


Abstract
Protein domain prediction is often the preliminary step in both experimental and computational protein research. Here we present a new method to predict the domain boundaries of a multidomain protein from its amino acid sequence using a fuzzy mean operator. Using the nr-sequence database together with a reference protein set (RPS) containing known domain boundaries, the operator is used to assign a likelihood value for each residue of the query sequence as belonging to a domain boundary. This procedure robustly identifies contiguous boundary regions. For a dataset with a maximum sequence identity of 30%, the average domain prediction accuracy of our method is 97% for one domain proteins and 58% for multidomain proteins. The presented model is capable of using new sequence/structure information without re-parameterization after each RPS update. When tested on a current database using a four year old RPS and on a database that contains different domain definitions than those used to train the models, our method consistently yielded the same accuracy while two other published methods did not. A comparison with other domain prediction methods used in the CASP7 competition indicates that our method performs better than existing sequence-based methods.
INTRODUCTION
The 3D structure of a protein holds the key to understanding the detailed function of a protein at the molecular level. However, the cost and time required for experimental structural characterization of larger (genomic) protein sets can be prohibitive, creating a need for developing accurate computational structure prediction approaches (1–3). Proteins can be considered to be built up from domains, where each domain can be thought of as a structural unit of a protein that is compact, local and constitutes a semi-independent unit capable of folding independently (4,5). Delineation of proteins into domains is often the first step in both experimental and computational protein research (6–9). Longhi and co-workers (10) suggest dividing large proteins into domains to increase the yield of protein crystals suitable for X-ray diffraction as large proteins are difficult to crystallize (11,12). Since the initial X-ray structure determinations of proteins were carried out for smaller, one domain proteins, the field of protein structure predictions was focused on one domain proteins. Thus, as a legacy, programs for protein structure prediction are still typically optimized for predicting structures for shorter one domain sequences. Moreover, a majority of eukaryotic proteins are multidomain proteins (13) and predicting the structure of long proteins continues to be a challenge (14). Copley and co-workers (15) present compelling arguments about analyzing genomes at the domain level rather than protein level. Also, reliable identification of domains influences the quality of multiple sequence alignments (16,17). Furthermore, the knowledge of domains is necessary for designing new chimeric proteins (18). Given the above listed applications, protein domain prediction continues to be an important area of research with broad utilities in protein science.
Most of the current approaches for protein domain boundary prediction can be classified into three broad categories (19): domain homology prediction, domain recognition and new domain prediction. Domain homology prediction methods take advantage of the close homology to known domain sequences. In this approach, databases, such as CATH (20), SCOP (21), Pfam (22), CDD (23) or SMART (24), are searched for a close match with the query sequence, and domains are assigned based on sequence similarities. Domain homology prediction is very efficient, provided homologs exist, e.g. the prediction method CHOP (25) uses this technique. In domain recognition methods, the database of proteins with known structures is searched for sequences that exhibit remote homology with the query sequence (26). In this approach, the remote homologs can be identified using sequence-based methods like PSI-BLAST (27) or using auxiliary information such as the predicted secondary structure (28,29). Both domain homology prediction and domain recognition methods rely on multiple sequence alignments (MSA). These methods, especially those based on artificial neural networks (NNs) (30–32), would still be unreliable for truly novel sequences, i.e. those that do not have detectable homology with protein sequences with known structures. For novel sequences, new domain prediction methods, which only use the amino acid sequence as the input, are often appropriate. Some examples in this direction include SnapDRAGON (33), RosettaDOM (34), DomCut (18) and Armadillo (35). The first two programs infer domains by initially predicting a coarse-grained tertiary structure that can be used to delineate domain boundaries. This methodology often gives good results, but typically requires significant computational resources. Other methods rely on machine learning or statistical models trained on biochemical properties of the amino acids, averaged over a window of the query protein. While these methods are fast and independent of homologs in the databases, they are rarely used because of their limited accuracies. Hybrid methods that combine several sources of information have been proposed in the past, but the performance gains have been modest. For example, in Biozon (31), the features derived from MSA, physiochemical properties of amino acids, secondary structures, exon boundary information, etc., are integrated using NNs. KemaDom (36) is another hybrid method that uses predicted secondary structure, predicted solvent accessibility, amino acid entropy and physiochemical properties of amino acids as input to an ‘ensemble’ of three support vector machines.
We propose a different method, which we call FIEFDom (Fuzzy Integration of Extracted Fragments for Domains), for predicting the domain boundaries of proteins from a given sequence and its sequence profile (a 2D matrix that represents the likelihood of each amino acid occurring at every position along the protein sequence) using a fuzzy mean operator (FMO). A FMO represents a special case of the fuzzy nearest neighbor algorithm (37), with the number of classes set to one. The choice of FMO was motivated by its simplicity, transparency, ease of updating the method and more abstractly for its asymptotic error bounds. FIEFDom is transparent, i.e. the choice of the program to designate a region as a domain boundary can be traced back to all proteins in the local database that contributed to the decision, offering additional insight. Also, our model need not be trained or tuned whenever new examples of domain boundaries become available. The sequences of newly determined boundaries can just be appended to the reference database file. In addition, the users can choose the domain definitions (e.g. CATH or SCOP) to suit their needs, just by replacing the reference protein set (RPS). As the available data approaches infinity, the upper bound of the maximum error rate is at most twice the optimal Bayes’ error rate (38). We show that our procedure works well for a wide range of proteins: from ones with many close homologs to ones with only remote homologs. We illustrate the effects of redundancy and the number of reference proteins in the database on the accuracy of our method. We compare the performance of our method with two other methods, PPRODO (32) and DOMpro (30), adjusting our reference database as necessary to ensure impartial comparisons of the underlying algorithms. Finally, we compare the performance of our method with six sequence-based domain prediction methods that participated in CASP7 (39), both in domain number prediction accuracy and domain position prediction accuracy. An executable of the FIEFDom software is freely available for download at http://www.bhsai.org/downloads/fiefdom.
METHODS AND MATERIALS
Databases
SCOP is a manually curated database that contains structural domains defined by Alexei Murzin and his colleagues. This database is generally accepted as a standard for protein structure classification (40). For analysis of various aspects of FIEFDom, we use the following ASTRAL SCOP (41) databases: SCOP 1.65 (30%) (i.e. the ASTRAL SCOP version 1.65 database containing domain sequences with 30% maximum sequence identity), SCOP 1.69 (20%), SCOP 1.69 (30%), SCOP 1.69 (40%), SCOP 1.73 (30%) and SCOP 1.73 (95%). Table 1 shows the domain compositions of the above databases. Since ASTRAL SCOP databases contain sequences of individual domains, we concatenate domain sequences from the same protein chain to reconstruct the original multidomain proteins. Due to the relative scarcity of proteins with more than four domains in the SCOP database, we only consider proteins that contain up to four domains in this study. Each of these databases, with domain and domain boundary residues labeled, constitutes a RPS. We choose every other version of the SCOP database for analysis to provide a larger increment in the number of newly observed domains as opposed to using consecutive versions. For multidomain proteins, 20 residues before and after the true domain boundary (as defined by SCOP) are designated as boundary residues. We use this widely used (19,28,30,32,33,36) labeling protocol to facilitate a fair comparison with other methods. The method developed is not strongly dependent on the number of boundary residues picked. Note that we do not address the issue of predicting domains with non-contiguous sequences and consequently we discard such proteins. We found that less than 7% of the domains in SCOP have non-contiguous sequences.
Table 1.
Domain composition of proteins contained in the SCOP databases used in this work
| Number of domains | SCOP database version (maximum percentage sequence identity) |
|||||
|---|---|---|---|---|---|---|
| 1.65 (30%) | 1.69 (20%) | 1.69 (30%) | 1.69 (40%) | 1.73 (30%) | 1.73 (95%) | |
| One | 3145 | 3449 | 4153 | 4724 | 5432 | 10 303 |
| Two | 533 | 494 | 627 | 789 | 826 | 1653 |
| Three | 107 | 96 | 123 | 157 | 148 | 267 |
| Four | 20 | 9 | 21 | 25 | 25 | 66 |
| Total | 3805 | 4048 | 4924 | 5695 | 6431 | 12 289 |
Data in the first row indicate the number of one-domain proteins in each database. The second row contains the number of two-domain proteins, etc. The last row indicates the total number of proteins included in each database.
Procedure
We use a three-step procedure to predict domain boundaries. First, we generate the position specific scoring matrix (PSSM, a profile generated by PSI-BLAST program) (27) of the query sequence using a large database of known sequences. Second, we use the generated profile to search for similar fragments in the RPS. Third, the matches with the proteins in RPS are parsed, and the domain boundary propensity (PB, the likelihood of an amino acid to be in domain boundary) of the query protein is predicted using a FMO. These steps are detailed below.
In the first step, the profile of the query sequence is calculated using the PSI-BLAST program and the non-redundant or nr (ftp://ftp.ncbi.nih.gov/blast/db) database (42). We generate the profile by running the PSI-BLAST program for three iterations. Default values are used for the remaining parameters. In the second step, we perform profile-sequence alignment between the query profile and the proteins in the RPS to search for matching fragments by running the PSI-BLAST program a second time. During this step, the expectation value threshold (e-value) is set at 10 000. This high threshold ensures that the alignments retrieved contain both large and small protein fragments. The parameters for this two-stage PSI-BLAST protocol were optimized in a previous work on secondary structure prediction (43). In the third step, the matching fragments found in the second step are parsed and scored using the following scoring scheme (43):
| 1 |
The score, S, is formulated as a ‘dissimilarity’ measure. For instance, the fragments of proteins in the RPS that have high sequence similarity with the subsequences of the query protein have high statistical significance (or low e-value), and therefore have low scores. Finally, the domain boundaries (if any) are predicted using the scored fragments. For each residue, the PB is calculated from the domain boundary memberships (B) of the residues in the fragments that are aligned with the current residue. The PB of the query protein is calculated using the following expression for the FMO:
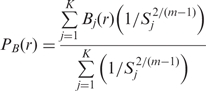 |
2 |
where, r is the current residue identifier, K is the number of fragments that have a residue aligned with the current residue r, Bj(r) ∈ (0 if the residue lies in the domain and 1 if the residue lies on the domain boundary) is the domain boundary membership of the residue in the jth fragment that has a residue aligned with the current residue r, Sj is the score for the jth fragment defined in Equation 1, and m is a fuzzifier (37) that controls the weight of the dissimilarity measure, S. The value of m was set to 1.5 based on previous work on secondary structure predictions (43). The boundary prediction results are not very sensitive to this parameter (data not shown). The values of PB(r) range from 0 to 1, where a value of 0 indicates that it is unlikely that r lies on a domain boundary, whereas a value of 1 indicates a strong likelihood that the residue is located in a boundary region. A typical alignment produced while searching for matching fragments in the RPS (step 3) is shown in Figure 1. The query protein is shown in the top line. The labels of residues that are aligned with residue ‘A’ (shaded box) are used to predict the PB(A) according to Equation 2, using the alignment scores shown on the right.
Figure 1.

The fragments retrieved when the RPS is searched for matching fragments with a typical protein. The fragments shown are labeled using their SCOP definitions. Residues labeled ‘D’ lie in protein domains, whereas residues labeled ‘B’™ lie on the domain boundary; ‘–’ is used to indicate that no residue in the current fragment is aligned with the query sequence. For the Alanine residue (A) in the shaded box, the domain boundary propensity is calculated using Equation 2 based on the five aligned residues (K = 5), four of which are found in non-boundary regions and one is found in a boundary region. The importance of these contributions is inversely weighted by their respective scores, S, shown on the right, as detailed in Equation 2. In this case, the likelihood PB that the alanine residue belongs to domain boundary is 0.0804.
Postprocessing
The values of PB are smoothed by averaging over a window of length W (W = 5, in this work) around each amino acid position in the query sequence. In the termini, the average is based only on those residues that are actually present in the window. The potential regions that contain domain boundaries are obtained by selecting those regions that have a PB value above a threshold value T, where T was set to 0.4. The details and the statistical measures underlying this choice are given in the next subsections. Once the potential regions are identified, the area under each identified sequence segment is calculated. We use this area to represent the confidence in the predicted domain boundary. If two regions lie within 40 residues of each other, the region with lower confidence is removed from further consideration. Also, predicted domain boundaries that fall within 40 residues of either the COOH or NH2 termini are discarded. The midpoint of each region is returned as the location of the domain boundary. As an example, the raw PB(r) output is illustrated for the Escherichia coli MurF protein [PDB: 1GG4, Chain A] in Figure 2. The predicted domain boundaries (residues 91 and 314) within two potential regions of interest are marked with dotted lines, agreeing very well with the actual boundaries centered on residues 98 and 313.
Figure 2.

The predicted raw domain boundary propensity (solid line) of the Escherichia coli MurF enzyme, PDB code 1GG4, chain A. Two regions that potentially contain domain boundaries are identified. The post-processing results in two predicted boundaries centered on residues 91 and 314 (dotted lines), whereas the true boundaries are centered on residues 98 and 313 (data not shown). The background noise that gets filtered out during the post-processing can be seen at the COOH- and NH2-terminal ends of the sequence.
Performance metrics
The performance is assessed in terms of three metrics: accuracy, specificity and sensitivity (29,35,44). These metrics are defined as follows:
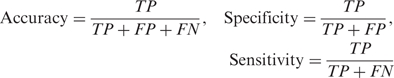 |
3 |
where TP denotes true positives (domain boundaries correctly predicted as domain boundaries), FP stands for false positives (regions incorrectly predicted as domain boundaries) and FN stands for false negatives (missed domain boundaries). Here we assume that if the predicted domain boundary is within 20 residues designated as boundary residues, the prediction is a true positive. Our definition of accuracy is appropriate since the term ‘true negative’ (all non-domain boundaries correctly predicted as non-domain boundaries) is not a practical concept in the context of domain boundary prediction. Also, for one-domain proteins, the accuracy is defined as the fraction of proteins in which no domain boundary is predicted.
Choice of threshold value, T
In this subsection we investigate the effect of the threshold, T, above which the regions on the PB curve are designated as potential regions containing domain boundaries. The post-processing step for the domain boundary prediction procedure involves applying a threshold T to filter the background noise and to designate potential regions that contain domain boundaries. We used SCOP 1.73 (30%) to study the effect of T on the sensitivity, specificity and accuracy of the domain boundary prediction. We systematically varied the value of T from 0 to 1 in increments of 0.1 and recorded the performance metrics as shown in Figure 3. We found that values of T in the range between 0.0 and 0.3 strongly influenced sensitivity, specificity and accuracy. For larger values, these measures remained relatively constant or had a plateau-like behavior in the region ∼0.3–0.5. Figure 3a illustrates the receiver operating characteristic (ROC) curve of the average multidomain predictions by varying T while Figure 3b illustrates the influence of T on the accuracy of one, two, three, four and all domain boundary predictions. Based on the plots in Figure 3, we fixed the value of T at 0.4 for all further analysis.
Figure 3.

The effect of threshold on the performance of FIEFDom for the SCOP 1.73 (30%) dataset. (a) Receiver operating characteristic (ROC) curve averaged over all of the domain sets is plotted as the threshold (T) is varied from 0 to 1 in intervals of 0.1. (b) One-domain (blue solid line), two-domain (pink dashed line), three-domain (black dotted line), four-domain (red dashed-dotted line) and the average domain boundary prediction accuracy are plotted as a function of the threshold value, T. Based on the maximum and slow variability of the accuracy values over a range of T values, we selected T = 0.4 as the appropriate value to be used in our model.
RESULTS
In this section, we analyze the performance of our method with varying levels of sequence/structure information availability in an attempt to simulate practical, real-life conditions. First, we present the results of the program under various conditions of homologous sequence availability for building a profile. Second, we investigate how growth of the RPS database affects accuracy. Third, we increase the redundancy of protein sequences (structure availability of related sequences) in the RPS and study its effect on our system's performance. We then compare the performance of our method with existing methods. We present results using a jack-knife procedure on the RPS, where each sequence in the RPS is used as a query protein, while the remaining proteins are used as the domain database for fragment searches.
Availability of homologs
In the nr database, some proteins have more homologs than others. The experiments described in this paragraph emulate various conditions under which homolog availability varies for the query protein using the SCOP 1.73 (30%) database. At one extreme, for query proteins that have many homologs in the nr database, the profile is rich in evolutionary information. Use of such profiles leads to more sensitive fragment searches in the RPS, resulting in higher prediction accuracy. The performance metrics when the query profile is used to indentify matching fragments are shown in Table 2 (first row, top section). On the other extreme, for proteins that do not have any homologs in nr, the profile returned is merely the scoring matrix [i.e. BLOSUM62 (45)] used in the alignment algorithm. A profile-sequence alignment in such a case is the same as a sequence-sequence alignment. To simulate the above scenario, for each protein, we perform sequence-sequence alignment using the query sequence directly (no profile is generated; only the second PSI-BLAST run is performed). The results are presented in Table 2 (second row, top section). These results help us draw the bottom line performance of our system, when the query sequences are truly novel and appear to have no known homologs. We can also infer that our system does not completely fail under these conditions; it only performs with reduced accuracy. The average accuracies on the SCOP 1.73 (30%) database using profile-sequence alignments for finding matching fragments in the RPS for one domain proteins and multidomain proteins are 97% and 58%, respectively while the average (specificity, sensitivity) for multidomain proteins is (91%, 61%). For sequence-sequence alignments, one and multidomain protein accuracies are 99% and 45%, respectively, and the average (specificity, sensitivity) for multidomain proteins is (92%, 48%). Note that, although average specificities of the two methods are comparable, the sensitivities of the method that uses profiles is significantly higher, reiterating the importance of evolutionary information (in the form of profiles) while searching for fragments. While the results clearly demonstrate the advantage of using a profile to aid the fragment search, they also indicate that the absence of profile, on average, reduces the multidomain accuracy of our method by 13%.
Table 2.
Studying the effect of homolog availability for building profiles, the number of proteins in the RPS and the effect of maximum sequence identity among the sequences in the RPS on the performance of FIEFDom
| Database | Alignment | Number of domains |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| One |
Two |
Three |
Four |
||||||||
| A | Sp | Sn | A | Sp | Sn | A | Sp | Sn | A | ||
| Homolog availability | |||||||||||
| SCOP 1.73 (30%) | PS | 97 | 88 | 60 | 55 | 95 | 61 | 59 | 90 | 63 | 59 |
| SCOP 1.73 (30%) | SS | 99 | 95 | 40 | 39 | 94 | 41 | 40 | 86 | 62 | 57 |
| Number of proteins in RPS | |||||||||||
| SCOP 1.65 (30%) | PS | 97 | 86 | 54 | 50 | 96 | 58 | 57 | 93 | 45 | 44 |
| SCOP 1.69 (30%) | PS | 97 | 90 | 57 | 54 | 93 | 58 | 56 | 91 | 49 | 47 |
| SCOP 1.73 (30%) | PS | 97 | 88 | 60 | 55 | 95 | 61 | 59 | 90 | 63 | 59 |
| Maximum sequence identity in RPS | |||||||||||
| SCOP 1.69 (20%) | PS | 97 | 86 | 43 | 41 | 90 | 42 | 40 | 71 | 19 | 17 |
| SCOP 1.69 (30%) | PS | 97 | 90 | 57 | 54 | 93 | 58 | 56 | 91 | 49 | 47 |
| SCOP 1.69 (40%) | PS | 97 | 91 | 67 | 63 | 92 | 66 | 62 | 93 | 56 | 54 |
A, accuracy; Sp, specificity; Sn, sensitivity. Alignment: PS profile-sequence, SS- sequence-sequence alignment. All values are percentages. Top: The availability of homology information for query sequences is simulated by using either the query profile (profile-sequence consistent with high availability) or the query sequence itself (sequence-sequence consistent with low availability) to search for identical fragments in the RPS. For multidomain proteins, the profile-sequence yields on average 13% higher overall accuracy, compared to the sequence-sequence alignment method. Middle: Every other version of the SCOP database, with 30% maximum sequence identity among the proteins, is used to study the effect of number of proteins in the RPS. The larger the size of the RPS (see Table 1 for the detailed breakdown in number of proteins and domain compositions), the higher is the average domain boundary prediction accuracy for multidomain proteins, presumably because the additional structure/sequence information uncovered as additional novel structures are added to the database. Bottom: Three simulations were conducted by experimenting with databases of three different maximum sequence identities among the reference proteins. The maximum sequence identity among the reference proteins varies from 20% to 40%.
Variability of the RPS database
We now turn to the performance of our method as new information is added to the RPS in the form of new protein sequences (for example, from newly sequenced genomes). We run our method on every other version of SCOP at the same sequence identity level, i.e. on SCOP 1.65 (30%), SCOP 1.69 (30%) and SCOP 1.73 (30%) databases. The same program is used to generate the alignments, parse the matches and calculate the PB curves. The only difference among the three experiments is the text file containing different RPSs, emphasizing the feature that updating the program amounts to merely appending (or replacing) the RPS text file. This advantage is unique to our approach due to the FMO-based model. The performance metrics of FIEFDom on various datasets for one and multidomain proteins are presented in Table 2 (middle section). The averages (specificity, sensitivity) for SCOP 1.65 (30%), SCOP 1.69 (30%) and SCOP 1.73 (30%) are (92%, 52%), (91%, 55%) and (87%, 63%), respectively. Note that as we move from an older database [SCOP 1.65 (30%)] to a newer database [SCOP 1.73 (30%)], the average specificity decreases while the average sensitivity increases. Concomitant with this trend, the average multidomain prediction accuracies increase from 50% for SCOP 1.65 to 58% for the SCOP 1.73 database, while the accuracy for one domain prediction remains at 97%. Quantitatively, we observed that, for every 1000 new protein sequences added to the RPS (while maintaining maximum sequence identity level), the overall accuracy (one domain and multidomain) increases roughly by 2.3%. Figure 4a shows one, two, three, four and average domain prediction accuracies plotted as a function of the database version. It is clear from Table 2 (middle section) and Figure 4a that, as time progresses, i.e. as additional sequence/structure information becomes available, the accuracy of FIEFDom increases due to availability of novel sequences that can be added to the RPS, without the need for retraining the model per se.
Figure 4.

(a) One-domain (red dashed line), two-domain (blue dashed-dotted line), three-domain (green dotted line), four-domain (solid magenta line) and average (bold solid black line) domain prediction accuracies are plotted as a function of database version. As time progresses, new information can be added to the prediction algorithm by updating the RPS. As the number of sequences in the database increases, the prediction accuracy improves. (b) The same domain prediction accuracies as in (a) are plotted as a function of maximum sequence identity cutoff in the RPS. More structural information is added to the prediction system by increasing the maximum sequence identity among proteins in the RPS.
The effect of protein sequence redundancy
Next, we study the dependency of the domain boundary likelihood, PB, on the redundancy of protein sequence information. This redundancy can be modeled by using RPSs of the same ASTRAL SCOP version, but with different sequence identity thresholds. Raising the maximum sequence identity among the sequences increases the number of available sequences in the RPS, thereby improving the chances of finding fragments in the RPS that are similar to the subsequences of the query sequence. We also simulate a real-life scenario where the RPS contains the sequences of all SCOP family members, but not the sequences that belong to same family as the query sequence. In this experiment, we run the jack-knife procedure with SCOP 1.69 (20%), SCOP 1.69 (30%) and SCOP 1.69 (40%). We did not experiment further with higher-identity thresholds for three reasons: higher thresholds might lead to bias in favor of highly sequenced protein families, 40% sequence identity is the lower limit after which comparative modeling for protein structure prediction becomes reliable (46), and the jack-knife procedure may not be objective beyond this threshold. Table 2 (bottom section) and Figure 4b summarize the results. For the database with lowest sequence identity [SCOP 1.69 (20%)], the average multidomain prediction accuracy is 33%. If we increase the maximum sequence identity in the RPS to 30%, the average multidomain prediction accuracy increases to 52%, while the one domain prediction accuracy remains constant at 97%. Further increasing the maximum sequence identity to 40% increases the average multidomain prediction accuracy to only 60%. The multidomain (specificity, sensitivity) for the SCOP 1.69 database with 20, 30 and 40% sequence identity cutoffs are (82%, 35%), (91%, 55%) and (92%, 63%), respectively. Both specificity and sensitivity increase with maximum sequence identity among the proteins in the RPS. Figure 4b clearly shows the substantial increase in accuracy seen for the multidomain proteins gained by looking at denser, or higher maximum sequence identity, databases.
Finally, we simulate a typical scenario where sequences of all SCOP family members are available in the RPS, but not the sequences that belong to same family as the query sequence. To simulate this case, we implement the following procedure. For each query sequence in the SCOP 1.73 (30%) database, we eliminate all sequences in the SCOP 1.73 (95%) database that belong to the same family as the query sequence and use the remaining proteins as the RPS. While we obtained an average one domain accuracy of 93%, the average multidomain accuracy is significantly lower at 14%. When we repeated the experiment with the super-family members of the query sequence removed from the RPS instead of family members, the one-domain prediction accuracy increased to 98%, but the accuracy of the multidomain accuracy is less then 1%. These results clearly indicate that FIEFDom is a domain recognition method that mainly predicts domain boundaries from alignments of the sub-sequences of the query sequence with its respective SCOP super-family members in the RPS.
Comparison with other domain-prediction programs
We now compare the performance of FIEFDom with two existing software programs, PPRODO (32) and DOMpro (30). We choose these two systems for comparison for multiple reasons. First, they are both relatively new and freely available for download. Second, like our method, both PPRODO and DOMpro are based on machine-learning methods that operate on protein profiles. Finally, the groups that developed these methods reported successful performance in CASP competitions (32,47). The first comparison is aimed at understanding how the three programs under consideration perform on a dataset that is more recent when compared to their training set (or RPS). The second comparison is aimed at understanding how the programs trained on SCOP domain definitions perform on proteins whose domain definitions are derived from the CATH database (20). PPRODO is an NN-based domain prediction system in which the profile extracted by the PSI-BLAST program is used as input to NNs for domain boundary prediction. A continuous signal is generated as output by the system, and the authors suggest a threshold of 0.25 above which an amino acid is designated as a domain boundary residue. DOMpro combines information from profiles, predicted secondary structures, and predicted relative solvent accessibility using recursive NNs. PPRODO was trained on two-domain proteins derived from SCOP 1.65 (released August 2003), and DOMpro was trained on the multidomain proteins in the CATH database version 2.5.1 (released January 2004). To make a fair comparison of different methodologies, we use FIEFDom with a RPS derived from the SCOP 1.65 (30%) (released August 2003) database. In the first comparison, we use the SCOP 1.73 (30%) (released September 2007) database as a test set, which was released about four years later than their respective training databases (PPRODO and DOMpro) or RPS (FIEFDom). Table 3 summarizes the performance characteristics of the three systems. The average multidomain prediction accuracy of FIEFDom on the SCOP 1.73 (30%) database is 80%, while the one domain prediction accuracy is 97%. The average multidomain accuracies of PPRODO and DOMpro are 36% and 13%, respectively. Their one domain accuracies are 56 and 80%, respectively. While testing PPRODO, we extracted the raw signal from the PPRODO output file and applied the cutoff suggested by the authors. One might argue that PPRODO used only two-domain proteins for training, and DOMpro used only multidomain proteins for training; hence, it is not fair to compare the results directly. To resolve these issues, we repeated the comparison twice with modified RPSs, once with the RPS containing only two-domain proteins and second time with the RPS containing only multidomain proteins. We summarize the results in Table 3. Thus, when FIEFDom uses the RPS that contains only two-domain proteins, the average multidomain prediction accuracy is 46%, and, when it uses the RPS with only multidomain proteins, the average accuracy is 79% while the respective one domain accuracies are 91% and 89%. From these results, it is clear that FIEFDom successfully maintains higher performance levels compared to these two programs when tested on a database that is more recent and even when a systematically domain-biased RPS is used. Note that PPRODO was optimized for predicting two-domain proteins only, and hence it has a tendency to divide many one-domain proteins into two-domain proteins. This tendency to overpredict domain boundaries is one of the main reasons for its lower accuracy compared to FIEFDom. On the other hand, the lower accuracies observed in the DOMpro model are due to its tendency to underpredict domain boundaries.
Table 3.
The performance metrics of the three programs on a dataset that is about four years further in time from the training or reference data
| Method | Number of domains |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
One |
Two |
Three |
Four |
|||||||
| A | Sp | Sn | A | Sp | Sn | A | Sp | Sn | A | |
| FIEFDom | 97 | 93 | 77 | 73 | 96 | 85 | 82 | 94 | 88 | 84 |
| PPRODO | 56 | 53 | 54 | 37 | 50 | 38 | 28 | 78 | 51 | 44 |
| DOMpro | 80 | 32 | 12 | 10 | 34 | 14 | 11 | 55 | 23 | 19 |
| FIEFDom (only two-domains) | 91 | 94 | 73 | 70 | 80 | 39 | 36 | 90 | 35 | 33 |
| FIEFDom (only multidomains) | 89 | 91 | 76 | 71 | 95 | 86 | 82 | 96 | 88 | 85 |
All values are percentages. Five prediction sets were generated to understand how FIEFDom (with three versions of the same RPS), PPRODO and DOMpro perform on the SCOP 1.73 (30%) database. The first row shows the performance of FIEFDom that uses the SCOP 1.65 (30%) database as the RPS. The second and third rows show the performance of PPRODO and DOMpro, respectively. The fourth and fifth rows show the performance of FIEFDom that uses a RPS containing only two-domain proteins or multidomain proteins, respectively.
For the second comparison, we predict the domain boundaries in the dataset used to develop DOMpro. The rationale here is to check how well the models trained on SCOP databases (FIEFDom and PPRODO) perform on proteins derived from the CATH database. The CATH-derived database used to train the DOMpro program contains 963 one-domain proteins and 354 multidomain proteins. Table 4 summarizes the results. Similar to the previous comparison, Table 4 also includes the performance of FIEFDom when using the RPS containing only two-domain proteins or multidomain proteins. The average domain prediction accuracies of FIEFDom, PPRODO and DOMpro on the CATH-derived database are 77%, 64% and 55%, respectively. If a RPS containing only two-domain proteins is used, then the accuracy of FIEFDom drops to 69%; when the RPS contains only multidomain proteins, the accuracy becomes 74%. It is clear from Table 4 that the application of FIEFDom on either of three different training sets (a RPS with one and multidomain proteins, a RPS with only two-domain proteins, and a RPS with multidomain proteins) yields, on average, better results compared with PPRODO and DOMpro. In this test, the slight variations (35,40,48,49) in domain definitions of the test database compared to the training database did not adversely affect the performance of our procedure.
Table 4.
The performance metrics of the three programs on a dataset that uses domain definitions derived from the CATH database
| Method | Number of domains |
|||
|---|---|---|---|---|
|
One |
Multi |
|||
| A | Sp | Sn | A | |
| FIEFDom | 92 | 91 | 65 | 61 |
| PPRODO | 90 | 58 | 51 | 37 |
| DOMpro | 91 | 58 | 21 | 18 |
| FIEFDom (only two domains) | 89 | 91 | 50 | 48 |
| FIEFDom (only multidomain) | 89 | 91 | 62 | 58 |
All values are percentages. Five prediction sets were generated to understand how FIEFDom (with three versions of the same RPS), PPRODO and DOMpro perform on a database that derives its domain definitions from the CATH database (version 2.5.1). The results for two-, three- and four-domain proteins have been averaged and are shown under ‘Multi’. The first row shows the performance of FIEFDom that uses the SCOP 1.65 (30%) database as the RPS. The second and third rows show the performance of PPRODO and DOMpro, respectively. The fourth and the fifth rows show the performance of FIEFDom that uses a RPS containing only two-domain proteins or multidomain proteins, respectively.
Comparison with other sequence-based methods in CASP7
We compared the domain number prediction accuracy of FIEFDom with six sequence-based methods (methods that do not use protein-fold information or ab initio processing) used in CASP7. The performance was measured across the 97 targets (70 one-domain proteins and 27 multidomain proteins) included in CASP7. In addition to domain number prediction accuracy, we also compared the ability of the methods to correctly predict both the domain number as well as the position of the domain boundary. For one-domain proteins we consider accuracy (A), and for multidomain number predictions, specificity (Sp), sensitivity (Sn) and accuracy (A) were determined. To rank the methods used in CASP7 we determined the average prediction accuracy of both one- and multidomain proteins for each method. If the position of at least one domain in a multidomain protein is not correctly predicted, the prediction is counted as a ‘partial’ success. If the positions of all domains in a multidomain protein are predicted correctly, it is counted as a ‘complete’ success. The results in Table 5 demonstrate that FIEFDom has comparable or better accuracy when compared to other methods. However, we caution that analyses based on small data sets, such as the target set used in CASP7, are less informative when compared to the large scale analyses shown in the previous section.
Table 5.
The performance of various sequence-based domain prediction methods on the 97 (70 one-domain proteins and 27 multidomain proteins) CASP7 targets
| Methods | Domain number |
Domain position |
|||||
|---|---|---|---|---|---|---|---|
|
One |
Multi |
Combined |
Multi |
||||
| A | Sp | Sn | A | A | Complete | Partial | |
| FIEFDom | 100 | 88.9 | 30.8 | 29.6 | 64.8 | 6 | 2 |
| CHOP (25) | 55.8 | 37.5 | 42.9 | 25.0 | 40.4 | 4 | 4 |
| DomSSEA (28) | 92.9 | 100 | 30.8 | 30.8 | 61.8 | 4 | 4 |
| DPSa | 80.5 | 100 | 42.3 | 42.3 | 61.4 | 5 | 2 |
| HHPred1a | 95.6 | 100 | 25.9 | 25.9 | 60.8 | 4 | 3 |
| HHPred3a | 95.7 | 100 | 25.9 | 25.9 | 60.8 | 4 | 3 |
| NNPutLaba | 78.5 | 80.0 | 15.4 | 14.8 | 46.6 | 2 | 3 |
All values under the domain number prediction are percentages. Sequence-based domain prediction methods that were used in the CASP7 are listed on left. For one domain number prediction, the accuracy (A) is listed. For multidomain number prediction, accuracy (A), specificity (Sp) and sensitivity (Sn) are listed. The domain number prediction accuracy for all targets in CASP7 set is listed under the ‘Combined’ heading. For the domain position prediction of multidomain proteins, the actual count of the proteins whose domain boundaries are predicted completely correct and partially correct is listed.
DISCUSSION AND CONCLUSION
We propose a new and transparent method to predict the domain boundaries for a given protein sequence. The method is based on finding fragments similar to the subsequences of the query sequence in the RPS and using a FMO to infer domain boundaries from these fragments. The query can either be a sequence or a sequence profile. Our algorithm provides a domain recognition method that mainly detects alignments to the super-family members (SCOP classification) of the query sequence in the RPS.
For sequences that have few or no homologs in the database, the profile of the sequence simply corresponds to the amino acid substitution matrix used in the construction of the profile. Use of such profiles in the profile-sequence alignment then becomes equivalent to performing the sequence-sequence alignment in the search of overlapping fragments. This, in effect, draws the lower boundary of our prediction accuracy in these cases. Conversely, if a query has a number of homologs in the database of known sequences, then the profile is well defined. Using a well-defined profile leads to more sensitive searches, resulting in higher prediction accuracy. A more rigorous implementation, using profile-profile alignment for finding similar fragments, is possible at the cost of increased computational time. In this way, our method can accommodate sequences that only have remote homologs with known boundaries (FIEFDom becomes a domain recognition method) and sequences that have many homologs with known domain boundaries (FIEFDom becomes a domain homology method).
One of the problems of many data-driven bioinformatics tools is that they quickly become outdated if developers do not take time to update or make use of new data that become available after the tool is released. Updating a tool generally involves training and fine tuning the system with new data. In our case, the implementation of the algorithm is separate from the data used by algorithm. Consequently, FMO in FIEFDom does not need any training. For example, a new sequence representing a novel fold, can be easily added to the system by appending to the existing sequence file, and such new information is readily accounted for in the subsequent queries. There are many other advantages of keeping the RPS separate from the algorithm itself. First, the user can add/remove sequences from the RPS, altering the number of homologous sequences available to the algorithm. Second, the user can define the domain boundaries using a different database (for example, CATH database). Third, the user may choose whether or not to label the termini of the proteins in the RPS as domain boundaries. One of the benefits of including N- and C-termini into the RPS is that domain boundaries can be recognized for proteins that contain segments similar to experimentally determined structural domains. For example, the structurally-characterized zinc-binding RING finger domain, which is typically 40–60 residues in length (50), is present in proteins from many eukaryotic and viral genomes. FIEFDom, with labeled termini in the RPS, can detect these domains within larger proteins and assign domain boundaries before and after the identified segment (results not shown). However, when we compared the results of the runs that used RPS with and without labeled termini, we found that the sensitivity of the termini-included run is increased at the cost of the specificity. Consequently, including the termini in the RPS results in lower one-domain accuracy and slightly higher multidomain accuracy. When we used the termini-included RPS on the SCOP 1.73 (30%) database, we obtained 81% one domain accuracy and (specificity, sensitivity, accuracy) of (78%, 71%, 59%) for multidomain proteins.
One of the advantages of our approach is the transparency of the system. All of the processing is done using plain text files. The PSI-BLAST algorithm returns a text file (default output format) that contains all of the information about matching fragments. This human readable file is parsed by our program for modeling domain boundaries. Looking into the PSI-BLAST output file, the user can trace the sequences whose fragments matched with stretches of the query protein and contributed to the current decision. Since each neighbor (match) is weighted by its e-value, the relative contribution of each neighbor is apparent. This is contrary to black-box models in which the decision made by the model cannot be attributed to specific training data. Regardless of the alignment strategy (sequence-sequence or profile-sequence), the PSI-BLAST program produces similar output, and the actual prediction algorithm is independent of the alignment method used.
Although the sensitivity of FIEFDom is comparatively higher than the programs we compared with, we note that an even higher sensitivity would be desirable. However, in contrast to other models FIEFDom has a relatively high specificity, i.e. if a boundary is predicted it is most likely correct. At this point, it is not clear to us what causes the modest sensitivity. Our future research will explore additional methods to increase the sensitivity of the query search with the RPS. We also caution that domain prediction at the genomic level may have reduced accuracy compared to our stated results because the RPS that we are using is heavily weighted by protein sequences that have been amenable to experimental structural determination.
FIEFDom is a flexible tool that can predict domain boundaries for both proteins that have only remote homologs and proteins from highly sequenced families with high accuracy. The transparent model of FIEFDom provides insight into the problem in contrast to the current machine learning-based models. Due to rapid improvements in sequencing technologies, many new complete genomes are available every year, and, since our method can readily absorb new information without the need for model training, FIEFDom should maintain its relevance in the future.
FUNDING
This work was supported by the U.S. Department of Defense High Performance Computing Modernization Program, under the High Performance Computing Software Applications Institutes initiative. Funding for open access charge is same as funding for work.
Conflict of interest statement. The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the US Army or of the US Department of Defense. This paper has been approved for public release with unlimited distribution.
ACKNOWLEDGEMENTS
We thank Drs. Jaques Reifman and Dong Xu for their critical review of the manuscript. We thank the anonymous reviewers for their valuable insight and suggestions.
REFERENCES
- 1.Dill KA, Ozkan SB, Weikl TR, Chodera JD, Voelz VA. The protein folding problem: when will it be solved? Curr. Opin. Struct. Biol. 2007;17:342–346. doi: 10.1016/j.sbi.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Buchete NV, Straub JE, Thirumalai D. Development of novel statistical potentials for protein fold recognition. Curr. Opin. Struct. Biol. 2004;14:225–232. doi: 10.1016/j.sbi.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008;18:342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richardson JS. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 5.Wetlaufer DB. Nucleation, rapid folding, and globular intrachain regions in proteins. Proc. Natl Acad. Sci. USA. 1973;70:697–701. doi: 10.1073/pnas.70.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta VK, Gowda LR. Alpha-1-proteinase inhibitor is a heparin binding serpin: molecular interactions with the Lys rich cluster of helix-F domain. Biochimie. 2008;90:749–761. doi: 10.1016/j.biochi.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Kosinski J, Plotz G, Guarne A, Bujnicki JM, Friedhoff P. The PMS2 subunit of human MutLalpha contains a metal ion binding domain of the iron-dependent repressor protein family. J. Mol. Biol. 2008;382:610–627. doi: 10.1016/j.jmb.2008.06.056. [DOI] [PubMed] [Google Scholar]
- 8.Egloff MP, Benarroch D, Selisko B, Romette JL, Canard B. An RNA cap (nucleoside-2'-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21:2757–2768. doi: 10.1093/emboj/21.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malmstrom L, Riffle M, Strauss CE, Chivian D, Davis TN, Bonneau R, Baker D. Superfamily assignments for the yeast proteome through integration of structure prediction with the gene ontology. PLoS Biol. 2007;5:e76. doi: 10.1371/journal.pbio.0050076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Longhi S, Ferron F, Egloff MP. Protein engineering. Methods Mol. Biol. 2007;363:59–89. doi: 10.1007/978-1-59745-209-0_4. [DOI] [PubMed] [Google Scholar]
- 11.Pang CN, Lin K, Wouters MA, Heringa J, George RA. Identifying foldable regions in protein sequence from the hydrophobic signal. Nucleic Acids Res. 2008;36:578–588. doi: 10.1093/nar/gkm1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horejs C, Pum D, Sleytr UB, Tscheliessnig R. Structure prediction of an S-layer protein by the mean force method. J. Chem. Phys. 2008;128:65106–66100. doi: 10.1063/1.2826375. [DOI] [PubMed] [Google Scholar]
- 13.Han JH, Batey S, Nickson AA, Teichmann SA, Clarke J. The folding and evolution of multidomain proteins. Nat. Rev. Mol. Cell Biol. 2007;8:319–330. doi: 10.1038/nrm2144. [DOI] [PubMed] [Google Scholar]
- 14.Schueler-Furman O, Wang C, Bradley P, Misura K, Baker D. Progress in modeling of protein structures and interactions. Science. 2005;310:638–642. doi: 10.1126/science.1112160. [DOI] [PubMed] [Google Scholar]
- 15.Copley RR, Doerks T, Letunic I, Bork P. Protein domain analysis in the era of complete genomes. FEBS Lett. 2002;513:129–134. doi: 10.1016/s0014-5793(01)03289-6. [DOI] [PubMed] [Google Scholar]
- 16.Gracy J, Argos P. DOMO: a new database of aligned protein domains. Trends Biochem. Sci. 1998;23:495–497. doi: 10.1016/s0968-0004(98)01294-8. [DOI] [PubMed] [Google Scholar]
- 17.Wheelan SJ, Marchler-Bauer A, Bryant SH. Domain size distributions can predict domain boundaries. Bioinformatics. 2000;16:613–618. doi: 10.1093/bioinformatics/16.7.613. [DOI] [PubMed] [Google Scholar]
- 18.Suyama M, Ohara O. DomCut: prediction of inter-domain linker regions in amino acid sequences. Bioinformatics. 2003;19:673–674. doi: 10.1093/bioinformatics/btg031. [DOI] [PubMed] [Google Scholar]
- 19.Bryson K, Cozzetto D, Jones DT. Computer-assisted protein domain boundary prediction using the DomPred server. Curr. Protein Pept. Sci. 2007;8:181–188. doi: 10.2174/138920307780363415. [DOI] [PubMed] [Google Scholar]
- 20.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH – a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 21.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 22.Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marchler-Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA, Geer LY, Bryant SH. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 2002;30:281–283. doi: 10.1093/nar/30.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Letunic I, Copley RR, Pils B, Pinkert S, Schultz J, Bork P. SMART 5: domains in the context of genomes and networks. Nucleic Acids Res. 2006;34:D257–D260. doi: 10.1093/nar/gkj079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Rost B. CHOP proteins into structural domain-like fragments. Proteins. 2004;55:678–688. doi: 10.1002/prot.20095. [DOI] [PubMed] [Google Scholar]
- 26.Chivian D, Kim DE, Malmstrom L, Bradley P, Robertson T, Murphy P, Strauss CE, Bonneau R, Rohl CA, Baker D. Automated prediction of CASP-5 structures using the Robetta server. Proteins. 2003;53(Suppl. 6):524–533. doi: 10.1002/prot.10529. [DOI] [PubMed] [Google Scholar]
- 27.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marsden RL, McGuffin LJ, Jones DT. Rapid protein domain assignment from amino acid sequence using predicted secondary structure. Protein Sci. 2002;11:2814–2824. doi: 10.1110/ps.0209902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gewehr JE, Zimmer R. SSEP-Domain: protein domain prediction by alignment of secondary structure elements and profiles. Bioinformatics. 2006;22:181–187. doi: 10.1093/bioinformatics/bti751. [DOI] [PubMed] [Google Scholar]
- 30.Cheng J, Sweredoski MJ, Baldi P. DOMpro: Protein Domain Prediction Using Profiles, Secondary Structure, Relative Solvent Accessibility, and Recursive Neural Networks. Data Min. Knowl. Discov. 2006;13:1–10. [Google Scholar]
- 31.Nagarajan N, Yona G. Automatic prediction of protein domains from sequence information using a hybrid learning system. Bioinformatics. 2004;20:1335–1360. doi: 10.1093/bioinformatics/bth086. [DOI] [PubMed] [Google Scholar]
- 32.Sim J, Kim SY, Lee J. PPRODO: prediction of protein domain boundaries using neural networks. Proteins. 2005;59:627–632. doi: 10.1002/prot.20442. [DOI] [PubMed] [Google Scholar]
- 33.George RA, Heringa J. SnapDRAGON: a method to delineate protein structural domains from sequence data. J. Mol. Biol. 2002;316:839–851. doi: 10.1006/jmbi.2001.5387. [DOI] [PubMed] [Google Scholar]
- 34.Kim DE, Chivian D, Malmstrom L, Baker D. Automated prediction of domain boundaries in CASP6 targets using Ginzu and RosettaDOM. Proteins. 2005;61(Suppl. 7):193–200. doi: 10.1002/prot.20737. [DOI] [PubMed] [Google Scholar]
- 35.Dumontier M, Yao R, Feldman HJ, Hogue CW. Armadillo: domain boundary prediction by amino acid composition. J. Mol. Biol. 2005;350:1061–1073. doi: 10.1016/j.jmb.2005.05.037. [DOI] [PubMed] [Google Scholar]
- 36.Chen L, Wang W, Ling S, Jia C, Wang F. KemaDom: a web server for domain prediction using kernel machine with local context. Nucleic Acids Res. 2006;34:W158–W163. doi: 10.1093/nar/gkl331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keller JM, Gray MR, Given JA. A Fuzzy K-Nearest Neighbor Algorithm. IEEE Trans. Syst. Man Cybernetics. 1985;15:580–585. [Google Scholar]
- 38.Ripley B. Pattern Recognition and Neural Networks. Cambridge: Cambridge University Press; 1996. p. 403. [Google Scholar]
- 39.Moult J, Fidelis K, Kryshtafovych A, Rost B, Hubbard T, Tramontano A. Critical assessment of methods of protein structure prediction - Round VII. Prot.: Struct. Funct. Bioinformatics. 2007;69:3–9. doi: 10.1002/prot.21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Day R, Beck DA, Armen RS, Daggett V. A consensus view of fold space: combining SCOP, CATH and the Dali Domain Dictionary. Protein Sci. 2003;12:2150–2160. doi: 10.1110/ps.0306803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brenner SE, Koehl P, Levitt M. The ASTRAL compendium for protein structure and sequence analysis. Nucleic Acids Res. 2000;28:254–256. doi: 10.1093/nar/28.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye J, McGinnis S, Madden TL. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 2006;34:W6–W9. doi: 10.1093/nar/gkl164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bondugula R, Xu D. MUPRED: a tool for bridging the gap between template based methods and sequence profile based methods for protein secondary structure prediction. Proteins. 2007;66:664–670. doi: 10.1002/prot.21177. [DOI] [PubMed] [Google Scholar]
- 44.CAFASP4. Critical Assessment of Fully Automated Structure Prediction (CAFASP) http://cafasp4.cse.buffalo.edu/dp/update.html (21 November 2008, date last accessed).
- 45.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wallner B, Elofsson A. All are not equal: a benchmark of different homology modeling programs. Protein Sci. 2005;14:1315–1327. doi: 10.1110/ps.041253405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng J. DOMAC: an accurate, hybrid protein domain prediction server. Nucleic Acids Res. 2007;35:W354–W356. doi: 10.1093/nar/gkm390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hadley C, Jones DT. A systematic comparison of protein structure classifications: SCOP, CATH and FSSP. Structure. 1999;7:1099–1112. doi: 10.1016/s0969-2126(99)80177-4. [DOI] [PubMed] [Google Scholar]
- 49.Holland TA, Veretnik S, Shindyalov IN, Bourne PE. Partitioning protein structures into domains: why is it so difficult? J. Mol. Biol. 2006;361:562–590. doi: 10.1016/j.jmb.2006.05.060. [DOI] [PubMed] [Google Scholar]
- 50.Borden KLB, Freemont PS. The RING finger domain: a recent example of a sequence–structure family. Curr. Opin. Struct. Biol. 1996;6:395–401. doi: 10.1016/s0959-440x(96)80060-1. [DOI] [PubMed] [Google Scholar]