

Abstract
The cAMP/Protein kinase A (PKA) signaling cascade is crucial for synaptic plasticity in a wide variety of species. PKA regulates Ca2+ permeation through NMDA receptors (NMDARs) and induction of NMDAR-dependent synaptic plasticity at the Schaffer-collateral to CA1 pyramidal cell synapse. Whereas the role of PKA in induction of NMDAR-dependent LTP at CA1 synapses is established, the identity of PKA isoforms involved in this phenomenon are less clear. Here we report that protein synthesis-independent NMDAR-dependent LTP at the Schaffer collateral-CA1 synapse in the hippocampus is deficient, but NMDAR-dependent LTD is normal, in young (postnatal day 10 (P10)-P14) mice lacking PKA RIIβ, the PKA regulatory protein that links PKA to NMDARs at synaptic sites. In contrast, in young adult (P21-P28) mice lacking PKA RIIβ, LTP is normal and LTD is abolished. These findings indicate that distinct PKA isoforms may subserve distinct forms of synaptic plasticity and are consistent with a developmental switch in the signaling cascades required for LTP induction.
Keywords: PKA, PKA type II regulatory subunit, PKA RIIβ knockout mice, NMDA receptors, synaptic plasticity, long term potentiation, long term depression, hippocampus, CA1 synapses
Introduction
N-methyl-D-aspartate (NMDA) receptor (NMDAR)-mediated Ca2+ influx is essential for synaptogenesis, experience-dependent synaptic remodeling and long-lasting changes in synaptic efficacy such as NMDAR-dependent long term potentiation and depression (Dingledine et al., 1999; Carroll and Zukin, 2002; Cull-Candy and Leszkiewicz, 2004; Malenka and Bear, 2004; Collingridge et al., 2004; Lau and Zukin, 2007). Synaptic NMDARs are localized to the postsynaptic density (PSD), where they are physically anchored and spatially restricted by scaffolding proteins such as the PSD protein of 95 kDa (PSD-95), which link upstream and downstream signaling molecules to NMDARs (Scannevin and Huganir, 2000; Lau and Zukin, 2007). Coupling of PKA and protein phosphatase-1 to synaptic NMDARs by A kinase adaptor proteins (AKAPs) enables bi-directional regulation of NMDAR channel activity by PKA (Westphal et al., 1999; Wong and Scott, 2004). The NMDAR-mediated rise in postsynaptic Ca2+ activates a network of kinases and phosphatases that promote persistent changes in synaptic strength.
The cAMP/PKA signaling cascade is crucial for synaptic plasticity in a wide variety of species. The role of cAMP/PKA signaling in the induction (Otmakhova et al., 2000; Skeberdis et al., 2006) and late, protein synthesis-dependent phase (Huang and Kandel, 1994; Qi et al., 1996; Abel et al., 1997) of NMDAR-dependent LTP at hippocampal Schaffer collateral-CA1 (Sch-CA1) synapses has attracted considerable attention. Moreover, pharmacological blockade of PKA (Yasuda et al., 2003) reduces induction of NMDAR-dependent LTP at Sch-CA1 synapses, suggesting a “gating” role for PKA in LTP (Blitzer et al., 1998). NMDARs are molecular targets of PKA phosphorylation (Leonard and Hell, 1997; Tingley et al., 1997); however, the effects of PKA on NMDAR function are less clear. Activation of PKA or inhibition of protein phosphatase I (PPI) potentiates NMDAR-mediated currents (Cerne et al., 1993; Wang et al., 1994; Raman et al., 1996; Blank et al., 1997; Snyder et al., 1998; Skeberdis et al., 2006). Moreover, targeted mutation or disruption of AKAP150, which links PKA to NMDARs and AMPARs, impairs protein synthesis-dependent (Nie et al., 2007) and independent LTP (Lu et al., 2007) at Sch-CA1 synapses. PKA promotes Ca2+ permeation through NMDARs, activity-dependent, NMDAR-mediated Ca2+ signaling in dendritic spines and induction of NMDAR-dependent LTP at Sch-CA1 synapses (Skeberdis et al., 2006). These findings link PKA-dependent synaptic plasticity to Ca2+ signaling in spines and provide a novel mechanism whereby PKA regulates induction of LTP and LTD.
An emerging view is that the specificity of PKA actions on multiple downstream proteins is dictated by the molecular diversity in PKA catalytic and regulatory subunits, which are differentially expressed in specific neuronal populations and spatially compartmentalized within differing microdomains of the neuron (Brandon et al., 1997). The PKA holoenzyme has four different regulatory subunits. The type II regulatory subunit of PKA (RII) localizes to the postsynaptic density (PSD) (Carr et al., 1992; Colledge and Scott, 1999; Colledge et al., 2000), where it links via A-kinase anchoring proteins (AKAPs) to NMDARs and AMPARs (Carr et al., 1992; Colledge et al., 2000; Oliveria et al., 2003) and modulates synaptic AMPAR phosphorylation and trafficking critical to synaptic plasticity and remodeling (Malinow and Malenka, 2002; Collingridge et al., 2004). Whereas the role of the PKA type I regulatory subunit in hippocampal synaptic plasticity is well-established (Brandon et al., 1995; Huang et al., 1995), the role of the type II regulatory subunit is less clear.
The present study was undertaken to define the role of the PKA RIIβ subunit in NMDAR-dependent synaptic plasticity at Sch-CA1 synapses of the hippocampus. Here we show that NMDAR-dependent LTD is impaired in adult (P21-28) PKA RIIβ−/− null mice; whereas presynaptic function (assessed by paired-pulse facilitation), excitatory synaptic transmission (assessed by input/output relations) and protein synthesis-independent, NMDAR-dependent LTP are normal. In contrast, in hippocampus from young (P10-P14) PKA RIIβ−/− null mice, protein synthesis-independent LTP is impaired, whereas LTD is normal. These findings indicate that distinct PKA isoforms may subserve distinct forms of synaptic plasticity and are consistent with a developmental switch in the signaling cascades required for LTP induction.
2. Materials and Methods
Animals
All animals (PKA RIIβ null/knockout (−/−) mice and wild-type (WT) littermates and non-littermates were housed and maintained in a temperature- and light-controlled environment with a 14/10-h light/dark cycle in a facility at the Institute for Animal Studies at the Albert Einstein College of Medicine, Bronx, NY. All animal experiments were carried out in accordance with the principles and guidelines of the National Institutes of Health guide for the care and use of laboratory animals. Protocols used for this study were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine. The generation of PKA RIIβ−/− mice was as described (Brandon et al., 1998; Wong et al., 1999); knockout mice were backcrossed seven or more times onto the C57BL/6 line (Taconic Farms Inc., Germantown, NY) for use in the study. Mice were a generous gift of Quentin Fischer (Yale University, New Haven CT). To maintain a colony of knockout and wild-type mice, PKA RIIβ+/− heterozygous mice were bred in-house. Additional animals were obtained by crossings of PKA RIIβ+/− heterozygous or PKA RIIβ−/− homozygous mice and genotyped by PCR before use as described (Fischer et al., 2004). The PKA RIIβ−/− mice were viable and exhibited no blatant phenotype.
Electrophysiology
Acute hippocampal slices were prepared from postnatal day (P) 10-14 or P21-28 PKA RIIβ−/− mice and wild-type littermates and non-littermate C57BL/6 mice. Mice were anesthetized with isoflurane and sacrificed by decapitation. Whole brains were rapidly removed and placed in an ice-cold cutting solution containing/consisting of (in mM): 234 sucrose, 2.5 KCl, 1.25 NaH2PO4, 10.0 MgSO4, 1 CaCl2, 26 NaHCO3, and 20 glucose, saturated with 95% O2 and 5% CO2. After 5 min incubation in the ice-cold sucrose solution, hippocampi were removed and glued on the stage of a DTK-1000 vibrating microslicer; Dosaka-EM, Kyoto, Japan) with an agar block, and immersed in ice-cold cutting solution (50%) and normal external (50%) solution. Coronal slices (400 μm thick) were cut with a microslicer and transferred to normal external solution containing (in mM): 124 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 26 NaHCO3, 1 NaH2PO4, and 10 glucose. All solutions were saturated with 95% O2 and 5% CO2 (pH 7.4). Slices were incubated for at least 1 h in the external recording solution at 32 ± 1.5°C prior to recording. For some experiments, CA3 was removed immediately after sectioning, but there was no difference between experimental data from CA3-removed slices and those from CA3-intact slices.
For field recordings, slices were transferred to a submersion-type recording chamber mounted on the stage of an upright microscope (BX50WI, Olympus), held fixed by a grid of parallel nylon threads and perfused with external solution at a rate of 2 ml/min. Slices were maintained at 32 ± 1.5°C. To record field EPSPs (fEPSPs), a patch electrode (1–2 MΩ) filled with external solution were positioned in the stratum radiatum of area CA1. fEPSPs were evoked by square pulses (10–100 μA, 200 μs) in Schaffer collateral afferents by means of a concentric bipolar tungsten stimulating electrode (MX21XEP, Frederick Haer). Baseline presynaptic stimulation was delivered once every 30 s using a stimulation intensity yielding 40–60% of the maximal response (for LTP and LTD experiments). The initial slope of the fEPSP was used to measure stability of synaptic responses and to quantify the magnitude of LTP and LTD.
For input/output curves, single-pulse monophasic test stimulation was applied with a Grass S88 stimulator (Grass Instruments, Quincy, MA) at 0.033 Hz, and electrode positions adjusted to maximize amplitude of the fEPSP. An input-output (I/O) relationship ranging from subthreshold to maximal response was established for 6 mice of each genotype. Slices in which the maximal fEPSP amplitude was < 2 mV were rejected.
Paired-pulse facilitation (PPF) was assessed in 6 mice of each genotype at interstimulus intervals ranging from 0.02 to 1 s. The PPF ratio was defined as the ratio of the amplitude of the second to the first fEPSP amplitude elicited by pairs of stimuli. Synaptic responses were monitored with stimuli consisting of constant current pulses of 0.1 ms duration at 0.067 Hz.
LTP was induced after stable baseline recording for at least 20 min by delivery of 2 trains of stimuli (2 trains of 100 pulses at 100 Hz separated by 20 s). LTD was induced by low frequency stimulation (LFS; 900 pulses at 1 Hz) or paired-pulse LFS (PP-LFS; 50-ms interstimulus interval). Field potentials were acquired using a Multiclamp 700A amplifier (Axon Instruments, Union City, CA, USA) and a PCI-MIO-16E-4 data acquisition device (National Instruments, Austin, TX, USA). Stimulation and acquisition were controlled by custom acquisition software run on Igor Pro 4.09A (Wavemetrics, Inc., Lake Oswego, OR, USA). To generate frequency-response curves, 900-pulse stimulation was also applied at 1, 3, 5, 10 and 100 Hz. Slices that did not show a stable baseline for at least 20 min prior to stimulation were discarded.
Drugs
D,L-AP5 (Tocris Cookson, Ballwin, MO) was prepared as a stock solution in DMSO or H2O, stored at −20°C and on the day of the experiment, diluted to a final concentration of 50 μM in external recording solution.
Data analysis
Every six responses were averaged, and fEPSP slopes were then normalized to baseline. To calculate the magnitude of long-term changes in synaptic strength (LTP, LTD), statistical comparisons were made between mean fEPSP slopes before and 45-60 min after tetanus (LTP) or 70-80 min after LFS (LTD). Data were digitized (5 kHz) and analyzed online using IgorPro. Results are reported as mean ± SEM. Statistical analysis was performed using both Student’s paired t-test and two-way ANOVA at the P < 0.05 significance level in Origin Pro 7.0 software (Origin-Lab Corporation, Northampton, MA); these tests yielded comparable P values.
3. Results
Basal synaptic transmission and presynaptic short-term plasticity are normal in area CA1 of juvenile PKA RIIβ−/− mice
To determine whether PKA RIIβ is essential at Sch-CA1 synapses, we examined short- and long-term synaptic plasticity at CA1 synapses in juvenile (P10 – P14) mice lacking PKA RIIβ. PKA RIIβ−/− mice are viable and fertile, but have markedly decreased white adipose tissue (Cummings et al., 1996). Moreover, PKA RIIβ−/− mice exhibit enhanced basal PKA activity and a compensatory increase in PKA RIα in tissues that normally express RIIβ (Cummings et al., 1996; Amieux et al., 1997). As at CA1 synapses in mice lacking PKA RIβ (Brandon et al., 1995), CA1 synapses are functional in PKA RIIβ−/− mice and synaptic responses could be elicited at wild-type and mutant synapses. We first monitored excitatory synaptic transmission onto CA1 neurons in hippocampal slices from wild-type and PKA RIIβ−/− mice using field-potential recording in the presence of a GABAA receptor inhibitor. The fEPSP and the NMDA component of the fEPSP, isolated by application of CNQX (10 μM) and low Mg2+ (0.1 mM), did not significantly differ at CA1 synapses of wild-type vs. mutant mice (Fig. 1A,B). The NMDA response was abolished by application of APV (100 μM; Fig. 1A).
Fig. 1. Young PKA RIIβ KO mice exhibit normal synaptic transmission and short term plasticity.
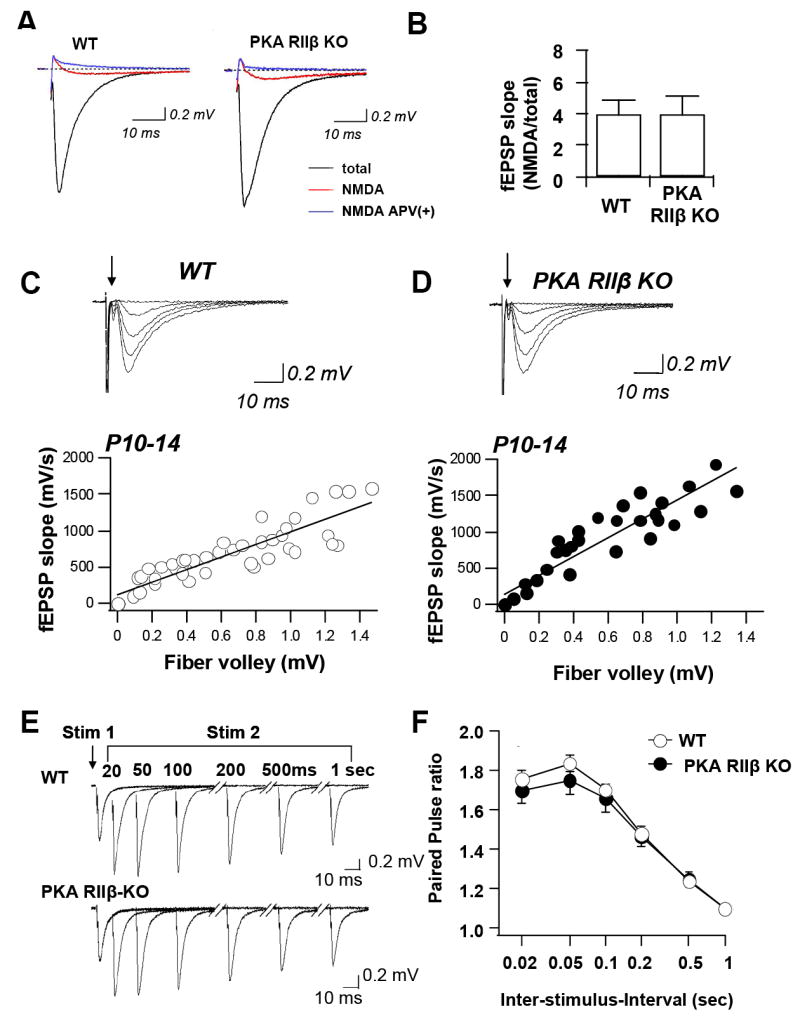
(A,B) Excitatory synaptic transmission at Schaffer collateral to CA1 synapses was monitored by extracellular field recordings in area CA1 of acute hippocampal slices from WT and PKA RIIβ KO mice aged P10 – P14. (A) Sample fEPSPs from WT and KO mice in the absence (black trace) and presence (red trace) of CNQX (10 μM) and low Mg2+ (0.1 mM) to isolate NMDA-receptor-mediated responses. Application of APV (100 μM) abolished the NMDA response (blue trace). (B) Summary data for the amplitude of the NMDA component of the fEPSP normalized to the total fEPSP for WT (left) and KO (right). The NMDA response did not differ significantly in slices of WT vs. KO mice. (C,D) The fEPSP input/output (I/O) relation was measured in area CA1 of acute hippocampal slices from WT and PKA RIIβ KO mice aged P10 – P14. Maximum slope was plotted as a function of the presynaptic volley size for different stimulus intensities. Representative superimposed responses and summary data for WT (C) and KO (D). The fiber volley is indicated by an arrow; initial deflections are stimulus artifact. Slopes of 3 consecutively obtained fEPSPs were averaged for each data point. The I/O function calculated from the slopes of the linear regression lines did not differ significantly between WT and PKA RIIβ KO mice (wild-type: 5 slices, 5 mice; knockout: 5 slices, 5 mice; 2-way ANOVA, P < 0.05). (E,F) Field excitatory postsynaptic potentials (fEPSPs) evoked by paired pulse stimulation of Schaffer collaterals at interstimulus intervals ranging from 0.02 to 1 s were recorded from stratum radiatum in area CA1 of acute hippocampal slices from juvenile wild-type and PKA RIIβ KO mice. (E) Sample records show equal facilitation for WT (upper) and KO (lower). (F) Summary data of paired pulse ratio as a function of interstimulus interval. The ratios did not differ significantly for WT vs. KO mice at any inter-stimulus interval, indicative of normal presynaptic function (wild-type: 6 slices, 6 mice; knockout: 6 slices, 6 mice; student’s t-test, P > 0.1). Data are reported as the mean ± sem.
To more rigorously examine excitatory synaptic transmission in PKA RIIβ−/− mice, we undertook two experimental approaches. First, we assessed basal synaptic transmission in PKA RIIβ−/− mice aged P10-P14 by monitoring field excitatory postsynaptic potential (fEPSP) input–output relations in slices from knockout and wild-type mice. To reduce the variation between slices, we measured input intensity as a function of fiber volley amplitude. The input/output relation (fEPSP slope vs. fiber volley amplitude) at Sch-CA1 synapses in hippocampal slices from PKA RII-β knockout mice was indistinguishable to that of wild-type mice (wild-type: n = 6 mice: knockout: n = 6 mice; P > 0.1; Student’s t-test; Fig. 1C, D). Thus, the loss of PKA RIIβ (and the compensatory increase of PKA RIβ (Amieux et al., 1997)) does not alter short-term plasticity or baseline synaptic transmission at Sch-CA1 synapses.
Second, we monitored field excitatory paired-pulse facilitation (PPF) at six interstimulus intervals (0.02, 0.05, 0.1, 0.2, 0.5 and 1 s) in slices of juvenile (P10 - P14) PKA RIIβ−/− and wild-type mice. PPF is a form of short-term plasticity that is attributed to enhanced transmitter release (Wu and Saggau, 1994; Zucker and Regehr, 2002). No differences in PPF were observed between the PKA RIIβ−/− and wild-type mice at any interstimulus interval tested (wild-type: n = 15 slices, 15 mice; knockout, n = 9 slices, 9 mice; P > 0.05; Fig. 1C, D) Fig. 1E,F). Thus, the loss of PKA RIIβ (despite the compensatory increase of PKA RIβ (Amieux, 1997)) does not alter short-term plasticity at Sch-CA1 synapses in juvenile mice.
LTP is impaired, whereas LTD is normal in juvenile PKA RIIβ KO mice
PKA is thought to play an important role in the induction (Otmakhova et al., 2000; Skeberdis et al., 2006) and late, protein synthesis-dependent phase (Huang and Kandel, 1994; Qi et al., 1996; Abel et al., 1997; Nguyen and Woo, 2003) of LTP at Sch-CA1 synapses. Most mammalian AKAPs bind PKA through the Type II regulatory subunit/PKA RII (Wong and Scott, 2004). In neurons, localization of PKA to dendritic structures is achieved via binding of the RII subunit to AKAPs (Wong and Scott, 2004), an interaction crucial for participation of PKA in LTP (Huang et al., 2006; Nie et al., 2007; Lu et al., 2007). Consistent with this notion, mice lacking the PKA RIβ subunit exhibit normal LTP (Brandon et al., 1995). To determine the role of the PKA RIIβ subunit in induction of NMDAR-LTP at the Sch-CA1 synapse, we examined synaptically-induced/NMDAR-dependent LTP in juvenile mice lacking PKA RIIβ, by applying high frequency stimulation (HFS, 2 trains at 100 Hz for 1 s, separated by 20 s) to acute hippocampal slices. Juvenile PKA RIIβ−/− mice exhibited markedly impaired LTP at Sch-CA1 synapses (wild-type: 134.77 ± 8.67; n = 11 slices, 9 mice; knockout: 106.21 ± 4.75%, n = 5 slices, 5 mice P < 0.01; Fig. 2).
Fig. 2. The PKA RIIβ subunit is required for LTP in young mice at P10-P14.
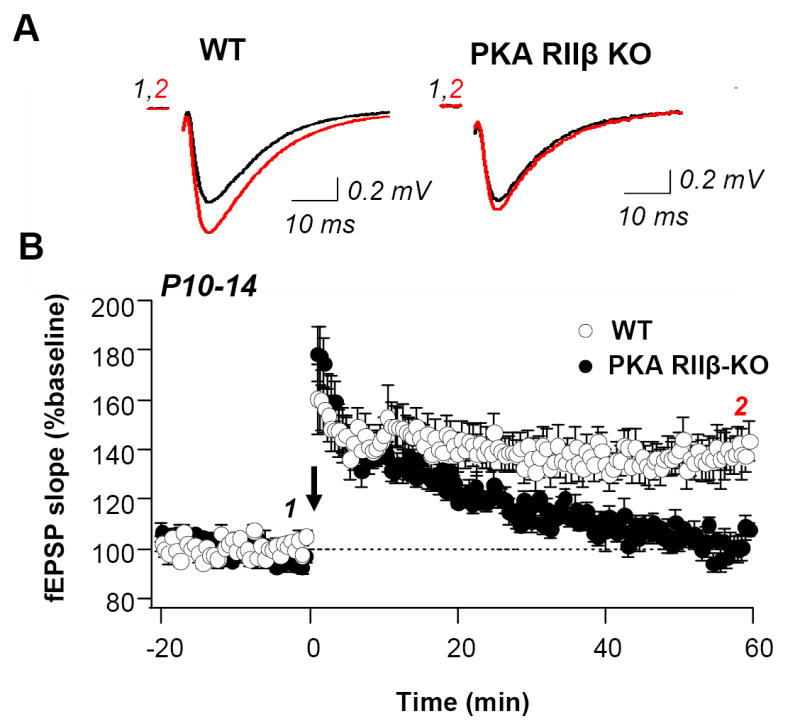
(A) Sample records of fEPSPs in area CA1 for WT (left) and PKA RIIß KO (right) mice aged P10 – P14 before (black trace) and at 60 min after (red trace) induction of LTP (two 100 Hz tetani, 1 s in duration separated by 20 s). (B) Summary data for LTP of fEPSPs from area CA1 of young WT and PKA RIIß KO mice. Open symbols represent wild-type slices; closed symbols are KO slices. KO slices were interleaved with WT slices; no differences were observed between them. For this and subsequent panels, arrow indicates LTP induction. In WT, LTP was robust (to 137.83 ± 7.48% of baseline, measured 50-60 min after HFS; n = 13 slices, 10 mice). In young KO mice, potentiation decays to near baseline by 60 min after tetanus (102.61 ± 3.38% of baseline, measured 50-60 min after HFS; n = 7 slices, 7 mice, P < 0.01 vs. WT, student’s t-test).
We next examined synaptically-induced LTD in area CA1 of PKA RIIβ−/− mice. NMDAR-dependent LTD (NMDAR-LTD) is a form of homosynaptic LTD that is mGluR-independent and is elicited in CA1 neurons by low-frequency stimulation (LFS) of Schaffer-collateral axons (Mulkey and Malenka, 1992; Oliet et al., 1997; Kemp et al., 2000). The role of PKA in LTD is relatively unclear. Pharmacological inhibition of cAMP/PKA signaling can either enhance or block the induction of NMDAR-dependent LTD at Sch-CA1 synapses (Qi et al., 1996; Kameyama et al., 1998; Santschi et al., 2006). Mice lacking PKA RIβ exhibit impaired LTD induction at Sch-CA1 synapses (Brandon et al. 1995). LFS LTD at Sch-CA1 synapses is prominent at an early stage in postnatal development (P10-P14) (Oliet et al., 1997; Kemp et al., 2000). Thus, we examined whether PKA RIIβ subunit might also participate in LFS LTD in juvenile rats. Juvenile PKA RIIβ KO mice exhibited robust LTD at Sch-CA1 synapses that was indistinguishable from that of age-matched wild-type mice (wild-type: 63.66 ± 3.91% of baseline; n = 8 slices, 6 mice; knockout: 60.47 ± 5.29% of baseline; n = 10 slices, 6 mice; P > 0.05 vs. wild-type; Fig. 3A). LTD induction in young animals was NMDAR-dependent in wild-type (n = 3 slices, 3 mice) and knockout (n = 3 slices, 3 mice), as indicated by block in the presence of APV (50 μM; Fig. 3B). Moreover, in a single experiment PP-LFS successfully induced LTD in both juvenile wild-type and knockout slices (data not illustrated).
Fig. 3. The PKA RIIβ subunit is not required for LTD in young mice.
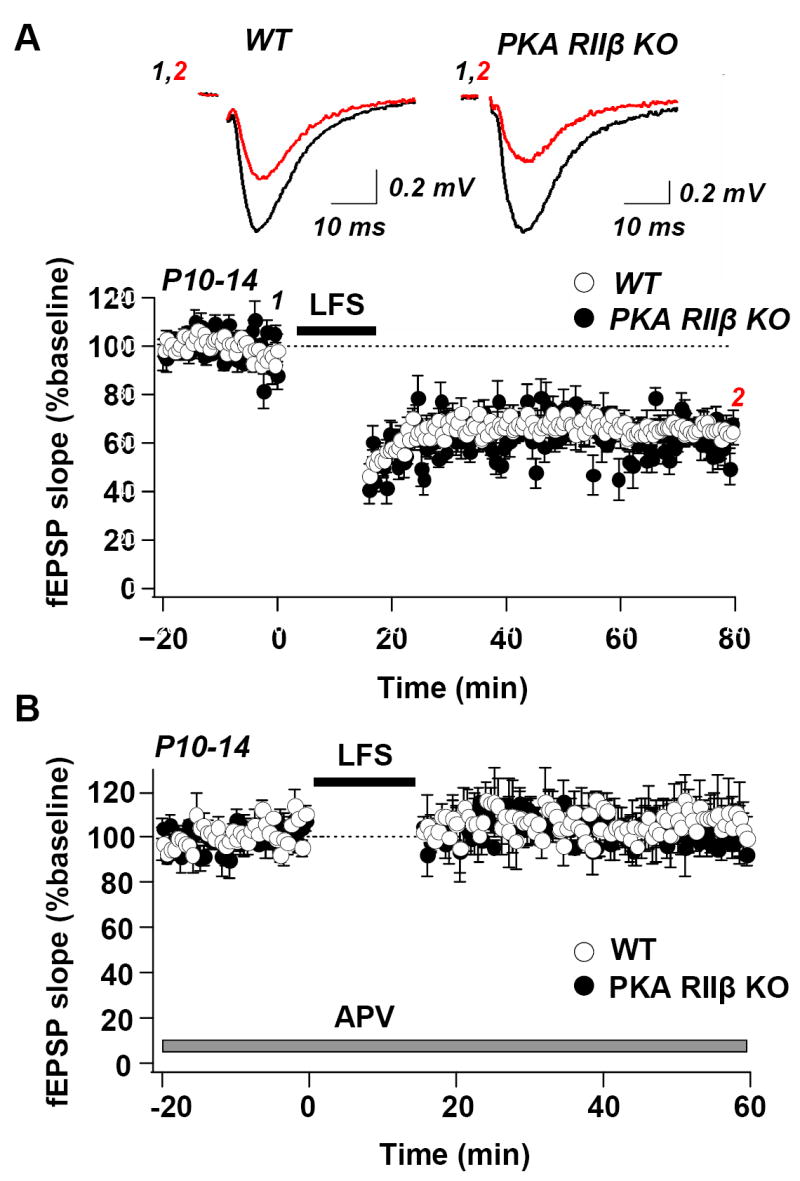
(A) Upper, sample records and lower, summary data show LTD of fEPSPs in area CA1 in hippocampal slices from WT mice (left) and PKA RIIß KO mice (right) before (black) and 60 min after (red) LFS (900 stimuli at 1 Hz). LTD was robust in WT (65.32 ± 3.75% of baseline; n = 8 slices, 6 mice) and KO mice (64.78 ± 3.76% of baseline; n = 10 slices, 6 mice). (B) LTD induction in young animals is NMDAR-dependent in WT (n = 3 slices, 3 mice) and KO (n = 3 slices, 3 mice), as indicated by block in the presence of APV (50 μM, application indicated by bar). As for LTP, KO slices were interleaved with WT. For this and subsequent figures an empty region in the fEPSP recordings and horizontal bar indicate time of low frequency stimulation (LFS).
The absence of PKA RIIβ might influence the magnitude of the rise in Ca2+ or the way that Ca2+ is handled after repetitive stimulation. We therefore determined whether altering the stimulus frequency, a manipulation that alters Ca2+ influx, could elicit LTP in the PKA RIIβ knockout mice. We tested different frequencies of stimulation (1, 3, 10, 30 and 100 Hz). Stimulation at 1Hz and 3Hz induced robust LTD in both wild-type and KO slices (wild-type: 65.3 ± 3.8 of baseline; n = 8 slices, 8 mice; knockout: 64.78 ± 3.8 of baseline; 10 slices, 10 mice; P > 0.05 vs. 1 Hz; wild-type: 75.0 ± 6.0 of baseline; n = 4 slices, 4 mice; knockout: 75.04 ± 6.0 of baseline; 4 slices, 4 mice;P > 0.05 vs. wild-type at 3 Hz; Fig. 4A, B, F). At 10 Hz, slices of both wild-type and RIIβ−/− mice exhibited very mild LTD, (wild-type: 80.0 ± 7.0% of baseline; n = 5 slices, 5 mice; knockout: 89.9 ± 5.0 of baseline; n = 5 slices, 5 mice; P > 0.05 vs. wild type at10 Hz; Fig. 4C,F). In wild-type mice, LTP could be triggered by repetitive stimulation at both 30Hz (Fig. 4D,F), and also at 100Hz (Fig. 4E,F). Increasing the stimulus frequency to 100 Hz (and presumably the tetanus-induced Ca2+ influx) elicited an enhancement of CA1 synaptic responses as expected (data not illustrated). At both frequencies LTP was abolished in the PKA RIIβ−/− mice (wild-type: 131.6 ± 8.9 of baseline; n = 5 slices, 5 mice; knockout: 108.4 ± 6.8 of baseline; 5 slices, 5 mice; P = 0.072 vs. wild type 30 Hz; wild-type: 137.8 ± 7.5 of baseline; n = 13 slices, 13 mice; knockout: 102.6 ± 3.4 of baseline; 7 slices, 7 mice; P < 0.005 vs. wild type at 100 Hz; Fig. 4D,E,F). These findings argue against a role for reduced NMDAR-mediated Ca2+ influx in the PKA RIIβ−/− mice and suggest that PKA RIIβ plays a more complex role in NMDAR-dependent LTP.
Fig. 4. The frequency response curve for induction of LTP and LTD is altered in young KO mice.
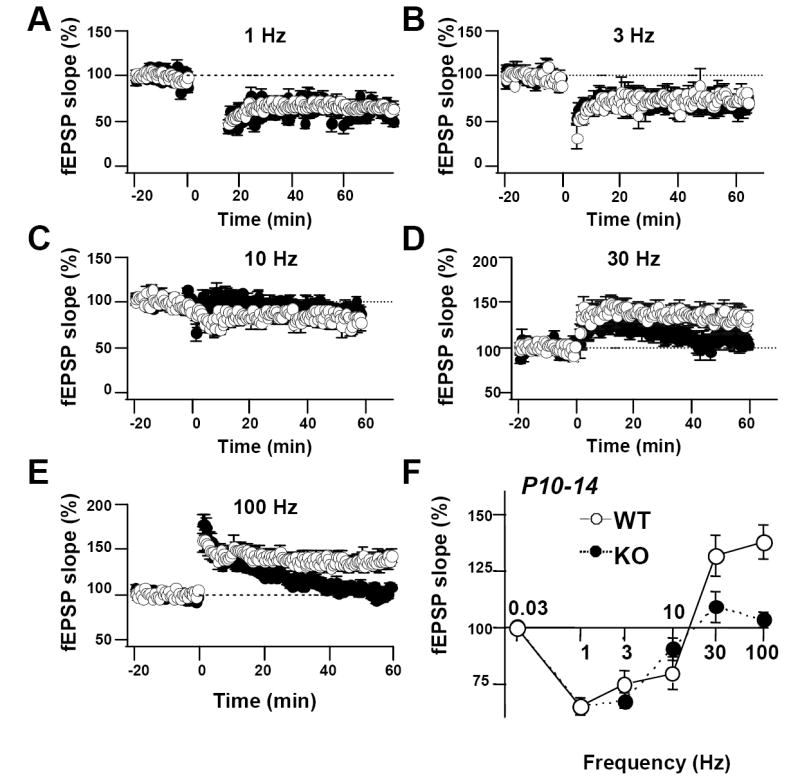
(A-E) Summary data for trains of stimuli given to young WT and KO mice at indicated frequencies. (Panel A is the same as panel A in Fig. 3; Panel E is the same as panel B in Fig. 2). (F) Frequency response plot showing magnitude of LTD or LTP at 50–60 min after stimulation for wild-type (white circles) and KO mice (black circles). PKA RIIβ KO mice exhibited normal LTD in response to 1, 3, and 10 Hz stimulation and deficient LTP in response to 30 Hz and 100 Hz stimulation (1 Hz: wild-type: 8 slices, 6 mice; KO: 10 slices, 6 mice; P = 0.92 vs. wild-type; 3 Hz: wild-type: 4 slices, 4 mice; KO: 4 slices, 4 mice; P = 0.29 vs. wild-type; 10 Hz: wild-type: 5 slices, 5 mice; KO: 5 slices, 5 mice; P = 0.28 vs. wild-type; 30 Hz: wild-type: 5 slices, 5 mice; KO: 5 slices, 5 mice; P < 0.05 vs. wild-type; 100 Hz: wild-type: 13 slices, 6 mice; KO: 7 slices, 6 mice; P < 0.005 vs. wild-type). Data reported as the mean ± sem normalized to baseline.
Basal synaptic transmission is normal in area CA1 of adult (P21 – P28) PKA RIIβ−/− mice
To examine a possible developmental regulation in the impact of RIIβ on synaptic plasticity, we next assessed basal synaptic transmission in slices of adult PKA RIIβ−/− and wild-type mice at P21 – P28. No differences in basal synaptic transmission were observed between the PKA RIIβ−/− and wild-type mice (Fig. 5A, B). Second, we monitored field excitatory PPF at various interstimulus intervals (0.05, 0.1, 0.2, 0.4 s). No differences in PPF were observed between the PKA RIIβ−/− and wild-type mice (wild-type: n = 15 slices, 4 mice; knockout: n = 11 slices, 6 mice; P > 0.1, Student’s t-test; Fig. 5C, D). These findings indicate that in young adult rats the lack of PKA RIIβ−/−, a postsynaptic protein, does not affect presynaptic function.
Fig. 5. Adult PKA RIIβ KO mice exhibit normal basal neural transmission and short term plasticity.
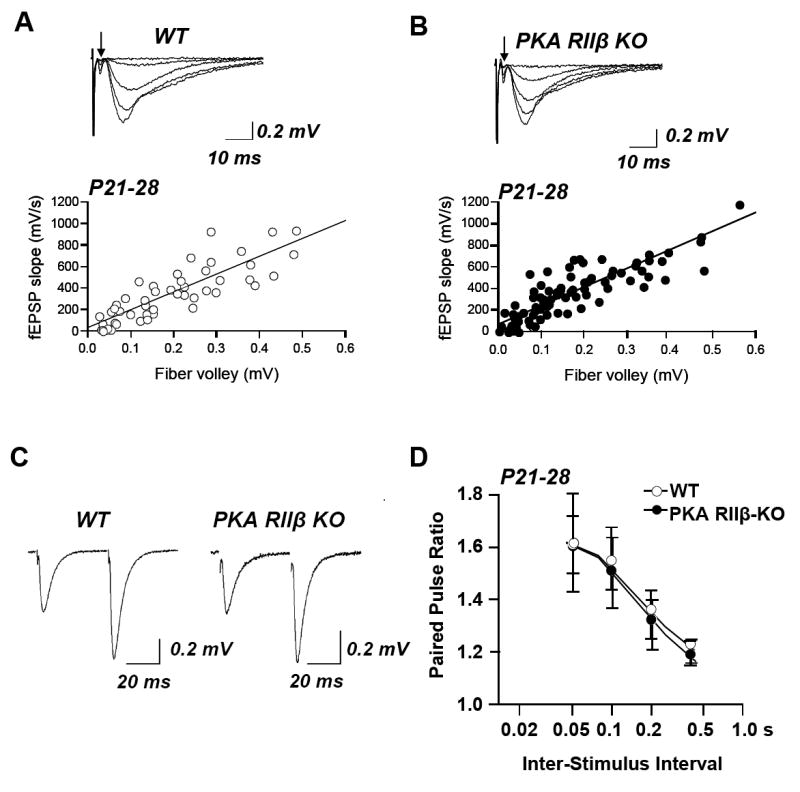
(A,B) Input/output relations of fEPSPs were determined as in Fig. 1. Representative superimposed responses and summary data for wild-type (A) and knockout (B) mice. The fiber volley is indicated by an arrow; initial deflections are stimulus artifact. Each data point represents the average slope of 3 consecutively obtained fEPSPs. The I/O function calculated from the slopes of the linear regression lines did not differ significantly between wild-type and PKA RIIβ knockout mice (wild-type: 6 slices, 6 mice; knockout: 6 slices, 6 mice; 2-way ANOVA, P < 0.05). (C,D) PPF was monitored in the stratum radiatum of area CA1 in acute hippocampal slices from wild-type and PKA RIIβ knockout mice aged P21 – P 28 as in Fig. 1. (C) Sample records for a 0.05 s interstimulus interval show equal facilitation for wild-type (left) and knockout (right). (D) Summary data showing PPF at interstimulus intervals ranging from 0.05 to 0.4 s in wild-type and PKA RIIβ knockout mice. PPF in the PKA RIIβ knockout mice did not differ significantly from that of wild-type mice, indicative of normal presynaptic function (wild-type: 15 slices, 4 mice; knockout: 11 slices, 6 mice; student’s t-test, P > 0.1). Data are reported as the mean ± sem.
NMDAR-dependent LTP at Sch-CA1 synapses is normal in adult PKA RIIβ−/− mice
LTP is thought to play a role not only in learning and memory, but also in activity-dependent synapse stabilization, retraction and maturation, and neural circuit formation and refinement (Katz and Shatz, 1996; Constantine-Paton and Cline, 1998). The signaling cascades required for LTP switch with development from PKA-dependent at young ages (< P9) to Ca2+/calmodulin-dependent protein kinase II (CaMKII)-dependent at more mature ages (> P17) (Yasuda et al., 2003). We next examined synaptically-induced/NMDAR-dependent LTP in young adult mice lacking PKA RIIβ. Whereas the magnitude of HFS-induced LTP in juvenile PKA RIIβ−/− mice exhibited markedly impaired LTP at Sch-CA1 synapses, LTP in young adult PKA RIIβ−/− mice was not significantly different from that in wild-type mice (wild-type: 158.3 ± 6.5, n = 15 slices, 6 mice; knockout: 144.9 ± 9.3, n = 9 slices, 5 mice; P = 0.23; Student’s t-test, Fig. 6A). These results point to a developmental switch in the role of RIIβ in synaptic plasticity.
Fig. 6. LTP is normal but LTD is deficient in slices from PKA RIIβ KO animals at P21 – P28.
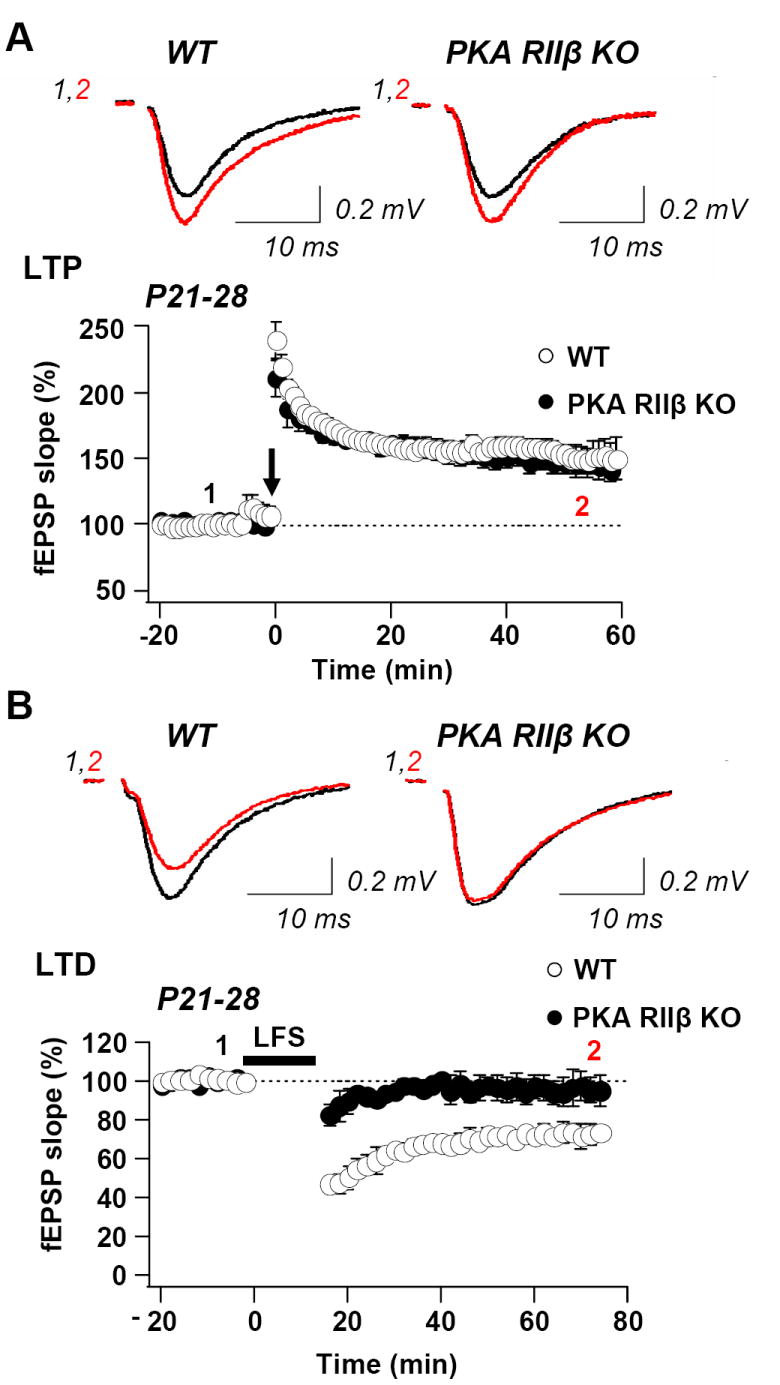
(A) Upper, samples records of fEPSPs for WT mice (left) and PKA RIIß KO mice (right) at times indicates before (black trace) and after (red) induction of LTP (two 100 Hz tetani, 1 s in duration separated by 20 s). Lower, summary data for LTP of fEPSPs from CA1 of WT (n = 15 slices, 6 mice) and KO (n = 8 slices, 6 mice). (B) Upper, sample records of fEPSPs for WT mice (left) and PKA RIIß KO mice (right) at indicated times before (black trace) and after (red trace) LTD-inducing stimulation was applied (900 stimuli at 1Hz). Lower, summary data for LTD of fEPSPs in WT and PKA RIIβ KO mice (n = 8 slices, 6 animals). In WT, LTD was robust (to 74.3 ± 3.7% of baseline, measured 50 to 60 min after LFS; n = 8 slices, 6 mice; P < 0.01 vs. baseline; Student’s t-test). In KO mice, LTD was absent (99.0± 6.2% of baseline, measured 50-60 min post-LFS; n = 7 slices, 6 mice, P < 0.01 vs. WT; Student’s t-test). As for LTP, KO slices were interleaved with WT.
NMDAR-dependent LTD is deficient in adult PKA RIIβ−/− mice
We next examined synaptically-induced LTD in area CA1 of adult (P21-P28) PKA RIIβ−/− mice. Whereas juvenile PKA RIIβ−/− mice exhibited robust LTD at Sch-CA1 synapses that was indistinguishable from that of age-matched wild-type mice, LTD in adult mice was significantly impaired. In control slices, LFS (900 stimuli at 1 Hz) elicited a robust depression in the field potential (FP) slope values to 82.2 ± 3.7 of baseline, measured 35 to 40 min after LFS (wild-type: n = 8 slices, 6 mice; Fig. 6B). By contrast, the same induction protocol did not elicit detectable LTD in slices from PKA RIIβ knockout mice (104.0 ± 6.2 of baseline response, 35 to 40 min post-LFS; n = 7 slices, 6 mice, P < 0.01 vs. wild-type: Student’s t-test, Fig. 6B).
As with the juvenile rats, we examined whether altering the stimulus frequency, a manipulation that alters Ca2+ influx, could elicit LTD in the PKA RIIβ knockout mice in young adult rats. We tested different frequencies of stimulation (1, 3, 5, 10 and 100 Hz). In wild-type mice, LTD could be triggered by repetitive stimulation not only at 1 Hz (Fig. 7A,F), but also at 3 Hz (Fig. 7B,F). In the PKA RIIβ−/− mice, increasing the stimulus frequency to 3 Hz (and presumably the tetanus-induced Ca2+ influx) elicited an enhancement of CA1 synaptic responses as expected (data not illustrated); however, LTD was still abolished in the PKA RIIβ−/− mice (wild-type: 89.4 ± 3.7 of baseline; n = 8 slices, 6 mice; knockout: 98.5 ± 4.2 of baseline; 8 slices, 8 mice; P = 0.56 vs. 1 Hz; P < 0.05 vs. wild-type at 3 Hz; Fig. 7B,F). 5Hz stimulation did not induce plasticity in either wild-type or KO slices (wild-type: 102.0 ± 6.6 of baseline; n = 7 slices, 6 mice; knockout: 96.6 ± 4.8 of baseline; 9 slices, 9 mice; P > 0.05 vs. 1 Hz; P > 0.05 vs. wild-type at 5 Hz; Fig. 7C,F). At 10 Hz, slices of wild-type mice exhibited robust LTP, but slices of RIIβ−/− mice exhibited a significantly reduced LTP relative to that of wild-type (wild-type: 119.6 ± 3.8% of baseline; n = 7 slices, 6 mice; knockout: 101.2 ± 5.4 of baseline; n = 10 slices, 10 mice; P < 0.01 vs. 1 Hz; P < 0.05 vs. wild-type at 5 Hz; Fig. 7D,F)). At 100 Hz stimulation, LTP induction was essentially normal in knockout vs. wild-type slices (Fig. 7E,F; see also Fig. 6A). These findings argue against a role for reduced NMDAR-mediated Ca2+ influx in the PKA RIIβ−/− mice and suggest that PKA RIIβ plays a more complex role in NMDAR-dependent LTD). Collectively, these findings indicate that the RIIβ subunit has a greater impact on long-term, postsynatpic vs. short-term, presynaptic plasticity and that the deficits in LTP and LTD observed in PKA RIIβ−/− mice are developmentally regulated.
Fig. 7. The frequency-response curve for induction of LTP and LTD is altered in adult KO mice.
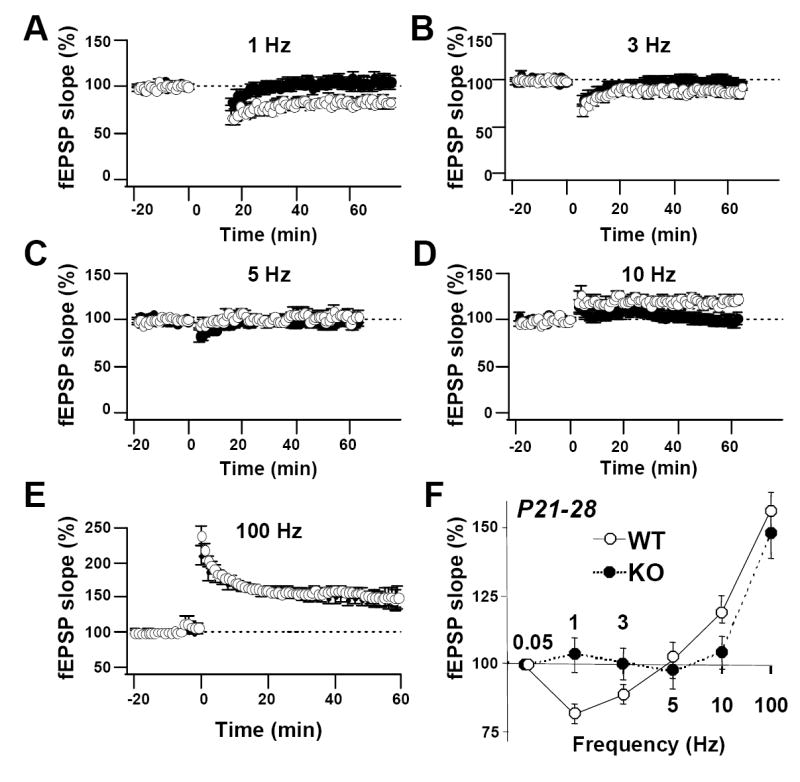
(A - E) Summary data for trains of stimuli given to WT and KO mice at indicated frequencies. (Panel E is the same as the graph in panel A in Fig. 6). (F) Frequency response plot showing magnitude of LTD or LTP at 45–60 min after stimulation for wild-type (white circles) and KO mice (black circles). PKA RIIβ KO mice exhibited no LTD in response to 1, 3, and 5 Hz stimulation, no significant effect in response to 10 Hz stimulation and normal LTP in response to 100 Hz stimulation (1 Hz: wild-type: 8 slices, 6 mice; KO: 7 slices, 6 mice; P = 0.01 vs. wild-type; 3 Hz: wild-type: 8 slices, 6 mice; KO: 8 slices, 6 mice; P = 0.056 vs. wild-type; 5 Hz: wild-type: 7 slices, 6 mice; KO: 9 slices, 9 mice; P = 0.38 vs. wild-type; 10 Hz: wild-type: 7 slices, 6 mice; KO: 10 slices, 10 mice; P = 0.01 vs. wild-type; 100 Hz: wild-type: 15 slices, 6 mice; KO: 8 slices, 5 mice; P = 0.23 vs. wild-type). Data reported as the mean ± sem normalized to baseline.
4. Discussion
The cAMP/PKA signaling cascade is crucial for synaptic plasticity in a wide variety of species. The role of cAMP/PKA signaling in the induction of NMDAR-dependent LTP at Sch-CA1 synapses is established (Otmakhova et al., 2000; Skeberdis et al., 2006), but the identity of the PKA isoform involved in LTP is, as yet unclear. We have shown here that in young hippocampus, PKA RIIβ, a regulatory subunit of PKA implicated in localization of PKA to the postsynaptic density in close proximity and direct physical association with NMDARs and AMPARs, is essential for inducing NMDAR-dependent LTP at Schaffer collateral to CA1 synapses. RIIβ is not required for normal NMDAR-mediated responses, excitatory synaptic transmission, presynaptic short-term plasticity or NMDAR-dependent LTD. This phenotype is unlikely to be caused by structural abnormalities, because light and electron microscopy both showed that the brain has an overall normal structure and organization in PKA RIIβ knockout mice (Cummings et al., 1996). In contrast, in young adult hippocampus, PKA RIIβ is critical to NMDAR-dependent LTD, but not LTP. These findings indicate that distinct PKA isoforms may subserve distinct forms of synaptic plasticity and are consistent with a developmental switch in the signaling cascades required for LTP induction.
Activity-dependent LTP is critical to formation, refinement and stabilization of new synapses in the young brain (Katz and Shatz, 1996; Constantine-Paton and Cline, 1998). Recent studies indicate that the signaling cascades involved in the induction phase of LTP at CA1 synapses are developmentally regulated. Whereas LTP is dependent upon CaMKII after postnatal day 20 (P20), it is dependent on PKA at P7-P8 (Yasuda et al., 2003). Our finding in the present study that PKA RIIβ is critical to LTP at young ages (P10-P14), but not at more mature ages (P21-P28) is consistent with this notion. Our finding extends the work of Malenka and colleagues in that it identifies for the first time a critical role for the PKA RIIβ subunit in LTP signalling in young, but not mature, rodent hippocampus. The PKA RIIβ subunit binds directly AKAP150 (Colledge et al., 2000; Oliveria et al.), which in turn binds via PSD-95 and/or SAP97 and tethers the PKA holoenzyme to the NMDAR NR2B subunit. At the time of birth, NMDARs in the hippocampus contain primarily NR1 and NR2B subunits. Over the course of postnatal development, there is a progressive inclusion of the NR2A subunit (Carroll and Zukin, 2002; Cull-Candy and Leszkiewicz, 2004; Lau and Zukin, 2007). A possible scenario is that at young ages, synaptic NMDARs are NR2B-containing and are more sensitive to regulation by PKA. At later ages, at a time when the number of spines has increased and synaptic connections have formed, NMDARs are primarily NR2A- or NR2A/NR2B-containing (Lau and Zukin) and become relatively less sensitive to regulation by PKA (Skeberdis et al., 2006).
Our finding of a critical role for the PKA RIIβ subunit in LTP signaling in young, but not mature, rodent hippocampus is also consistent with findings of others that early in development, PKA phosphorylation of GluR4-long is a primary mediator of activity-dependent synaptic incorporation of AMPARs, a mechanism thought to underlie enhanced synaptic strength (Zhu et al., 2000; Esteban et al., 2003). GluR4 delivery requires PKA-dependent phosphorylation at serine residue 842 in its carboxy-terminal tail (Zhu et al., 2000; Esteban et al., 2003). Thus, a deficit in RIIβ, which is required for correct targeting of PKA to AMPARs could account for impaired LTP in the CA1 of RIIβ knockout mice. Our finding that LTP is not completely abolished even at young ages could be explained by a role for CaMKII-dependent, PKA-independent delivery of GluR2-long. In contrast, in adult hippocampus, activity drives PKA-independent insertion of GluR2-long, as well as PKA-dependent delivery of GluR1 (Man et al., 2007). These findings predict a slight impairment of LTP at CA1 synapses of adult RIIβ−/− mice. Thus, PKA phosphorylation of AMPARs mediates plasticity through synaptic incorporation of AMPARs early in development; in mature hippocampus, PKA is thought to play a “gating” role through phosphorylation and delivery of GluR1 (Esteban et al., 2003). Our finding that LTD is impaired in PKA RIIβ−/− mice in young adulthood, but not early in development is novel. Further experiments are warranted to clarify the role of PKA in LTD.
In visual cortex, regulatory subunits type I and type II of PKA differentially regulate synaptic plasticity. Whereas the PKA RI subunit is required for LTP and LTD at synapses onto layer II/III visual cortex cells, but not ocular dominance plasticity (Hensch et al. 1998), the PKA RIIβ subunit is required for ocular dominance plasticity and LTD, but not LTP (Fischer et al., 2004). Pharmacological blockade by PKA abolishes all three forms of plasticity (Beaver et al., 2001; Liu et al., 2003). Targeted deletion of RIβ severely reduces LTP at mossy fiber-CA3 synapses (Brandon et al., 1995) and homosynaptic LTD and depotentiation (but not LTP) at Sch-CA1 synapses of adult mice at 4-6 weeks of age (Huang et al., 1995). Our findings are consistent with and extend these studies in that we identify a role not only for RIβ, but also for RIIβ in LTD at CA1 synapses of adult mice. These studies establish PKA subunit-specific roles in different forms of plasticity within a given region.
Our finding that LTP is impaired at developing Schaffer collateral-CA1 synapses of PKA RIIβ−/− mice is consistent with findings of others that LTP is impaired at developing thalamocortical synapses in the barrel cortex (Inan et al., 2006). Thus, the role of RIIß in NMDAR-dependent LTP at developing synapses is not limited to CA1 synapses. Our finding that LTP is normal at CA1 synapses of PKA RIIβ−/− mice at 3-4 weeks of age is consistent with findings that mice deficient in AKAP150-PKA coupling exhibit normal LTP at 4 weeks, but impaired LTP at 7-12 weeks (Lu et al., 2007). Our finding that LTD is impaired at mature CA1 synapses is consistent with findings LTD is impaired at synapses of mature visual cortex of PKA RIIβ−/− mice (Fischer et al., 2004). Our findings extend previous findings in that we show that PKA RIIβ subserves distinct forms of plasticity at CA1 synapses at different developmental stages. Interestingly, mice deficient in PKA anchoring exhibit impaired protein-synthesis dependent LTP at CA1 synapses at 8-12-weeks of age (Nie et al., 2007), suggesting a possible second “developmental switch” in LTP signaling.
The cAMP/PKA signaling cascade is crucial for synaptic plasticity in a wide variety of species. Our earlier finding that PKA activity increases Ca2+ permeability links Ca2+ signaling to NMDAR-dependent synaptic plasticity. Findings in the present study identify a role for PKA RIIβ in NMDAR-dependent synaptic plasticity at Sch-CA1 synapses. Our findings that 3 – 4 week old mice lacking PKA RIIβ exhibit normal LTP at Sch-CA1, but deficient LTD, indicate that distinct PKA isoforms may subserve distinct forms of synaptic plasticity. Our findings that young mice lacking PKA RIIβ exhibit deficient LTP and normal LTD are consistent with a developmental switch in the signaling cascades required for LTP induction. Given the widespread distribution of NMDARs and PKA throughout the CNS, the developmentally-dependent, subunit-specific regulation of NMDAR-dependent synaptic plasticity by PKA potentially represents a powerful mechanism to modulate synaptic efficacy.
Acknowledgments
The authors thank Dr. Pablo E. Castillo for helpful comments on the manuscript and Ms. Adrianna Latuszek-Barrantes for help in maintaining the mice and genotyping. The PKA RIIν knockout mice are a gift of Quentin Fischer (Yale University School of Medicine, New Haven, CT). This study was supported by NIH grants NS20752 (to R.S.Z.) and NS45287 (to M.V.L.B.) and Training Grant T32 DK07513. M.V.L.B. is the Sylvia and Robert S. Olnick Professor of Neuroscience and Distinguished Professor of the College.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Amieux PS, Cummings DE, Motamed K, Brandon EP, Wailes LA, Le K, Idzerda RL, McKnight GS. Compensatory regulation of RIalpha protein levels in protein kinase A mutant mice. J Biol Chem. 1997;272:3993–3998. doi: 10.1074/jbc.272.7.3993. [DOI] [PubMed] [Google Scholar]
- Beaver CJ, Ji Q, Fischer QS, Daw NW. Cyclic AMP-dependent protein kinase mediates ocular dominance shifts in cat visual cortex. Nat Neurosci. 2001;4:159–163. doi: 10.1038/83985. [DOI] [PubMed] [Google Scholar]
- Blank T, Nijholt I, Teichert U, Kugler H, Behrsing H, Fienberg A, Greengard P, Spiess J. The phosphoprotein DARPP-32 mediates cAMP-dependent potentiation of striatal N-methyl-D-aspartate responses. Proc Natl Acad Sci U S A. 1997;94:14859–14864. doi: 10.1073/pnas.94.26.14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1942. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- Brandon EP, Idzerda RL, McKnight GS. PKA isoforms, neural pathways, and behaviour: making the connection. Curr Opin Neurobiol. 1997;7:397–403. doi: 10.1016/s0959-4388(97)80069-4. [DOI] [PubMed] [Google Scholar]
- Brandon EP, Zhuo M, Huang YY, Qi M, Gerhold KA, Burton KA, Kandel ER, McKnight GS, Idzerda RL. Hippocampal long-term depression and depotentiation are defective in mice carrying a targeted disruption of the gene encoding the RI beta subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1995;92:8851–8855. doi: 10.1073/pnas.92.19.8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser ID, Cone RD, Scott JD. Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J Biol Chem. 1992;267:16816–16823. [PubMed] [Google Scholar]
- Carroll RC, Zukin RS. NMDA-receptor trafficking and targeting: implications for synaptic transmission and plasticity. Trends Neurosci. 2002;25:571–577. doi: 10.1016/s0166-2236(02)02272-5. [DOI] [PubMed] [Google Scholar]
- Cerne R, Rusin KI, Randic M. Enhancement of the N-methyl-D-aspartate response in spinal dorsal horn neurons by cAMP-dependent protein kinase. Neurosci Lett. 1993;161:124–128. doi: 10.1016/0304-3940(93)90275-p. [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Colledge M, Scott JD. AKAPs: from structure to function. Trends Cell Biol. 1999;9:216–221. doi: 10.1016/s0962-8924(99)01558-5. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Constantine-Paton M, Cline HT. LTP and activity-dependent synaptogenesis: the more alike they are, the more different they become. Curr Opin Neurobiol. 1998;8:139–148. doi: 10.1016/s0959-4388(98)80017-2. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Cummings DE, Brandon EP, Planas JV, Motamed K, Idzerda RL, McKnight GS. Genetically lean mice result from targeted disruption of the RII beta subunit of protein kinase A. Nature. 1996;382:622–626. doi: 10.1038/382622a0. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Fischer QS, Beaver CJ, Yang Y, Rao Y, Jakobsdottir KB, Storm DR, McKnight GS, Daw NW. Requirement for the RIIbeta isoform of PKA, but not calcium-stimulated adenylyl cyclase, in visual cortical plasticity. J Neurosci. 2004;24:9049–9058. doi: 10.1523/JNEUROSCI.2409-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, McDonough CB, Abel T. Compartmentalized PKA signaling events are required for synaptic tagging and capture during hippocampal late-phase long-term potentiation. Eur J Cell Biol. 2006;85:635–642. doi: 10.1016/j.ejcb.2006.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem. 1994;1:74–82. [PubMed] [Google Scholar]
- Huang YY, Kandel ER, Varshavsky L, Brandon EP, Qi M, Idzerda RL, McKnight GS, Bourtchouladze R. A genetic test of the effects of mutations in PKA on mossy fiber LTP and its relation to spatial and contextual learning. Cell. 1995;83:1211–1222. doi: 10.1016/0092-8674(95)90146-9. [DOI] [PubMed] [Google Scholar]
- Inan M, Lu HC, Albright MJ, She WC, Crair MC. Barrel map development relies on protein kinase A regulatory subunit II beta-mediated cAMP signaling. J Neurosci. 2006;26:4338–4349. doi: 10.1523/JNEUROSCI.3745-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama K, Lee HK, Bear MF, Huganir RL. Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron. 1998;21:1163–1175. doi: 10.1016/s0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kemp N, McQueen J, Faulkes S, Bashir ZI. Different forms of LTD in the CA1 region of the hippocampus: role of age and stimulus protocol. Eur J Neurosci. 2000;12:360–366. doi: 10.1046/j.1460-9568.2000.00903.x. [DOI] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Hell JW. Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-D-aspartate receptors at different sites. J Biol Chem. 1997;272:12107–12115. doi: 10.1074/jbc.272.18.12107. [DOI] [PubMed] [Google Scholar]
- Liu S, Rao Y, Daw N. Roles of protein kinase A and protein kinase G in synaptic plasticity in the visual cortex. Cereb Cortex. 2003;13:864–869. doi: 10.1093/cercor/13.8.864. [DOI] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Man HY, Sekine-Aizawa Y, Huganir RL. Regulation of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci U S A. 2007;104:3579–3584. doi: 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401–437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Nie T, McDonough CB, Huang T, Nguyen PV, Abel T. Genetic disruption of protein kinase A anchoring reveals a role for compartmentalized kinase signaling in theta-burst long-term potentiation and spatial memory. J Neurosci. 2007;27:10278–10288. doi: 10.1523/JNEUROSCI.1602-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Oliveria SF, Gomez LL, Dell’Acqua ML. Imaging kinase--AKAP79--phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol. 2003;160:101–112. doi: 10.1083/jcb.200209127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhova NA, Otmakhov N, Mortenson LH, Lisman JE. Inhibition of the cAMP pathway decreases early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 2000;20:4446–4451. doi: 10.1523/JNEUROSCI.20-12-04446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi M, Zhuo M, Skalhegg BS, Brandon EP, Kandel ER, McKnight GS, Idzerda RL. Impaired hippocampal plasticity in mice lacking the Cbeta1 catalytic subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1996;93:1571–1576. doi: 10.1073/pnas.93.4.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Tong G, Jahr CE. Beta-adrenergic regulation of synaptic NMDA receptors by cAMP- dependent protein kinase. Neuron. 1996;16:415–421. doi: 10.1016/s0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Santschi LA, Zhang XL, Stanton PK. Activation of receptors negatively coupled to adenylate cyclase is required for induction of long-term synaptic depression at Schaffer collateral-CA1 synapses. J Neurobiol. 2006;66:205–219. doi: 10.1002/neu.20213. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Huganir RL. Postsynaptic organization and regulation of excitatory synapses. Nat Rev Neurosci. 2000;1:133–141. doi: 10.1038/35039075. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP- regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–10303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- Wang LY, Orser BA, Brautigan DL, MacDonald JF. Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature. 1994;369:230–232. doi: 10.1038/369230a0. [DOI] [PubMed] [Google Scholar]
- Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. J Neurosci. 1994;14:645–654. doi: 10.1523/JNEUROSCI.14-02-00645.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, Malenka RC. A developmental switch in the signaling cascades for LTP induction. Nat Neurosci. 2003;6:15–16. doi: 10.1038/nn985. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]