

Abstract
A kinetically controlled diastereoselective cycloaddition between a chiral enol ether and an ortho-quinone methide (o-QM) produces a chroman spiroketal motif that is found in the core of berkelic acid, a novel matrix metalloproteinase (MMP) inhibitor and potent anticancer agent. The transformation lays the groundwork for preparation of future inhibitors aimed at distinguishing among the active sites of the twenty-three known MMP. Experimental findings suggest that the stereochemistry that emerges from cycloaddition is opposite that which results from thermodynamic ketalization.
Keywords: o-quinone methide, chroman spiroketal, berkelic acid, diastereoselective synthesis, MMP, anticancer agent, ketalization
(-)-Berkelic acid (1), a unique chroman spiroketal, was isolated from green Penicillium slime thriving in the largest superfund clean-up site in America in Butte, Montana (Scheme 1).1 The organism that produces 1 flourishes in a witches’ brew of mercury, cadmium, copper, and other heavy-metal contaminants dissolved in a 1.5 mile long and 1,800 foot-deep waste pit filled with acidic water (pH 2.0). (–)-Berkelic acid (1) displays potent anticancer activity toward OVCAR-3 in the NCI-60 panel screen (GI50 = 91.0 nM) and it inhibits MMP-3 and caspase-1 (GI50 = 1.87 mM; GI50 = 98.0 mM, respectively). Perhaps the unusual bioactivity of 1 is related to its ability to protect its parent organism from the surrounding hostile environment, which is as extreme as the nearby volcanic estuaries of Yellowstone park.
Scheme 1.
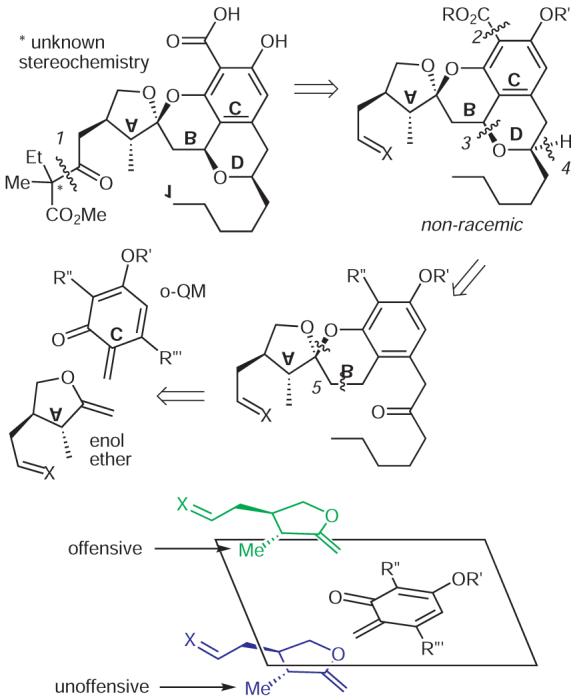
Our retrosynthetic strategy for 1 suggests starting with a derivative of a chiral enol ether corresponding to ring A of 1
The MMP are zinc-dependent endopeptidases that cause degradation of peptide bonds within the extracellular matrix (ECM). These enzymes play a crucial role in angio-genesis and tumor invasion.2 In the case of ovarian cancer, tumors originate from the ovarian surface epithelium (OSE) and require MMP activity for infiltration of the surrounding connective tissues of OSE. Thus, the implications are that MMP inhibition should correlate with anticancer activity towards ovarian carcinomas. Of the twenty-three MMP identified thus far, several (−3, −8, −9, and −12) are proven antitargets that when inhibited enhance angiogenesis and metastasis.3 This paradoxical property likely led to the clinical failure of the first generation of broad-spectrum MMP inhibitors. Future generations must spare antitargets,4 yet inhibit validated cancer targets,5 such as MMP (−1, −2, and −7). Diminished zinc chelation might enable better differentiation among the subtle steric differences found with the side pockets of the twenty-three MMP. Compound 1 is enormously different from prior MMP inhibitors,6 and it may possess sufficient structural complexity to probe the subtle differences among the active sites of these related enzymes.
A first step in improving selectivity among MMP in our minds was the development of a straightforward synthesis of this rare chroman spiroketal (Scheme 1). The unassigned quaternary center, which is far removed from other stereocenters, however, complicates planning. This fixture would have to be introduced near the end of the synthesis through enolate addition to the -CH=X residue masked in earlier intermediates as either a nitrile or an ether so that both stereochemical possibilities could be accessed with minimal effort (disconnection 1). Our plan, therefore, simplified to the diastereoselective synthesis of the tetracyclic (ABCD) core with the added provision that the salicylic acid involved with zinc coordination could be readily modified so that the effects of other residues on MMP selectivity might be studied (disconnection 2). We suspected that the chroman spiroketal ABC would be conformationally locked and thus would enable the construction of the isobenzopyran ring D by stereoselective oxidation and reduction (disconnections 3 and 4). Therefore, in our mind the entire strategy hinged on whether the a-methyl residue in the enol ether A would control the stereochemistry accruing in its cycloaddition with C (disconnections 5) instead of the b-substituent in A, which we saw as too distant from the forming spiroketal carbon to exert much influence.
To resolve this question, we chose a short, versatile entry point, which began by the addition of lithiated acetonitrile to the unsaturated lactone 2 (Scheme 2). This reaction avoids the problem of an undesirable proton transfer during the course of the 1,4-conjugate addition, enables a one-pot procedure to secure the two stereocenters, and provides access to several functional groups which could serve as a surrogate for -CH=X. The intermediate enolate expectedly undergoes in situ C-methylation with methyl iodide and affords the trans-substituted lactone 3 in 73% yield (>10:1 dr). After chromatographic purification, refluxing lactone 3 (0.1 M 3:2 v/v ratio of toluene—THF) with Petasis’ reagent (2.5 equiv, 4 h) affords the enol ether 5 in an acceptable 40–50% yield after sequential purification by flash chromatography and distillation.7 No isomerization was evident after purification. Another derivative of A can be alternatively accessed by exposure of the nitrile 3 to a solution of acetic acid and concentrated HCl (1:10 ratio); these conditions produce the carboxylic acid 4 in 95% yield. Subsequent reduction of the acid 5 (0.06 M in THF, 2.0 equiv BH3 ·THF), protection of the resulting alcohol (0.1 M in Et2O, 1 equiv BnBr, 1 equiv Ag2O), and methylenation of the lactone (0.5 M in toluene, 2.5 equiv Petasis, 75 °C, 4 h) affords after distillation the enol ether 6 as an alternative entry point. We were now ready to examine the role of 5 and, if needed, 6, in determining the diastereoselectivity of cycloaddition reactions with various o-quinone methides.
Scheme 2.
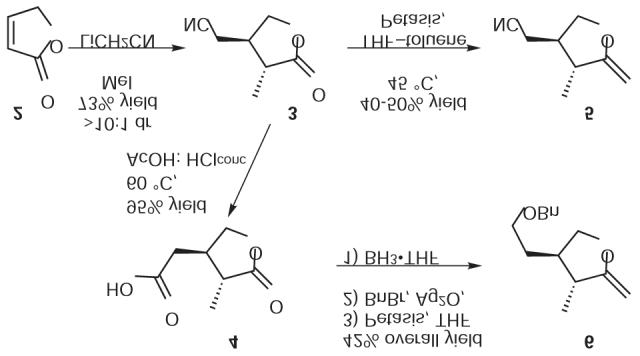
Synthesis of some enol ethers with -CH=X equivalency
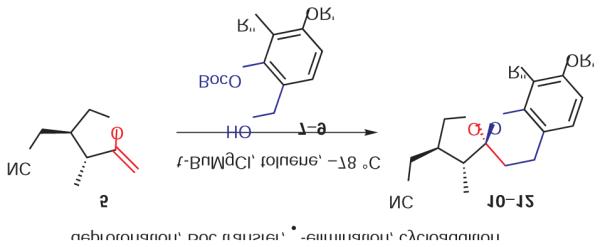
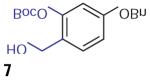
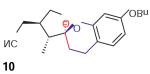
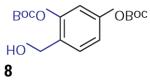
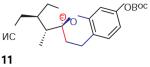
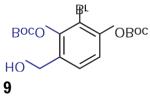
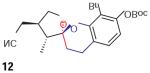
We found that treatment of the benzyl alcohol 7 (0.1 M in toluene at −78 °C with 2–4 equiv of the enol ether 5) with tert-butylmagnesium chloride followed by slow warming to room temperature over eight hours resulted in the formation of the chroman spiroketal 10 (Table 1). Flash chromatography allowed isolation of the major diastereomer in 61% yield. Exchange of the benzyl ether for a more electron-deficient OBoc residue as in 88 led to a small increase in diastereoselectivity (3.5:1 for 11), but the diastereomers proved inseparable. On the other hand, introduction of a bromine atom to the o-QM generated from 9 resulted in still higher selectivity (4.5:1 for 12). The bromine atom also facilitates chromatographic separation of the diastereomers enabling isolation of the major spiroketal 12 (72% yield) and minor diastereomer 12¢ (8% yield).
Table 1.
Diastereoselective Cycloadditions
The dr is based on 1H NMR analysis of crude product mixtures.
The combined yield of two isomers.
In order to confirm the relative stereochemistry of the predominant chroman spiroketal produced in these studies, we had attempted to crystallize compounds 10–12, but were unsuccessful. Therefore, we decided to cleave the Boc group from compound 12 so that the phenol functional group might allow for more straightforward crystallization. This task was accomplished under mild conditions by treatment of 12 (0.1 M in THF) with 5% LiOH (10 equiv) to furnish the phenol 13, which gratifyingly crystallizes from dichloromethane and methanol (1:1) to provide X-ray quality crystals (Figure 1). The relative configuration of the major product suggests that the reaction proceeds through an endo orientation on the face of the enol ether opposite the a-methyl residue and on the same side as the b-methylenenitrile. This outcome was what we had expected from our earlier investigation of diastereoselective cycloadditions of o-quinone methides generated at low temperature by this pKa -driven cascade.9
Figure 1.

X-ray crystal structure of 13
Next, we performed Macromodel calculations with MM3 and Monte Carlo to determine the relative energies of the simplified analogues (R1 = R2 = R4 = H) corresponding to diastereomers 12 and 12¢, which emerge from the corresponding endo and exo transition states (Figure 2). We find that the lowest energy for each conformer is obtained by formation of a half-chair spiroketal, whereby the furan oxygen atom is oriented in axial fashion to benefit from anomeric stabilization. Remarkably, the endo product is calculated to be 5 kJ/mol less stable than the exo product. The cause is quite clear; the methyl group adjacent to the spiroketal carbon atom prefers to reside in proximity to the chroman oxygen atom (exo product) as opposed to the methylene (endo product).
Figure 2.
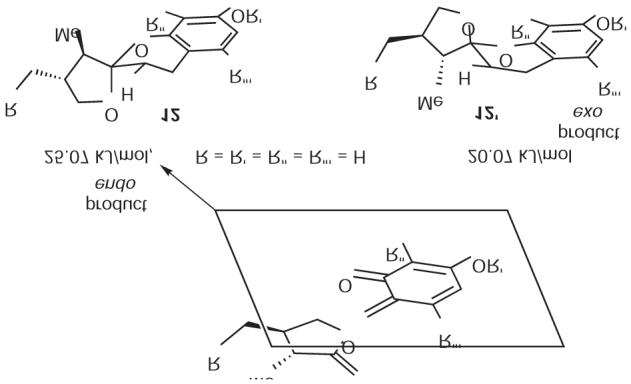
Relative stabilities of two diastereomers; endo is 5 KJ/mol less stable than exo
To experimentally confirm the meaning of this calculation, the major diastereomer 12 and the minor diastereomer 12¢ that had emerged from the earlier Diels—Alder reaction were separated and independently subjected to acidic THF (0.2% TFA). Over time (48 h), the major isomer 12 equilibrates to a 1:2 mixture of 12:12¢ (Scheme 3). Similarly, the minor diastereomer 12¢ equilibrates to a 1:2 mixture of 12:12¢. This result indicates that the endo product 12 is the kinetic product, whereas the minor diastereomer 12¢ is the thermodynamic product. Moreover the ratio (1:2, 12/12¢) reflects the energy difference of the previous calculation.
Scheme 3.
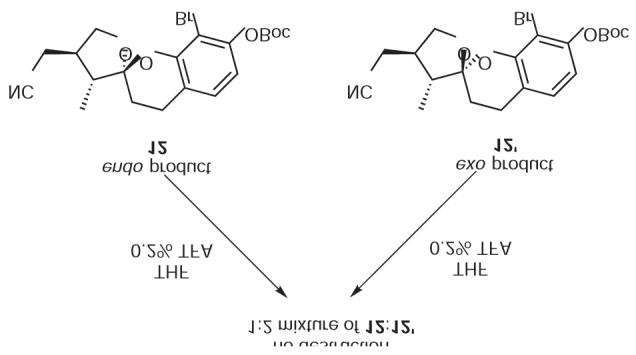
Equilibration of 12 or 12¢ into a 1:2 mixture
In conclusion, we have shown that several b-unsubstituted o-QM undergo diastereoselective [4+2] cycloaddition re-Br actions with a range of chiral furanoid enol ethers whereby the stereocenter closest to the site of bond formation determines the diastereoselective outcome of the reaction. The stereochemistry that resides within the major product proves the reaction proceeds through a kinetically favored endo-boat transition state. This core strategy could prove useful in the eventual diastereoselective synthesis of 1 by enabling formation of the unknown quaternary center near the end of its total synthesis. Most significantly, however, the equilibration study raises the question as to whether a recently proposed strategy for 1 can reach the fully elaborated natural product since the effect of stereochemistry within the furan component was not examined in the earlier thermodynamic ketalization strategy.10
Compound 3
To a stirring solution of n-BuLi (4.45 mL, 7.125 mmol) in THF (12.5 mL) at −78 °C was added MeCN (1.50 mL, 4.0 equiv) over 2 min. The reaction was stirred for an additional 2 min at −78 °C, and then lactone 2 (500 mL, 7.125 mmol) dissolved in THF (3 mL) was added dropwise. After 2 h stirring, a solution of MeI (440 mL, 7.125 mmol) and HMPA (1.25 mL, 7.125 mmol) in THF (6 mL) was added slowly via cannula. The reaction was allowed to warm up to r.t. over 6 h and quenched with 0.5 M HCl. The mixture was extracted with EtOAc (3•). The combined organics were then washed with sat. NaHSO3 , brine, H2 O, dried (MgSO4 ), filtered, and concentrated to give crude product 3 (720 mg) in 73% yield. No further purification was required.
1H NMR (400 MHz, CDCl3 ): d = 4.50 (dd, J1 = 9.3 Hz, J2 = 7.3 Hz, 1 H), 3.97 (t, J = 9.2 Hz, 1 H), 2.62 (t, J = 5.7 Hz, 2 H), 2.58–2.40 (m, 2 H), 1.35 (d, J = 7.0 Hz, 3 H) ppm. 13C NMR (100 MHz, CDCl3 ): d = 177.8, 116.5, 69.0, 39.8, 39.6, 19.5, 13.8 ppm.
Compound 5
To a stirring solution of compound 3 (100 mg, 0.7168 mmol) in THF (2.7 mL) was added the Petasis reagent (3.5 mL, 0.5 M in toluene). The reaction was warmed up to 75 °C and stirred for 4.5 h. The reaction was then cooled down to r.t. and treated with NaHCO3 (100 mg), MeOH (1.63 mL), and H2 O (60 mL), followed by warming to 45 °C and stirred overnight. The reaction mixture was diluted with hexane (40 mL), filtered through Celite (washing with hexane), and concentrated to give the crude product. Purification by column chromatography [i-PrOH–hexanes (1:10) with trace Et3N and using KMnO4 stain) followed by bulb-to-bulb distillation (0.16 torr, 80 °C) provided enol ether 5 (40 mg) in 40% yield.
1H NMR (400 MHz, CDCl3 ): d = 4.29 (t, J = 2.0 Hz, 1 H), 4.29 (t, J = 8.0 Hz, 1 H), 3.87 (t, J = 2.0 Hz, 1 H), 3.81 (t, J = 8.0 Hz, 1 H), 2.57–2.43 (m, 3 H), 2.26–2.18 (m, 1 H), 1.24 (d, J = 6.8 Hz, 3 H) ppm. 13C NMR (100 MHz, CDCl3 ): d = 166.3, 117.7, 80.5, 72.0, 42.0, 41.0, 19.0, 16.9 ppm.
Compound 7
To a stirring solution of 4-benzyl-2-hydroxy benzaldehyde (300 mg, 1.316 mmol) in DMF (20 mL) at r.t. was added NaH (58 mg, 60% in mineral oil), followed by Boc anhydride (315 mg). The reaction was allowed to stir overnight and quenched with cold H2O. The reaction mixture was extracted with Et2O (3•). The combined Et2O extracts were washed with brine, dried over MgSO4 , filtered, and concentrated to give the crude product. Flash chromatography on silica gel afforded tert-butyl 5-(benzyloxy)-2-formyl phenyl carbonate (400 mg) in 93% yield.
1H NMR (400 MHz, CDCl3 ): d = 10.0 (s, 1 H), 7.82 (d, J = 8.8 Hz, 1 H), 7.43–7.37 (m, 5 H), 6.96 (dd, J1 = 8.6 Hz, J2 = 2.4 Hz, 1 H), 6.85 (d, J = 2.4 Hz, 1 H), 5.13 (s, 2 H), 1.59 (s, 9 H) ppm. 13C NMR (100 MHz, CDCl3 ): d = 187.6, 164.4, 153.7, 151.3, 135.6, 133.0, 128.9, 128.6, 127.8, 122.2, 113.1, 109.5, 84.7, 70.8, 27.8 ppm.
To a stirring solution of tert-butyl 5-(benzyloxy)-2-formyl phenyl carbonate (400 mg, 1.219 mmol) in THF at 0 °C was added BH3 ·SMe2 (640 mL, 2 M in THF, 1.05 equiv). The reaction was allowed to warm up to r.t. over 3 h and quenched by 0.1 M HCl. The mixture was extracted with EtOAc (3•), and the combined extracts were then washed with brine, H2 O, dried (MgSO4 ), filtered, and concentrated to give product 7 (382 mg) in 95% yield. No further purification was required.
1H NMR (400 MHz, CDCl3 ): d = 7.44–7.34 (m, 6 H), 6.89 (dd, J1 = 8.5 Hz, J2 = 2.6 Hz, 1 H), 6.80 (d, J = 2.6 Hz, 1 H), 5.05 (s, 2 H), 4.56 (s, 2 H), 2.05 (br s, 1 H), 1.57 (s, 9 H) ppm. 13C NMR (100 MHz, CDCl3 ): d = 159.6, 152.6, 149.9, 136.6, 131.0, 128.8, 128.3, 127.7, 125.5, 113.2, 109.0, 84.3, 70.5, 60.4, 27.8 ppm.
General Procedure for Compounds 10–12
Enol ether (2.0 equiv) was dissolved in anhyd toluene (0.1 M) and added to a stirring solution of the benzyl alcohol (1.0 equiv) in anhyd toluene (0.1 M) at −78 °C. Next, t-BuMgCl (1.1 equiv 1 M in toluene) was added slowly, and the reaction was allowed to warm up to r.t. over 3 h. Upon completion by TLC, the reaction was then quenched by 0.1 M HCl, extracted with EtOAc, washed with brine, H2 O, dried (Mg2 SO4 ), and concentrated.
Compound 10
1H NMR (500 MHz, CDCl3 ): d = 7.43–7.31 (m, 5 H), 6.97 (d, J = 8.4 Hz, 1 H), 6.54 (dd, J1 = 8.4 Hz, J2 = 2.6 Hz, 1 H), 6.45 (d, J = 2.5 Hz, 1 H), 5.04 (s, 2 H), 4.23 (dd, J1 = J2 = 8.6 Hz, 1 H), 3.68 (dd, J1 = 8.7 Hz, J2 = 7.6 Hz, 1 H), 3.00 (ddd, J1 = 15.0 Hz, J2 = 14.0 Hz, J3 = 6.0 Hz, 1 H), 2.71–2.61 (m, 2 H), 2.59 (dd, J1 = 17.0 Hz, J2 = 5.5 Hz, 1 H), 2.51 (dd, J1 = 17.0 Hz, J2 = 7.0 Hz, 1 H), 2.01 (ddd, J1 = J2 = 13.0 Hz, J3 = 6.0 Hz, 1 H), 1.92 (ddd, J1 = 13.0 Hz, J2 = 6.0 Hz, J3 = 3.0 Hz, 1 H), 1.17 (d, J = 6.8 Hz, 1 H) ppm. 13C NMR (125 MHz, CDCl3 ): d = 158.5, 153.3, 137.3, 129.7, 128.7, 128.1, 127.6, 117.9, 114.4, 108.3, 106.8, 103.2, 70.4, 70.2, 48.1, 40.7, 28.2, 21.3, 19.8, 12.0 ppm. IR (CH2 Cl2 solution): nmax = 2958, 2929, 2885, 2360, 2341, 1622, 1583, 1506, 1458, 1380, 1267, 1150, 1114, 1014, 987, 906, 877, 738, 696 cm–1. HRMS (ESI+/TOF): m/z calcd for C22 H23 NO3 Na [M + Na]+ : 372.1570; found: 372.1573.
Compound 11
1H NMR (500 MHz, CDCl3): d = 7.05 (d, J = 3.0 Hz, 1 H), 6.70 (dd, J1 = 6.0 Hz, J2 = 3.0 Hz, 1 H), 6.62 (d, J = 3.0 Hz, 1 H), 4.22 (dd, J1 = 8.0 Hz, J2 = 1.5 Hz, 1 H), 3.72 (dd, J1 = 8.0 Hz, J2 = 1.5 Hz, 1 H), 2.94 (m, 1 H), 2.80–2.78 (m, 1 H), 2.69–2.57 (m, 1 H), 2.33–2.27 (m, 2 H), 2.02–1.98 (m, 1 H), 1.88–1.83 (m, 1 H), 1.55 (s, 9 H), 1.13 (d, J = 7.2 Hz, 3 H).
Compound 12
1H NMR (500 MHz, CDCl3 ): d = 7.03 (d, J = 8.5 Hz, 1 H), 6.76 (d, J = 8.5 Hz, 1 H), 4.24 (dd, J1 = J2 = 8.7 Hz, 1 H), 3.71 (dd, J1 = 8.8 Hz, J2= 7.7 Hz, 1 H), 3.08 (ddd, J1 = 16.0, J2 = 13.0 Hz, J3 = 6.0 Hz, 1 H), 2.80–2.71 (m, 2 H), 2.62 (dd, J1 = 17.0, J2 = 5.0 Hz, 1 H), 2.55 (dd, J1 = 17.0, J2 = 6.5 Hz, 1 H), 2.03–1.90 (m, 3 H), 1.57 (s, 9 H), 1.22 (d, J = 6.8 Hz, 2 H) ppm. 13C NMR (125 MHz, CDCl3 ): d = 151.3, 150.2, 147.8, 128.1, 121.2, 117.8, 115.1, 107.6, 106.5, 84.2, 70.5, 48.0, 40.6, 27.86, 27.42, 22.0, 19.7, 12.1 ppm. IR (CH2 Cl2 solution): nmax = 2977, 2933, 1760, 1477, 1423, 1286, 1236, 1151, 1062, 1010, 989, 894, 879 cm–1. HRMS (ESI+/TOF): m/z calcd for C20 H24 NO5 NaBr [M + Na]+ = 460.0730; found: 460.0737.
Compound 13
To a stirring solution of 12 (20 mg) in THF (3 mL) and MeOH (3 mL), LiOH (5%, 3 mL) was added at r.t. The reaction mixture was stirred at r.t. for 12 h. The reaction mixture was diluted with EtOAc and neutralized with HCl (0.1 M) until the pH of the solution changed to 7. The reaction mixture was extracted with EtOAc. The combined EtOAc extracts was washed with brine, dried over MgSO4 , filtered, and concentrated to give the crude product 13 (15.5 mg, 99%) without further purification. The product 13 can be recrystallized from a mixture of MeOH and CH2 Cl2 (1:1); mp 124–126 °C.
1H NMR (400 MHz, CDCl3 ): d = 6.94 (d, J = 8.3 Hz, 1 H), 6.77 (d, J = 8.4 Hz, 1 H), 5.31 (br s, 1 H), 4.28 (dd, J1 = 9.1 Hz, J2 = 8.3 Hz, 1 H), 3.72 (dd, J1 = 9.2 Hz, J2 = 5.7 Hz, 1 H), 3.06 (dd, J1 = 16.7 Hz, J2 = 8.3 Hz, 1 H), 2.96 (ddd, J1 = 16.8 Hz, J2 = 13.8 Hz, J3 = 6.0 Hz, 1 H), 2.87 (dd, J1 = 16.7 Hz, J2 = 7.5 Hz, 1 H), 2.74 (ddd, J1 = 15.8 Hz, J2 = 5.7 Hz, J3 = 2.3 Hz, 1 H), 2.44 (qd, J1 = 7.4 Hz, J2 = 2.6 Hz, 1 H), 2.40–2.32 (m, 1 H), 2.05 (ddd, J1 = 13.5 Hz, J2 = 6.1 Hz, J3 = 2.6 Hz, 1 H), 1.88 (ddd, J1 = J2 = 13.0 Hz, J3 = 6.0 Hz, 1 H), 1.15 (d, J = 7.3 Hz, 2 H).
Acknowledgment
A research grant from the University of California, Cancer Research Coordinating Committee [http://crcc.ucdavis.edu/] for the investigations leading to the synthesis of 1 is greatly appreciated.
References
- (1).Stierle AA, Stierle DB, Kelly K. J. Org. Chem. 2006;71:5357. doi: 10.1021/jo060018d. [DOI] [PubMed] [Google Scholar]
- (2).Wasserman ZR. Chem. Biol. 2005:143. doi: 10.1016/j.chembiol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- (3).Overall CM, Kleifeld O. Br. J. Cancer. 2006;94:941. doi: 10.1038/sj.bjc.6603043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Itoh Y, Nagase H. Essays Biochem. 2002;38:21. doi: 10.1042/bse0380021. [DOI] [PubMed] [Google Scholar]
- (5).Zucker S, Cao J, Chen WT. Oncogene. 2000;19:6642. doi: 10.1038/sj.onc.1204097. [DOI] [PubMed] [Google Scholar]
- (6).Engel CK, Pirad B, Schimanski S, Kirsch R, Habermann J, Kingler O, Schlotte V, Weithmann KU, Wendt KU. Chem. Biol. 2005:181. doi: 10.1016/j.chembiol.2004.11.014. [DOI] [PubMed] [Google Scholar]
- (7).Petasis NA, Lu S. J. Am. Chem. Soc. 1995;117:6394. [Google Scholar]
- (8).Mejorado LH, Hoarau C, Pettus TRR. Org. Lett. 2004;6:1535. doi: 10.1021/ol0498592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9)(a).Selenski C, Pettus TRR. J. Org. Chem. 2004;69:9196. doi: 10.1021/jo048703c. [DOI] [PubMed] [Google Scholar]
- Selenski C, Mejorado LH, Pettus TRR. Synlett. 2004:1101. [Google Scholar]
- (10).Zhou J, Snider BB. Org. Lett. 2007;9:2071. doi: 10.1021/ol0704338. [DOI] [PMC free article] [PubMed] [Google Scholar]