduced T-cell stimulatory capacity and reduced IL-12 production.12 Although in the latter study this might be more an effect of HBV on monocytes than on DC, in the present study also HBV was able to reduce IL-12 production by DC. Since IL-12 is a strong T helper type 1 polarizing cytokine, the inhibitory effect of HBV on IL-12 production might partially explain the impaired T helper type 1 response observed in patients with chronic HBV. This hypothesis is supported by the fact that a 5-log reduction in viral load induced by the nucleoside analogue adefovir strongly increased the capacity of mDC from patients with chronic HBV to produce IL-12.23 In the latter study, the T-cell stimulatory capacity of mDC also increased, which might be the result of a decreased viral load as well as decreased HBsAg levels in adefovir-treated patients.
The additional tolerogenic effect of HBV over HBsAg with regard to IL-12 production might be explained by the presence of the preS1 antigen on the viral envelope of the HBV particles, which is not present in purified HBsAg, or by the presence of capsid protein or viral DNA. Nevertheless, both HBsAg and HBV diminished the T-cell stimulatory capacity of mDC. Whether the reduction of costimulatory molecules on mDC could completely explain their decreased T-cell stimulatory capacity, or that additional proinflammatory signals are decreased and/or regulatory signals are increased by HBsAg and HBV remains to be elucidated.
It has been previously reported that peripheral blood of patients with chronic HBV contains an increased proportion of regulatory T cells compared to both individuals who have resolved their HBV infection and healthy controls.30 This observation is apparently not caused by the effect of HBV and HBsAg on mDC, because both HBV-treated and HBsAg-treated mDC did not significantly induce regulatory T cells in an allogeneic MLR. The observed reduction in T-cell outgrowth and T-cell activation as defined by high expression of CD25 might therefore be the result of T-cell anergy or deletion induced by mDC treated with HBV or HBsAg.
The results of the present study might seem to contrast with several studies showing activation of DC after HBsAg loading or during vaccine therapy, but these studies either added cytokines or uric acid to induce DC activation,31,32 or used an HBsAg vaccine containing an alum adjuvant.33 Of note, even though the routinely used HBsAg vaccination contains aluminium salts as an immune stimulatory adjuvant, about 5% of all HBV-vaccinated individuals do not develop a protective immune response, which is still not understood.34 Therefore, it seems that only in the presence of immune-activating stimuli a proper response to HBsAg could be induced, although even under this condition some individuals fail to develop an HBsAg-specific response. Further proof of the low immunogenicity of HBsAg is derived from studies on HBV-specific CD8+ T-cell populations: in contrast to HBV core-specific CD8+ T cells which are associated with viral control, HBV envelope-specific CD8+ T cells are characterized by an altered, HBV-tolerant, phenotype. In addition, the frequency of HBV envelope-specific CD8+ T cells is very low and they can only occasionally be found in the setting of high levels of HBV replication.35,36
Next to HBV and HBsAg, serum of a subgroup of chronic HBV patients also contains the envelope antigen HBeAg that could, in theory, also contribute to impaired mDC function. Although HBeAg has been described as being involved in T-cell tolerance in a mouse model37 the exact function of HBeAg is still controversial.38 The presence of either baculovirus-derived or Escherichia coli-derived recombinant HBeAg did not lead to a change in mDC function (data not shown).
In conclusion, these results show that both HBV particles and purified HBsAg have an immune modulatory capacity and may directly contribute to the dysfunction of mDC in chronic HBV patients. The direct immune regulatory effect of HBV and circulating HBsAg particles on the function of DC can be considered as part of the mechanism by which HBV escapes immunity. Successful immunotherapy of patients with chronic HBV should therefore aim first to reduce the viral load and viral antigens to restore the immune system and thereafter to boost the immune system to clear the chronic infection.
Acknowledgments
The authors thank Bettina Hansen for help with statistical analysis. The present study is financially supported by NWO VENI grant 916·66·015 to A.W. and NWO VIDI grant 917·59·329 to H.J.
References
- 1.Maini MK, Boni C, Ogg GS, et al. Direct ex vivo analysis of hepatitis B virus-specific CD8(+) T cells associated with the control of infection. Gastroenterology. 1999;117:1386–96. doi: 10.1016/s0016-5085(99)70289-1. [DOI] [PubMed] [Google Scholar]
- 2.Webster GJ, Reignat S, Maini MK, et al. Incubation phase of acute hepatitis B in man: dynamic of cellular immune mechanisms. Hepatology. 2000;32:1117–24. doi: 10.1053/jhep.2000.19324. [DOI] [PubMed] [Google Scholar]
- 3.Marinos G, Torre F, Chokshi S, et al. Induction of T-helper cell response to hepatitis B core antigen in chronic hepatitis B: a major factor in activation of the host immune response to the hepatitis B virus. Hepatology. 1995;22:1040–9. doi: 10.1016/0270-9139(95)90607-x. [DOI] [PubMed] [Google Scholar]
- 4.Maini MK, Boni C, Lee CK, et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med. 2000;191:1269–80. doi: 10.1084/jem.191.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 6.Moretta A. Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat Rev Immunol. 2002;2:957–64. doi: 10.1038/nri956. [DOI] [PubMed] [Google Scholar]
- 7.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 8.Lanzavecchia A, Sallusto F. Antigen decoding by T lymphocytes: from synapses to fate determination. Nat Immunol. 2001;2:487–92. doi: 10.1038/88678. [DOI] [PubMed] [Google Scholar]
- 9.van der Molen RG, Sprengers D, Binda RS, de Jong EC, Niesters HG, Kusters JG, Kwekkeboom J, Janssen HL. Functional impairment of myeloid and plasmacytoid dendritic cells of patients with chronic hepatitis B. Hepatology. 2004;40:738–46. doi: 10.1002/hep.20366. [DOI] [PubMed] [Google Scholar]
- 10.Duan XZ, Zhuang H, Wang M, Li HW, Liu JC, Wang FS. Decreased numbers and impaired function of circulating dendritic cell subsets in patients with chronic hepatitis B infection (R2) J Gastroenterol Hepatol. 2005;20:234–42. doi: 10.1111/j.1440-1746.2004.03529.x. [DOI] [PubMed] [Google Scholar]
- 11.Beckebaum S, Cicinnati VR, Dworacki G, et al. Reduction in the circulating pDC1/pDC2 ratio and impaired function of ex vivo-generated DC1 in chronic hepatitis B infection. Clin Immunol. 2002;104:138–50. doi: 10.1006/clim.2002.5245. [DOI] [PubMed] [Google Scholar]
- 12.Beckebaum S, Cicinnati VR, Zhang X, Ferencik S, Frilling A, Grosse-Wilde H, Broelsch CE, Gerken G. Hepatitis B virus-induced defect of monocyte-derived dendritic cells leads to impaired T helper type 1 response in vitro: mechanisms for viral immune escape. Immunology. 2003;109:487–95. doi: 10.1046/j.1365-2567.2003.01699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tavakoli S, Schwerin W, Rohwer A, et al. Phenotype and function of monocyte derived dendritic cells in chronic hepatitis B virus infection. J Gen Virol. 2004;85:2829–36. doi: 10.1099/vir.0.80143-0. [DOI] [PubMed] [Google Scholar]
- 14.Arima S, Akbar SM, Michitaka K, Horiike N, Nuriya H, Kohara M, Onji M. Impaired function of antigen-presenting dendritic cells in patients with chronic hepatitis B: localization of HBV DNA and HBV RNA in blood DC by in situ hybridization. Int J Mol Med. 2003;11:169–74. [PubMed] [Google Scholar]
- 15.Op den Brouw ML, Van Roosmalen MH, Kusters JG, Janssen HLA, van der Molen RG. Functional impairment of mDC in chronic HBV patients: the role of HBV proteins. Abstract. Hepatology. 2005;42:712A. [Google Scholar]
- 16.Untergasser A, Zedler U, Langenkamp A, et al. Dendritic cells take up viral antigens but do not support the early steps of hepatitis B virus infection. Hepatology. 2006;43:539–47. doi: 10.1002/hep.21048. [DOI] [PubMed] [Google Scholar]
- 17.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bozdayi AM, Uzunalimoglu O, Turkyilmaz AR, et al. YSDD: a novel mutation in HBV DNA polymerase confers clinical resistance to lamivudine. J Viral Hepat. 2003;10:256–65. doi: 10.1046/j.1365-2893.2003.00435.x. [DOI] [PubMed] [Google Scholar]
- 19.Paulij WP, de Wit PL, Sunnen CM, van Roosmalen MH, Petersen-van Ettekoven A, Cooreman MP, Heijtink RA. Localization of a unique hepatitis B virus epitope sheds new light on the structure of hepatitis B virus surface antigen. J Gen Virol. 1999;8:2121–6. doi: 10.1099/0022-1317-80-8-2121. [DOI] [PubMed] [Google Scholar]
- 20.Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci U S A. 1987;84:1005–9. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirschman SZ, Price P, Garfinkel E, Christman J, Acs G. Expression of cloned hepatitis B virus DNA in human cell cultures. Proc Natl Acad Sci U S A. 1980;77:5507–11. doi: 10.1073/pnas.77.9.5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 23.van der Molen RG, Sprengers D, Biesta PJ, Kusters JG, Janssen HL. Favorable effect of adefovir on the number and functionality of myeloid dendritic cells of patients with chronic HBV. Hepatology. 2006;44:907–14. doi: 10.1002/hep.21340. [DOI] [PubMed] [Google Scholar]
- 24.Tavakoli S, Mederacke I, Herzog-Hauff S, et al. Peripheral blood dendritic cells are phenotypically and functionally intact in chronic hepatitis B virus (HBV) infection. Clin Exp Immunol. 2008;151:61–70. doi: 10.1111/j.1365-2249.2007.03547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okamoto H, Imai M, Tsuda F, Tanaka T, Miyakawa Y, Mayumi M. Point mutation in the S gene of hepatitis B virus for a d/y or w/r subtypic change in two blood donors carrying a surface antigen of compound subtype adyr or adwr. J Virol. 1987;61:3030–4. doi: 10.1128/jvi.61.10.3030-3034.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyakawa Y, Mizokami M. Classifying hepatitis B virus genotypes. Intervirology. 2003;46:329–38. doi: 10.1159/000074988. [DOI] [PubMed] [Google Scholar]
- 27.Loirat D, Mancini-Bourgine M, Abastado JP, Michel ML. HBsAg/HLA-A2 transgenic mice: a model for T cell tolerance to hepatitis B surface antigen in chronic hepatitis B virus infection. Int Immunol. 2003;15:1125–36. doi: 10.1093/intimm/dxg117. [DOI] [PubMed] [Google Scholar]
- 28.Vanlandschoot P, Van Houtte F, Roobrouck A, Farhoudi A, Leroux-Roels G. Hepatitis B virus surface antigen suppresses the activation of monocytes through interaction with a serum protein and a monocyte-specific receptor. J Gen Virol. 2002;83:1281–9. doi: 10.1099/0022-1317-83-6-1281. [DOI] [PubMed] [Google Scholar]
- 29.Vanlandschoot P, Roobrouck A, Van Houtte F, Leroux-Roels G. Recombinant HBsAg, an apoptotic-like lipoprotein, interferes with the LPS-induced activation of ERK-1/2 and JNK-1/2 in monocytes. Biochem Biophys Res Commun. 2002;297:486–91. doi: 10.1016/s0006-291x(02)02243-x. [DOI] [PubMed] [Google Scholar]
- 30.Stoop JN, van der Molen RG, Baan CC, van der Laan LJ, Kuipers EJ, Kusters JG, Janssen HL. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology. 2005;41:771–8. doi: 10.1002/hep.20649. [DOI] [PubMed] [Google Scholar]
- 31.Shimizu Y, Guidotti LG, Fowler P, Chisari FV. Dendritic cell immunization breaks cytotoxic T lymphocyte tolerance in hepatitis B virus transgenic mice. J Immunol. 1998;161:4520–9. [PubMed] [Google Scholar]
- 32.Ma XJ, Tian DY, Xu D, Yang DF, Zhu HF, Liang ZH, Zhang ZG. Uric acid enhances T cell immune responses to hepatitis B surface antigen-pulsed-dendritic cells in mice. World J Gastroenterol. 2007;13:1060–6. doi: 10.3748/wjg.v13.i7.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horiike N, Akbar SMF, Ninomiya T, Abe M, Michitaka K, Onji M. Activation and maturation of antigen-presenting dendritic cells during vaccine therapy in patients with chronic hepatitis due to hepatitis B virus. Hepatol Res. 2002;23:38–47. doi: 10.1016/s1386-6346(01)00165-6. [DOI] [PubMed] [Google Scholar]
- 34.Milich DR, Leroux-Roels GG. Immunogenetics of the response to HBsAg vaccination. Autoimmun Rev. 2003;2:248–57. doi: 10.1016/s1568-9972(03)00031-4. [DOI] [PubMed] [Google Scholar]
- 35.Reignat S, Webster GJ, Brown D, et al. Escaping high viral load exhaustion: CD8 cells with altered tetramer binding in chronic hepatitis B virus infection. J Exp Med. 2002;195:1089–101. doi: 10.1084/jem.20011723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webster GJ, Reignat S, Brown D, et al. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: implications for immunotherapy. J Virol. 2004;78:5707–19. doi: 10.1128/JVI.78.11.5707-5719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen M, Sallberg M, Hughes J, Jones J, Guidotti LG, Chisari FV, Billaud JN, Milich DR. Immune tolerance split between hepatitis B virus precore and core proteins. J Virol. 2005;79:3016–27. doi: 10.1128/JVI.79.5.3016-3027.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milich D, Liang TJ. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology. 2003;38:1075–86. doi: 10.1053/jhep.2003.50453. [DOI] [PubMed] [Google Scholar]