

To the Editor:
The prevalence of significant hearing loss (≥ 25 db HL) is 15-20% in young adults and rises to approximately 50% in individuals 80 years of age or older [Morton 1991]. Autosomal dominant nonsyndromic hearing loss (ADSNHL) accounts for approximately 15% of inherited hearing loss. To date, 22 genes have been implicated as causative for ADNSHL, and a further 30 loci mapped to autosomal chromosomal regions [Van Camp and Smith 2007].
ADSNHL at the DFNA9 locus is unusual amongst ADNSHL forms of deafness in that it is also associated with vestibular dysfunction. This type of hearing loss is caused by mutations in the COCH gene (chromosome 14q12), which encodes cochlin, a protein that consists of a signal peptide, an LCCL module, and two von Willebrand factor A (vWFA) domains (Table I) [de Kok et al., 1999]. Cochlin is the most highly expressed protein in the human and mouse inner ear, but its precise function remains unclear [Dessens et al., 2004; Robertson et al., 2006]. A possible role in either structural integrity or antimicrobial activity is predicted by its protein structure [Liepinsh et al., 2001] and immunohistochemistry of a temporal bone from a person with DFNA9-related deafness showing abundant aggregation of homogeneous acellular eosinophilic deposits with loss of cellularity in the cochlea and the vestibular labyrinth [Robertson et al., 2006].
Table I.
DFNA9-causing mutations in the COCH gene
| Family | Mutation | Domain | Type | Progressive | Frequency Loss | Vestibular symptoms | Age of onset | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 USA | p.V66G | LCCL | SNHL | Yes | Intially high Progresses to all | Present in one third | 2nd-3rd decade | [Robertson et al., 1998] |
| 1 USA | p.G88E | LCCL | SNHL | Yes | Intially high Progresses to all | Present in some | 5th decade | [Robertson et al., 1998] |
| 1 USA | p.W117R | LCCL | SNHL | Yes | Intially high Progresses to all | Present in some | 5th decade | [Robertson et al., 1998] |
| 1 Belgian 2 Dutch |
p.P51S | LCCL | SNHL | Yes | Intially high Progresses to all | Present in all | 4th-6th decade | [Fransen et al., 1999] |
| 4 Dutch | p.P51S | LCCL | SNHL | Yes | Intially high Progresses to all | Present in all | 4th-7th decade | [de Kok et al., 1999] |
| 8 Belgian | p.P51S | LCCL | SNHL | Yes | Intially high Progresses to all | Present in most | 4th-7th decade | [Fransen et al., 2001] |
| 1 Australian | p.I109N | LCCL | SNHL | Yes | Intially high Progresses to all | Present in all | 2nd-3rd decade | [Kamarinos et al., 2001] |
| 1 Belgian | p.P51S | LCCL | SNHL | Yes | Intially high Progresses to all | Present in most | 4th-5th decade | [Lemaire et al., 2003] |
| 1 Japanese | p.A119T | LCCL | SNHL | Yes | Intially high Progresses to all | Present in all | 5th decade | [Usami et al., 2003] |
| 1 Hungarian | p.V104del | LCCL | SNHL | Yes | Intially high Progresses to all | Present in all | 4th decade | [Nagy et al., 2004] |
| 1 Dutch | p.G88E | LCCL | SNHL | Yes | Intially high Progresses to all | Present in some | 5th-7th decade | [Kemperman et al., 2005] |
| 1 Dutch | p.P51S | LCCL | SNHL | Yes | Intially high Progresses to all | Present in all | 4th-7th decade | [Bischoff et al., 2005] |
| 1 USA | p.C542F | vWFA2 | SNHL | Yes | Intially high Progresses to all | Present in some | 2nd-3rd decade | [Street et al., 2005] |
| 1 Dutch | p.G87W | LCCL | SNHL | Yes | Intially high Progresses to all | Present in all | 5th-7th decade | [Collin et al., 2006] |
| 1 Dutch | p.I109T | LCCL | SNHL | Yes | Intially high Progresses to all | Present in all | 5th decade | [Pauw et al., 2007b] |
| 2 Chinese | p.M512T p.C542Y |
vWFA2 | SNHL | Yes | Intially high Progresses to all | Present in some | 2nd-5th decade | [Yuan et al., 2008] |
| 1 USA | p.P51S | LCCL | SNHL | Yes | Intially high Progresses to all | Present in all | 4th-7th decade | This study |
Most DFNA9-causing mutations are located within the LCCL domain, which is predicted to be involved in protein folding or a host-defense function (Table I) [Trexler et al., 2000]. The p.P51S variant that affects the LCCL module is the most frequently observed mutation in DFNA9 families [Bischoff et al., 2005; de Kok et al., 1999; Fransen et al., 2001; Fransen et al., 1999; Lemaire et al., 2003]. The identification of a large number of DFNA9 families carrying this variant has facilitated a detailed longitudinal analysis of the audiometric and vestibular impairment in 74 mutation carriers, which has shown that vestibular failure precedes and progresses more rapidly than hearing impairment [Bischoff et al., 2005].
In this study (approved by the University of Iowa Institutional Review Board), we recruited American subjects with ADNSHL and obtained DNA samples and audiograms. Audiograms were formatted for audioprofile analysis, which was conducted using the AudioGene v2.0 system [Hildebrand et al., 2008]. This system predicts the likely underlying genetic cause of hearing loss based on a number of different phenotypic parameters, including auditory thresholds, and rate of decrease in hearing relative to age and frequency. AudioGene v2.0 was trained with audiometric data from families with known deafness-causing mutations in KCNQ4 (DFNA2), DFNA5 (DFNA5), WFS1 (DFNA6/14/38), TECTA (DFNA8/12), COCH (DFNA9) and COL11A2 (DFNA13).
To validate AudioGene v2.0 as a clinical and research tool, we studied a cohort of 77 families segregating presumed ADSNHL represented by audiograms from 160 individuals. Individuals from seven families were identified by AudioGene v2.0 as having a DFNA9 profile, and in six families, mutation screening of the COCH gene was completed (genomic DNA was unavailable for screening in one family). While no mutations were identified in five families, one family was found to segregate the p.P51S mutation (positive predictive value, ∼16.7%) (Figs 1, 2). To our knowledge, this family is the first outside Western Europe to be reported with this mutation. However by comparing highly heterozygous short tandem repeat polymorphisms (STRPs) tightly linked to the COCH gene, we could show that this family is related to five previously reported Belgian p.P51S families [Fransen et al., 2001] (data not shown), providing further evidence of a founder effect in the Benelux region of Western Europe.
Figure 1.

Pedigree of the five-generation American family with nonsyndromic autosomal dominant HFSNHL, vestibular dysfunction and SSCD. Genotypes for nuceotide c.151 in the COCH gene are shown for those individuals included in the genetic analysis. Individuals IV:5 and IV:6 were reported to have hearing loss, however they are likely to represent phenocopies. Diagonal line deceased.
Figure 2.
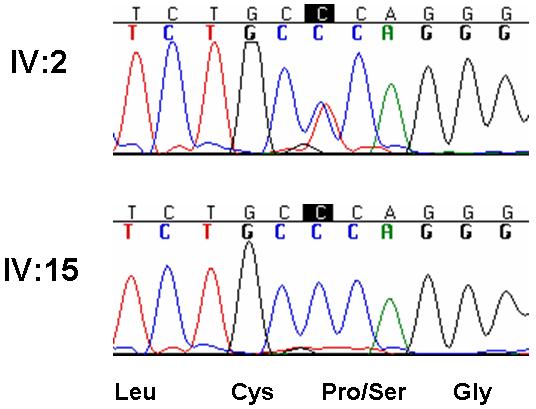
The causative mutation in the COCH gene in the American family. The mutation is a heterozygous C-to-T base change in exon 3 (c.151C→T) that results in substitution of a proline residue for a serine residue at position 51 (p.P51S).
The clinical presentation of all p.P51S families is very similar (Table I) [de Kok et al., 1999; Fransen et al., 2001; Fransen et al., 1999; Lemaire et al., 2003]. The hearing loss initially preferentially affects high frequencies (down-sloping) and progresses to become profound across all frequencies at a rate of ∼1.8 dB annually [Bom et al., 1999]. In a few exceptional cases, however, hearing at low frequencies declines much more rapidly, at greater than 24 dB annually [Bom et al., 1999]. In the American family, the self-reported age-of-onset of hearing loss ranged from 20 to 60 years. The hearing loss was progressive, with a general trend of initial moderate-to-severe high frequency (> 2000 Hz) hearing loss (HFSNHL) that became profound (Fig.3). In younger persons, the audioprofile was down-sloping, preserving hearing in the low and mid frequencies; with advancing age, low and mid frequency loss developed and the audioprofile flattened. Individuals IV:5 and IV:6 represent phenocopies.
Figure 3.
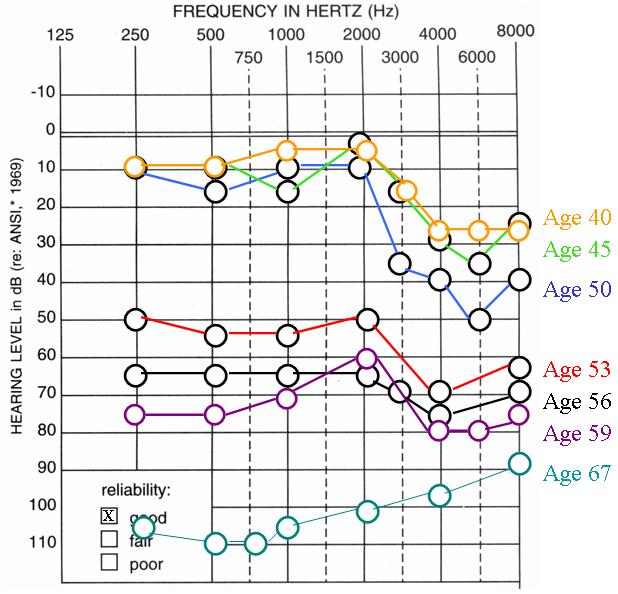
Audiograms of representative affected family members at various ages. Affected individuals initially exhibit high frequency (> 2000 Hz) mild-to-moderate SNHL (HFSNHL) that progresses to become severe-to-profound before flattening out later in life.
All hearing impaired persons in this family excluding IV:5 and IV:6 also had vestibular impairment characterized by intermittent unsteadiness, particularly in the dark, and sporadic periods of dizziness. In addition, one person was diagnosed with Meniere’s-like symptoms and another with autoimmune inner ear disease requiring treatment with a diuretic and a low-salt diet. This variety of complaints is similar to that reported in other p.P51S families [Bischoff et al., 2005]. In general, p.P51S mutation carriers over 40 years of age usually have prominent vestibular symptoms however these complaints only emerge from the medical history if a targeted vestibular history is obtained.
Computed tomography (CT) scanning on a number of family members revealed intact ossicles, cochleae and semicircular canals with the exception of individual V:1. This person presented at age 31 in October, 2006, for an audiogram because of the family history of hearing loss. He has no complaints and by audiometry had 10 dB thresholds bilaterally through 2 kHz, a drop to 25 dB between 3-6 kHz, and normal hearing again at 8 kHz (Fig.4A). In February, 2008, he returned with a complaint of left aural fullness and persistent otalgia (6-9 months) and underwent temporal bone CT, which revealed bilateral superior semicircular canal dehiscence (SSCD) (Fig.4B).
Figure 4.


Audiometry and temporal bone CT of individual V:1. A Audiogram recorded at 31 years of age showing slightly elevated hearing thresholds at high frequencies. B Coronial high-resolution CT scan of the temporal bones showing bilateral superior semicircular canal dehiscence (SSCD).
SSCD is a rare disorder. Originally described by Minor and colleagues, it is characterized by the absence of bone overlying the superior semicircular canal, which creates a third labyrinthine window (with the oval and round windows) [Minor 2000; Minor 2005]. The consequence is the loss of acoustic energy and abnormal stimulation of the vestibular system; the clinical manifestations include Valsalva- and exercise-induced vertigo, sound-induced vertigo (Tullio phenomenon), and variable conductive hearing loss [Cox et al., 2003; Mikulec et al., 2004; Minor et al., 2003]. Diagnosis requires high resolution temporal bone CT [Belden et al., 2003; Hirvonen et al., 2003].
The genetics of SSCD have not been studied, and our finding of a COCH mutation in a single patient with SSCD is noteworthy since both DFNA9-related deafness and SSCD are rare. Although it seems unlikely that SSCD is directly related to the well-documented type of vestibular impairment that is part of the DFNA9 phenotype, SSCD may be present in other DFNA9 patients. We therefore recommend high resolution temporal bone CT in patients with DFNA9-related deafness and screening of COCH in sporadic or familial cases of SSCD. This diagnostic algorithm is important because SSCD and its associated symptoms can be partially or fully corrected with surgery [Carey et al., 2007; Limb et al., 2006; Mikulec et al., 2005; Wilkinson et al., 2008].
A remarkable finding in the process of identifying DFNA9/COCH as the underlying cause of deafness in this American family was not only that a dedicated computer program, AudioGene v2.0, hinted at this possibility, but that this occurred in the course of a screening procedure for DFNA2 families [Hildebrand et al., 2008]. It is reassuring to find that AudioGene v2.0 can distinguish between the audioprofiles of genetically different types of ADNSHL, especially when both DFNA2 and DFNA9 show remarkably similar high frequency hearing loss with steeply downsloping thresholds.
Admittedly, in the case of p.P51S mutation carriers genetic screening should be preferentially directed to DFNA9 and DFNA11, on the basis of the vestibular impairment findings. However, it is realistic to acknowledge the possibility that vestibular symptoms can go unnoticed. Furthermore, with other DFNA9/COCH mutations vestibular failure can be less prominent. It is not invariably penetrant and/or occurs at a more advanced age [Kemperman et al., 2005; Pauw et al., 2007a].
ACKNOWLEDGMENTS
The authors sincerely thank the family for their participation in this study. RJH Smith is the Sterba Hearing Research Professor, University of Iowa College of Medicine, who supported the project with National Institutes of Health (NIH) grant RO1 DCOO3544. No researchers involved in this study report a conflict of interest.
Abbreviations
- ADSNHL
autosomal dominant sensorineural hearing loss
- COCH
cochlin gene
- CT
computed tomography
- HFSNHL
high frequency sensorineural hearing loss
- SSCD
superior semicircular canal dehiscence
REFERENCES
- Belden CJ, Weg N, Minor LB, Zinreich SJ. CT evaluation of bone dehiscence of the superior semicircular canal as a cause of sound- and/or pressure-induced vertigo. Radiology. 2003;226:337–343. doi: 10.1148/radiol.2262010897. [DOI] [PubMed] [Google Scholar]
- Bischoff AM, Huygen PL, Kemperman MH, Pennings RJ, Bom SJ, Verhagen WI, Admiraal RJ, Kremer H, Cremers CW. Vestibular deterioration precedes hearing deterioration in the P51S COCH mutation (DFNA9): an analysis in 74 mutation carriers. Otol Neurotol. 2005;26:918–925. doi: 10.1097/01.mao.0000185048.84641.e3. [DOI] [PubMed] [Google Scholar]
- Bom SJ, Kemperman MH, De Kok YJ, Huygen PL, Verhagen WI, Cremers FP, Cremers CW. Progressive cochleovestibular impairment caused by a point mutation in the COCH gene at DFNA9. Laryngoscope. 1999;109:1525–1530. doi: 10.1097/00005537-199909000-00031. [DOI] [PubMed] [Google Scholar]
- Carey JP, Migliaccio AA, Minor LB. Semicircular canal function before and after surgery for superior canal dehiscence. Otol Neurotol. 2007;28:356–364. doi: 10.1097/01.mao.0000253284.40995.d8. [DOI] [PubMed] [Google Scholar]
- Collin RW, Pauw RJ, Schoots J, Huygen PL, Hoefsloot LH, Cremers CW, Kremer H. Identification of a novel COCH mutation, G87W, causing autosomal dominant hearing impairment (DFNA9) Am J Med Genet Part A. 2006;140A:1791–1794. doi: 10.1002/ajmg.a.31354. [DOI] [PubMed] [Google Scholar]
- Cox KM, Lee DJ, Carey JP, Minor LB. Dehiscence of bone overlying the superior semicircular canal as a cause of an air-bone gap on audiometry: a case study. Am J Audiol. 2003;12:11–16. doi: 10.1044/1059-0889(2003/004). [DOI] [PubMed] [Google Scholar]
- de Kok YJ, Bom SJ, Brunt TM, Kemperman MH, van Beusekom E, van der Velde-Visser SD, Robertson NG, Morton CC, Huygen PL, Verhagen WI, Brunner HG, Cremers CW, Cremers FP. A Pro51Ser mutation in the COCH gene is associated with late onset autosomal dominant progressive sensorineural hearing loss with vestibular defects. Hum Mol Genet. 1999;8:361–366. doi: 10.1093/hmg/8.2.361. [DOI] [PubMed] [Google Scholar]
- Dessens JT, Sinden RE, Claudianos C. LCCL proteins of apicomplexan parasites. Trends Parasitol. 2004;20:102–108. doi: 10.1016/j.pt.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Fransen E, Verstreken M, Bom SJ, Lemaire F, Kemperman MH, De Kok YJ, Wuyts FL, Verhagen WI, Huygen PL, McGuirt WT, Smith RJ, Van Maldergem LV, Declau F, Cremers CW, Van De Heyning PH, Cremers FP, Van Camp G. A common ancestor for COCH related cochleovestibular (DFNA9) patients in Belgium and The Netherlands bearing the P51S mutation. J Med Genet. 2001;38:61–65. doi: 10.1136/jmg.38.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen E, Verstreken M, Verhagen WI, Wuyts FL, Huygen PL, D’Haese P, Robertson NG, Morton CC, McGuirt WT, Smith RJ, Declau F, Van de Heyning PH, Van Camp G. High prevalence of symptoms of Meniere’s disease in three families with a mutation in the COCH gene. Hum Mol Genet. 1999;8:1425–1429. doi: 10.1093/hmg/8.8.1425. [DOI] [PubMed] [Google Scholar]
- Grabski R, Szul T, Sasaki T, Timpl R, Mayne R, Hicks B, Sztul E. Mutations in COCH that result in non-syndromic autosomal dominant deafness (DFNA9) affect matrix deposition of cochlin. Hum Genet. 2003;113:406–416. doi: 10.1007/s00439-003-0992-7. Epub 2003 Aug 20. [DOI] [PubMed] [Google Scholar]
- Hildebrand MS, Tack D, McMordie SJ, DeLuca A, Hur IA, Nishimura C, Huygen P, Casavant TL, Smith RJ. Audioprofile-directed screening identifies novel mutations in KCNQ4 causing hearing loss at the DFNA2 locus. Genet Med. 2008 doi: 10.1097/GIM.0b013e318187e106. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirvonen TP, Weg N, Zinreich SJ, Minor LB. High-resolution CT findings suggest a developmental abnormality underlying superior canal dehiscence syndrome. Acta Otolaryngol. 2003;123:477–481. doi: 10.1080/0036554021000028099. [DOI] [PubMed] [Google Scholar]
- Huygen P, Pauw R, Cremers C. In: Genes, Hearing and Deafness. From Molecular Biology to Clinical Practice - Audiometric profiles associated with genetic nonsyndromal hearing impairment: a review and phenotype analysis. Martini M, Stephens D, Read AP, Martini M, Stephens D, Read AP, editors. Informa Healthcare; London, UK: 2007. pp. 185–204. Chapter 12. [Google Scholar]
- Kamarinos M, McGill J, Lynch M, Dahl H. Identification of a novel COCH mutation, I109N, highlights the similar clinical features observed in DFNA9 families. Hum Mutat. 2001;17:351. doi: 10.1002/humu.37. [DOI] [PubMed] [Google Scholar]
- Kemperman MH, De Leenheer EM, Huygen PL, van Duijnhoven G, Morton CC, Robertson NG, Cremers FP, Kremer H, Cremers CW. Audiometric, vestibular, and genetic aspects of a DFNA9 family with a G88E COCH mutation. Otol Neurotol. 2005;26:926–933. doi: 10.1097/01.mao.0000185062.12458.87. [DOI] [PubMed] [Google Scholar]
- Lemaire FX, Feenstra L, Huygen PL, Fransen E, Devriendt K, Van Camp G, Vantrappen G, Cremers CW, Wackym PA, Koss JC. Progressive late-onset sensorineural hearing loss and vestibular impairment with vertigo (DFNA9/COCH): longitudinal analyses in a belgian family. Otol Neurotol. 2003;24:743–748. doi: 10.1097/00129492-200309000-00009. [DOI] [PubMed] [Google Scholar]
- Liepinsh E, Trexler M, Kaikkonen A, Weigelt J, Banyai L, Patthy L, Otting G. NMR structure of the LCCL domain and implications for DFNA9 deafness disorder. EMBO J. 2001;20:5347–5353. doi: 10.1093/emboj/20.19.5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limb CJ, Carey JP, Srireddy S, Minor LB. Auditory function in patients with surgically treated superior semicircular canal dehiscence. Otol Neurotol. 2006;27:969–980. doi: 10.1097/01.mao.0000235376.70492.8e. [DOI] [PubMed] [Google Scholar]
- Mikulec AA, McKenna MJ, Ramsey MJ, Rosowski JJ, Herrmann BS, Rauch SD, Curtin HD, Merchant SN. Superior semicircular canal dehiscence presenting as conductive hearing loss without vertigo. Otol Neurotol. 2004;25:121–129. doi: 10.1097/00129492-200403000-00007. [DOI] [PubMed] [Google Scholar]
- Mikulec AA, Poe DS, McKenna MJ. Operative management of superior semicircular canal dehiscence. Laryngoscope. 2005;115:501–507. doi: 10.1097/01.mlg.0000157844.48036.e7. [DOI] [PubMed] [Google Scholar]
- Minor LB. Superior canal dehiscence syndrome. Am J Otol. 2000;21:9–19. [PubMed] [Google Scholar]
- Minor LB. Clinical manifestations of superior semicircular canal dehiscence. Laryngoscope. 2005;115:1717–1727. doi: 10.1097/01.mlg.0000178324.55729.b7. [DOI] [PubMed] [Google Scholar]
- Minor LB, Carey JP, Cremer PD, Lustig LR, Streubel SO, Ruckenstein MJ. Dehiscence of bone overlying the superior canal as a cause of apparent conductive hearing loss. Otol Neurotol. 2003;24:270–278. doi: 10.1097/00129492-200303000-00023. [DOI] [PubMed] [Google Scholar]
- Morton NE. Genetic epidemiology of hearing impairment. Ann N Y Acad Sci. 1991;630:16–31. doi: 10.1111/j.1749-6632.1991.tb19572.x. [DOI] [PubMed] [Google Scholar]
- Nagy I, Horvath M, Trexler M, Repassy G, Patthy L. A novel COCH mutation, V104del, impairs folding of the LCCL domain of cochlin and causes progressive hearing loss. J Med Genet. 2004;41:e9. doi: 10.1136/jmg.2003.012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauw RJ, Collin RW, Huygen PL, Hoefsloot LH, Kremer H, Cremers CW. Clinical characteristics of a Dutch DFNA9 family with a novel COCH mutation, G87W. Audiol Neurootol. 2007a;12:77–84. doi: 10.1159/000097794. Epub 2006 Dec 6. [DOI] [PubMed] [Google Scholar]
- Pauw RJ, Huygen PL, Collin RW, Cruysberg JR, Hoefsloot LH, Kremer H, Cremers CW. Phenotype description of a novel DFNA9/COCH mutation, I109T. Ann Otol Rhinol Laryngol. 2007b;116:349–357. doi: 10.1177/000348940711600506. [DOI] [PubMed] [Google Scholar]
- Platt J. In: Fast Training of Support Vector Machines using Sequential Minimal Optimization. Schoelkopf B, Burges C, Smola A, editors. MIT Press; 1998. [Google Scholar]
- Robertson NG, Cremers CW, Huygen PL, Ikezono T, Krastins B, Kremer H, Kuo SF, Liberman MC, Merchant SN, Miller CE, Nadol JB, Jr., Sarracino DA, Verhagen WI, Morton CC. Cochlin immunostaining of inner ear pathologic deposits and proteomic analysis in DFNA9 deafness and vestibular dysfunction. Hum Mol Genet. 2006;15:1071–1085. doi: 10.1093/hmg/ddl022. Epub 2006 Feb 15. [DOI] [PubMed] [Google Scholar]
- Robertson NG, Hamaker SA, Patriub V, Aster JC, Morton CC. Subcellular localisation, secretion, and post-translational processing of normal cochlin, and of mutants causing the sensorineural deafness and vestibular disorder, DFNA9. J Med Genet. 2003;40:479–486. doi: 10.1136/jmg.40.7.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson NG, Lu L, Heller S, Merchant SN, Eavey RD, McKenna M, Nadol JB, Jr., Miyamoto RT, Linthicum FH, Jr., Lubianca Neto JF, Hudspeth AJ, Seidman CE, Morton CC, Seidman JG. Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat Genet. 1998;20:299–303. doi: 10.1038/3118. [DOI] [PubMed] [Google Scholar]
- Street VA, Kallman JC, Robertson NG, Kuo SF, Morton CC, Phillips JO. A novel DFNA9 mutation in the vWFA2 domain of COCH alters a conserved cysteine residue and intrachain disulfide bond formation resulting in progressive hearing loss and site-specific vestibular and central oculomotor dysfunction. Am J Med Genet Part A. 2005;139A:86–95. doi: 10.1002/ajmg.a.30980. [DOI] [PubMed] [Google Scholar]
- Trexler M, Banyai L, Patthy L. The LCCL module. Eur J Biochem. 2000;267:5751–5757. doi: 10.1046/j.1432-1327.2000.01641.x. [DOI] [PubMed] [Google Scholar]
- Usami S, Takahashi K, Yuge I, Ohtsuka A, Namba A, Abe S, Fransen E, Patthy L, Otting G, Van Camp G. Mutations in the COCH gene are a frequent cause of autosomal dominant progressive cochleo-vestibular dysfunction, but not of Meniere’s disease. Eur J Hum Genet. 2003;11:744–748. doi: 10.1038/sj.ejhg.5201043. [DOI] [PubMed] [Google Scholar]
- Van Camp G, Smith RJ. Hereditary Hearing Loss Homepage. 2007 [Google Scholar]
- Wilkinson EP, Liu GC, Friedman RA. Correction of progressive hearing loss in superior canal dehiscence syndrome. Laryngoscope. 2008;118:10–13. doi: 10.1097/MLG.0b013e31814b8d67. [DOI] [PubMed] [Google Scholar]
- Witten I, Frank E. Practical machine learning tools and techniques. Morgan Kaufmann; San Francisco: 2005. [Google Scholar]
- Yuan HJ, Han DY, Sun Q, Yan D, Sun HJ, Tao R, Cheng J, Qin W, Angeli S, Ouyang XM, Yang SZ, Feng L, Cao JY, Feng GY, Wang YF, Dai P, Zhai SQ, Yang WY, He L, Liu XZ. Novel mutations in the vWFA2 domain of COCH in two Chinese DFNA9 families. Clin Genet. 2008;73:391–394. doi: 10.1111/j.1399-0004.2008.00972.x. Epub 2008 Feb 27. [DOI] [PubMed] [Google Scholar]