

Abstract
Background and Purpose
Nortriptyline, an antidepressant, was identified as a strong inhibitor of mitochondrial permeability transition by our screening of a library of 1040 drugs. Because mitochondrial permeability transition and consequent mitochondrial dysfunction have been implicated in acute neuronal death, we proposed to investigate the possible neuroprotective effects of nortriptyline in cerebral ischemia.
Methods
The effects of nortriptyline were first studied in oxygen/glucose deprivation-induced death of primary cerebrocortical neurons, a cellular model of cerebral ischemia. Mitochondrial membrane potential, mitochondrial factor release, and caspase 3 activation were evaluated after its treatment. Nortriptyline was also studied in a mouse model, which was established by occlusion of the middle cerebral artery. The infarct volume, neurological function, and biochemical events were examined in the absence or the presence of nortriptyline.
Results
Nortriptyline inhibits oxygen/glucose deprivation-induced cell death, loss of mitochondrial membrane potential, downstream release of mitochondrial factors, and activation of caspase 3 in primary cerebrocortical neurons. Furthermore, it decreases infarct size and improves neurological scores after middle cerebral artery occlusion in mice.
Conclusions
The ability of nortriptyline to inhibit mitochondrial factor release and caspase activation and further protect the animals correlates to its inhibitory effect on mitochondrial permeability transition in isolated mitochondria. This study indicated that nortriptyline is neuroprotective against cerebral ischemia. It also suggested mitochondrial permeability transition might be a valuable therapeutic target for acute neurodegeneration.
Keywords: cell death, cerebral ischemia, mitochondrial permeability transition, neuroprotection, nortriptyline
Mitochondrial permeability transition (mPT) is the opening of pores in the inner mitochondria membrane, thereby allowing free exchange of all solutes <1.5 kDa.1-4 mPT has been proposed as an important event leading to the release of mitochondrial factors and the subsequent activation of caspase-dependent and caspase-independent cell death pathways.5-8 Because release of mitochondrial factors is known to correlate with acute neurological damage, eg, cerebral ischemia, traumatic brain injury, and spinal cord injury,9-11 we hypothesize that its upstream event mPT is crucial in the pathogenesis of these diseases and it could be a potential therapeutic target for acute neurological injury. These considerations led us to screen a library of 1040 compounds for their ability to inhibit the induction of mPT in isolated mitochondria. Medium throughput screening of this library and subsequent analysis of initial “hits” identified 23 compounds that are mPT inhibitors. The chemical structure of 12 of these compounds, notably trifluoperazine, nortriptyline, and promethazine, form a specific subclass of heterocyclics, tricyclics, and phenothiazines. We studied one of the most promising mPT inhibitors, tricyclic antidepressant nortriptyline, as a proof of principle. We tested its effectiveness as a neuroprotective agent in both in vitro and in vivo models of cerebral ischemia. Nortriptyline indeed proves significantly neuroprotective in both systems. Our work also demonstrates that mPT might contribute, at least partially, to the molecular pathogenesis of cerebral ischemia.
Materials and Methods
Mitochondrial Physiology
Rat liver mitochondria were isolated using differential centrifugation as described previously.6,12 Mitochondrial membrane potential (ΔΨm) was measured using a TPP2+; Ca2+ and absorbance (A660) were determined with a Ca2+-sensitive electrode and a light-emitting diode, respectively. These measurements were made simultaneously using a time-resolved multiparameter chamber.6,13,14 Buffers and additions were described in the figure legend. All experiments were performed in triplicate, of which representative plots were shown.
Primary Cerebrocortical Neuron Culture and Oxygen and Glucose Deprivation Treatment
Cerebral cortex from Swiss Webster mouse embryos at day 15 (E15) were freed from the meninges and separated from the olfactory bulb and hippocampus. The cells were dissociated by treatment with trypsin and cultured in poly-d-lysine-coated dishes in neurobasal medium supplemented with 2% B27 (Invitrogen), 2 mmol/L glutamine (Invitrogen), and 100 U/mL penicillin, and streptomycin (Invitrogen). Experiments on primary cerebrocortical neurons (PCNs) were performed after 7 days in culture. PCNs were preincubated with 0 to 5 μmol/L nortriptyline (Sigma) for 2 hours before challenging them with oxygen and glucose deprivation (OGD). OGD was conducted by culturing the cells for 3 hours in glucose-free Earle’s balanced salt solution, an isotonic solution buffered to pH 7.3 with sodium bicarbonate/CO2 that lacks CaCl2 and MgSO4 (Sigma). Cell cultures were incubated in an anaerobic chamber. The atmosphere therein was depleted of oxygen using a palladium catalyst (BBL GasPak Plus; BD Pharmingen), causing O2 to drop below 100 ppm within 90 minutes. Control cultures were incubated in Earle’s balanced salt solution supplemented with 5.5 mmol/L glucose in a normoxic atmosphere for the same period. OGD was terminated by transferring the cells to normal culture conditions.
Measurement of Lactate Dehydrogenase Activity
When cells die, they release their complement of lactate dehydrogenase (LDH). Measuring this activity in the resulting supernatant reveals the extent of cell death. Such assays were conducted according to the manufacturer’s instructions (Roche). PCNs were preincubated with nortriptyline for 2 hours and challenged with: (1) OGD, (2) 1 mmol/L H2O2, or (3) 500 μmol/L NMDA. The culture medium of each well was collected 18 hours later and centrifuged. The supernatant was then mixed with the lactate dehydrogenase assay reaction mixture. After 15 minutes of incubation at room temperature, the absorbance at 490 nm was measured with an enzyme-linked immunosorbent assay reader (Biorad). The same volume of medium was used as background control.
Annexin v/PI Staining and Fluorescence-Activated Cell-Sorting Analysis
PCNs were cultured as described previously. On the seventh day in culture, PCNs were preincubated with 2.5 μmol/L nortriptyline for 2 hours, subjected to 3 hours of OGD, and transferred to normal culture conditions. Cells were trypsinized 18 hours later and collected by centrifugation. They were then stained with annexin v/propidium iodide (Roche) and analyzed using a Beckman flow cytometer (Dana-Farber Cancer Institute Flow Cytometry Core Facility, Boston, Mass). Total of 10 000 cells were counted for each sample. The percentage of apoptosis or necrosis was calculated by the formula: [(apoptotic or necrotic events/total events)×100%].
Rhodamine 123 Staining
PCNs were pretreated with 2.5 μmol/L nortriptyline for 2 hours. For staining, the cultured (living) PCNs were directly incubated with 2 μmol/L rhodamine 123 (Molecular Probes) for 5 minutes at room temperature followed by the rinse through several changes of phosphate-buffered saline (5 minutes per rinse). Digital images were taken with a Nikon ECLIPSE TE 200 fluorescence microscope and processed with IP LAB Software (Spectra Services). Reduced green rhodamine 123 fluorescence indicates dissipated ΔΨm.
Cytosolic Fractionation and Immunoblotting
PCNs (3 to 3.6×106) or brain tissue were homogenized using a Dounce homogenizer (Kontes Glass) in a buffer containing 250 mmol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 2 mmol/L EDTA, 20 mmol/L HEPES, 1 mmol/L dithiothreitol, 50 μmol/L N-benzoxycarbonyl-Val-Ala-Asp-fluoromethylketone (zVAD-fmk; Sigma) plus a protease inhibitor cocktail (Sigma). After removing unlyzed cells by a low-speed spin (500g), the clarified supernatant was collected and centrifuged at 13 000g. Because of their dominant constitution, the pellet was considered to be the mitochondrial fraction and the supernatant the cytosolic fraction.15 β-actin was used as a loading control for cytosolic fraction. The primary antibodies used in the immunoblots were a mouse monoclonal for cytochrome c (BD Pharmingen), a rat monoclonal for smac/Diablo (Novus Biologicals), a rabbit polyclonal for apoptosis-inducing factor (AIF; Sigma), a rabbit polyclonal for caspase 3 (Cell Signaling), and a mouse monoclonal for β-actin (Sigma).
Middle Cerebral Artery Occlusion and Drug Treatment
Male C57/BL6 mice (≈20 g, 5 to 6 weeks old) were obtained from Jackson Laboratories (Bar Harbor, Maine) and housed under a natural light/dark cycle with food and water available ad libitum. All experiments were conducted in accordance with protocols approved by the Harvard Medical School Animal Care Committee. Focal cerebral ischemia was induced by insertion of an intraluminal nylon thread into the cervical internal carotid artery. Beforehand, animals were divided at random into a treated (n=12) and a control group (n=13). Treated animals received 2 intraperitoneal injections, each of 2 mg/kg nortriptyline. The first injection was given 30 minutes before and the second 12 hours after ischemia. Control animals were injected with equivalent volumes of the saline vehicle. Mice were anesthetized with 1.5% (vol/vol) isoflurane in 70% N2O/30% O2 before the start of the operation and were held in an atmosphere with 0.5% isoflurane during the entire procedure. Rectal temperature was maintained between 37 and 37.5°C with a heating pad (Harvard Apparatus). The right common carotid artery was exposed, and the external carotid artery was ligated. A 7–0 monofilament nylon thread with a silicone-blunted tip was inserted 9±1.0 mm into the internal carotid artery up to the middle cerebral artery. The rate of cerebral blood flow was continually monitored with an infrared Doppler (Perimed AB).10 The cerebral blood flow should reduce 85% to 90% in 5 minutes after occlusion. In the meantime, we also recorded arterial blood pressure. After 2 hours of occlusion, the thread was removed, restoring blood flow through the middle cerebral artery.
Determination of Neurological Score and Infarct Volume
After either 30 minutes or 24 hours of reperfusion, each mouse was assigned a neurological score according to the following scale: 0, no neurological deficits; 1, failure to extend the left forepaw; 2, circling to the contralateral side; 3, inability to walk or loss of the righting reflex.16 Subsequently, brains were rapidly removed and cut into 6 coronal sections, each being 2 mm thick. The sections were stained with 2, 3, 5-triphenyltetrazolium chloride (Sigma) for 30 minutes at 37°C. Brain slices were scanned, and the stained areas in the ipsilateral and contralateral hemispheres were measured with a HP Scanjet 4200C. Within each slice, the ischemic volume was calculated according to the following formula: 2 mm×[stained area in the left side (normal side)–stained area in the right side (ischemic side)]. These 6 values were summed to yield the total volume of the infarct. The scoring of neurological function and infarct size was done by blinded observers to avoid bias.
Results
Selection of Nortriptyline by Mitochondrial Screening
We screened a library of 1040 compounds for their ability to inhibit mPT induction in isolated rat liver mitochondria. The design and overall results of the screen have recently been published.13,17 We selected nortriptyline as a representative agent of the heterocyclics, tricyclics, and phenothiazines class for further study because it is a strong mPT inhibitor and it is well tolerated by patients.13 In this study, we evaluated the effects of nortriptyline on mitochondrial physiology by assessing ΔΨm and Ca2+ uptake–release capacity after challenge with a series of bolus additions of Ca2+. Nortriptyline did not impair normal mitochondrial function, but did delay induction of mPT (Figure 1A). Furthermore, nortriptyline increased the Ca2+ retention capacity of mitochondria, and it delayed Ca2+-induced Ca2+ release and loss of ΔΨm (Figure 1). Thus, nortriptyline protected against Ca2+overload-mediated mitochondrial dysfunction while not detectably affecting mitochondrial physiology. Similar to their effects on liver mitochondria, nortriptyline is also protective in isolated brain mitochondria (data not shown).
Figure 1.

Nortriptyline increases Ca2+ retention capacity, delays Ca2+-induced Ca2+ release, slows dissipation of ΔΨm, and reduces swelling in isolated mitochondria. A, Ca2+ capacity was studied using a calcium-selective electrode. The signal is inversely proportional to the concentration of free Ca2+ in the ambient solution. Addition of nortriptyline caused a shift in the baseline signal. On correcting for this effect, it was apparent that the initial calcium uptake was the same in the presence as in the absence of the compound. B, ΔΨm was measured using a TPP2+ electrode. The signal is directly proportional to ΔΨm. Again, addition of nortriptyline shifts the baseline signal from the TPP2+ electrode but does not change ΔΨm. The rate of TPP2+ uptake by the mitochondria is invariant, compelling the same conclusion as with Ca2+. C, Mitochondrial swelling was determined by measuring the resulting change in the absorbance of infrared light, ie, a diminished value of A660. All these assays were performed in buffer containing 300 mmol/L sucrose, 2.5 mmol/L KH2PO4, 3 mmol/L HEPES, pH 7.4, 2.5 μmol/L of TPP2+, and 5 mmol/L K+-succinate. We started recording at “0.” X-axis started at “≈200” when we added mitochondria. For each assay, 1 mg/mL mitochondria was added and the total volume was 1 mL. The calcium (30 nmol/mg protein) challenge began 3 minutes after addition of mitochondria and the establishment of steady-state 4 conditions. Decreasing calcium was due to mitochondria uptake. Each bolus of Ca2+ (indicated by an arrow) increased the concentration of Ca2+ by 30 μmol/L. Although the baseline signal changes on addition of nortriptyline,13 this effect is independent of the presence of mitochondria. Consequently, it is easily corrected and does not confound interpretation of the experiment. The apparent visual discrepancies are due to the nonlinear nature of the responses of these electrodes. Experiments were terminated when Ca2+ was spontaneously released from mitochondria and no further uptake occurs.
Nortriptyline Inhibits Neuronal Cell Death
By analogy with other inhibitors of mPT,17,18 nortriptyline is apt to be neuroprotective after acute neurological injury. Before beginning trials in an animal model, we first evaluated whether nortriptyline could protect PCNs from OGD, a cellular model of cerebral ischemia.10 Indeed, nortriptyline significantly inhibited neuronal cell death, as indicated by decreased lactate dehydrogenase release (Figure 2A). Phase-contrast micrograph also showed that OGD caused PCN loss of processing and shrinkage of the cell body, whereas nortriptyline partially restored their normal morphology (Figure 2D). We continued our investigation by evaluating neuroprotection by nortriptyline in other cell death paradigms. Nortriptyline inhibited PCN death induced by the oxidant H2O2 (Figure 2B) or by the glutamate receptor agonist NMDA (an excitotoxic model19-21; Figure 2C, E). Fluorescence-activated cell-sorting analysis confirmed the inhibitory effect of nortriptyline on OGD-induced cell death (Figure 3). It further demonstrated that apoptosis is the dominant type of cell death in this model. Nortriptyline significantly inhibited apoptosis but only slightly reduced necrosis.
Figure 2.

Effects of nortriptyline on the cell death of PCNs. PCNs were pretreated with serial concentrations of nortriptyline (1, 2.5, 5 μmol/L) and then subjected to different cell death inducers: (A) OGD; (B) 1 mmol/L H2O2; and (C) 500 μmol/L NMDA. Cell death was evaluated by lactate dehydrogenase assay and expressed as the percentage of the control condition in which neither nortriptyline nor death inducers was included. Data are presented as mean±SE of 3 independent experiments. *P<0.05 as compared with stress-only cells. Phase-contrast light micrographs of PCNs and their Hoechst 33342-stained nuclei are shown before and after (D) OGD or (E) NMDA treatment in the presence or absence of nortriptyline (2.5 μmol/L). Blue, nuclei stained by Hoechst 33342 (1:1000).
Figure 3.
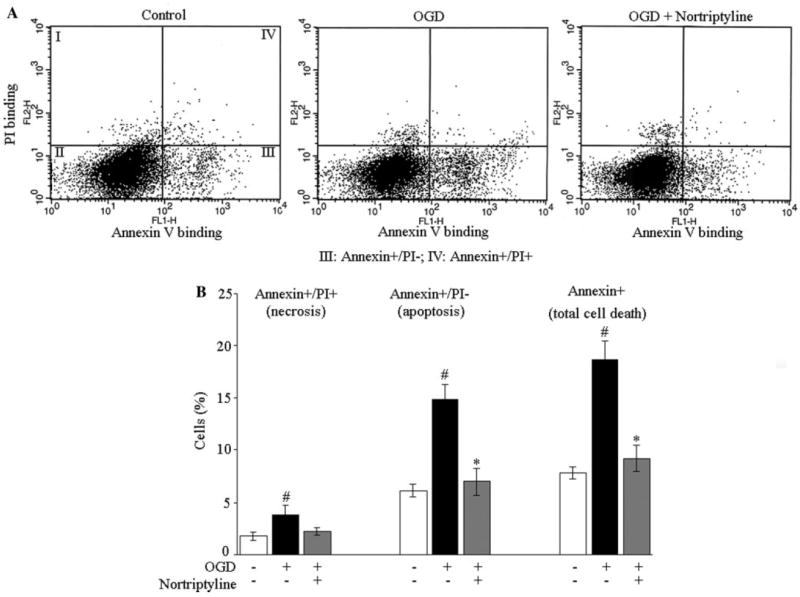
Fluorescence-activated cell-sorting analysis of the effects of nortriptyline on OGD-induced death of PCNs. A, Representative fluorescence-activated cell-sorting analysis. The data points in quadrant III correspond to apoptotic cells (annexin+/PI−), which expose phosphatidylserine from the inner part of membrane to bind annexin v. However, these cells still preserve membrane integrity and thereby exclude PI. Points in quadrant IV represent necrotic cells (annexin+/PI+), in which PI passes through the degraded membrane and binds their complement of DNA. Total cell death includes both apoptotic and necrotic cell death. B, A bar graph of the analytical data from fluorescence-activated cell-sorting. The data are presented as mean±SE of 3 independent experiments, in which triplicate is performed. #P<0.05 as compared with control cells in the absence of both OGD and nortriptyline; *P<0.05 as compared with OGD-only cells.
Nortriptyline Inhibits Oxygen/Glucose Deprivation-Mediated Release of Apoptogenic Factors From the Mitochondria of Primary Cerebrocortical Neurons
Hypoxic injury to PCNs causes the mitochondria to release apoptogenic factors, which in turn activates caspases.10,11,22 It is believed that depolarization of the mitochondria with concomitant induction of mPT commits the host neuron to cell death.23 We therefore investigated whether loss of ΔΨm correlates with OGD-induced death of PCNs and whether that molecular change is forestalled by treatment with nortriptyline. In healthy cells, rhodamine 123 staining showed a punctuate pattern due to preferential staining of negatively charged mitochondria. After OGD, the fluorescent signal from rhodamine 123 staining became diffuse, presumably due to mitochondrial depolarization. Nortriptyline inhibited the dissipation of ΔΨm in living PCN as it did in the isolated mitochondria (Figure 4A). In addition, nortriptyline significantly inhibited OGD-induced release of cytochrome c, AIF, and smac/Diablo from mitochondria (Figure 4B) and countered the activation of caspase 3 (Figure 4C).
Figure 4.

Nortriptyline reduces OGD-induced death of PCNs by inhibiting mitochondrial cell death pathways. A, Nortriptyline retains ΔΨm in PCNs after OGD. The green fluorescence results from the accumulation of rhodamine 123 within negatively charged (functioning) mitochondria. The scale bar is 5 μm. B, OGD-induced release of cytochrome c, AIF, and smac/Diablo is reduced by nortriptyline. PCNs were fractionated to obtain cytosolic component. Samples were analyzed by Western blotting with cytochrome c, AIF, and smac/Diablo antibodies. β-actin was a loading control. C, Caspase 3 activation induced by OGD is decreased by nortriptyline in PCNs.
Nortriptyline Improves Neurological Recovery and Reduces Lesion Size After Ischemia
We next investigated the ability of nortriptyline to reduce ischemic damage in mice. The model consists of 2 hours of middle cerebral artery occlusion (MCAO) followed by 24 hours of reperfusion. Resulting changes in the animal’s behavior were rated on a scale of 0 to 3 (the “neurological score”) 30 minutes or 24 hours after reperfusion. Treated animals were administered nortriptyline before and after MCAO; controls received the saline vehicle. Nortriptyline is found to have little effect on the neurological score 30 minutes after the reperfusion. This finding is consistent with that reported for other neuroprotective agents. It presumably reflects the lack of change in the volume of ischemic/nonfunctional cerebral territory.16,24 At this relatively early time, however, nonfunctional brain territory is not necessarily dead and still could be salvaged. Nortriptyline significantly improves the neurological score 24 hours after reperfusion (Figure 5A). The infarct volume was also quantified by staining with 4% 2, 3, 5-triphenyltetrazolium chloride. Nortriptyline-treated mice have 55% reduction in the lesion size as compared with saline-treated controls (Figure 5B). The reduction in infarct volume correlates well with the improved neurological scores in nortriptyline-treated mice.
Figure 5.

Nortriptyline inhibits MCAO-mediated ischemic brain injury by decreasing release of mitochondrial apoptogenic factors. A, Neurological grading. B, infarct volume. The data are presented as mean±SE for the saline (n=13) and nortriptyline groups (n=12). *P<0.05, **P<0.01 as compared with saline. C, Effects of nortriptyline on the release of mitochondrial factors in the MCAO model of ischemia. Mice brains from each group were homogenized and fractionated. The cytosolic components were analyzed by Western blot with cytochrome c, smac/Diablo, and AIF antibodies. β-actin was used as a loading control. The blot is representative of 3 independent experiments.
Nortriptyline Inhibits Postischemic Cytochrome c, Smac/Diablo, and Apoptosis-Inducing Factor Release In Vivo
When compared with untreated controls, primary neurons that undergo OGD in the presence of nortriptyline exhibit a reduction of release of mitochondrial apoptogenic factors (Figure 4B). This effect of nortriptyline correlates with its ability to block mPT in purified mitochondria. Consequently, we evaluated whether nortriptyline inhibits release of these factors from mitochondria in areas affected by cerebral ischemia. We and others have previously demonstrated that the release of mitochondrial apoptogenic factors occur after experimental cerebral ischemia.10,11 Our results confirmed that ischemia causes release of cytochrome c, smac/Diablo, and AIF into the cytosol. Furthermore, in nortriptyline-treated as opposed to control mice, these proteins are translocated from mitochondria to cytosol to a less degree (Figure 5C).
Discussion
Nortriptyline was originally selected from a library screening of 1040 drugs by using rat isolated liver mitochondria. Mitochondria treated with nortriptyline showed delayed mPT induction and significant resistance to calcium overload-induced injury. Similar results were also obtained from brain mitochondria (IGS, BSK, unpublished data). Our work further indicates that nortriptyline is neuroprotective in both in vitro and in vivo models of cerebral ischemia. The delineated mechanism is that nortriptyline acts on the mitochondria to inhibit mPT-mediated release of apoptogenic factors, thereby forestalling caspase cascades leading to cell death.
In the cellular models, its optimal concentration for neuroprotection is 1 to 5 μmol/L. We have tested other concentrations; however, nortriptyline could not inhibit the death at such concentrations (Supplement), suggesting that it has a relatively narrow window of effectiveness in these models. Thus, drug cocktails might be required to interfere with the cell death pathways that are involved in the acute neurological injury. One choice is creatinine. We previously reported that creatinine indirectly inhibits cytochrome c release from mitochondria by delaying ischemia-induced cerebral ATP depletion.25 Further investigation is needed to examine whether the combination of these drugs will show additive or synergetic effects.
Nortriptyline displayed significant protection against cerebral ischemia in our mouse MCAO model. To exclude the possibility that this protection is mainly due to vascular rather than neuronal effects, we continuously monitored cerebral blood flow and arterial blood pressure in our experiments. Our recordings demonstrated that there was no difference between the treated and control groups in the vascular variables both before and after the occlusion (Table). In addition, nortriptyline is a US Food and Drug Administration-approved antidepressant, penetrates the blood–brain barrier, is frequently used in patient care, and has a high oral availability. These advantages further prepare nortriptyline as a potential therapeutic for acute neurodegeneration.
Table.
Physiological Variables in MCAO Model
| MABP, mm Hg | CBF, mL/100 g per min | |
|---|---|---|
| Control groups | ||
| Preocclusion | 101±2 | 123±6 |
| Postocclusion | 107±3 | 11±3 |
| Treated groups | ||
| Preocclusion | 105±4 | 126±5 |
| Postocclusion | 110±6 | 12±4 |
Values are mean±SE for each group.
MABP indicates mean arterial blood pressure; CBF, cerebral blood flow; Preocclusion, 5 minutes before MCAO; Postocclusion, 5 minutes after MCAO.
Our observations also provide circumstantial evidence for the role of mPT in the pathology of cerebral ischemia. Recently, several groups used a genetic approach to investigate the potential involvement of mPT in the mechanism of cell death after ischemia. They note that neurons from a mouse lacking cyclophilin D resist cell death. After cerebral ischemia, cycloaphilin D-deficient mice develop smaller infarcts than do wild-type controls.28-31 These findings are consistent with our studies, in which the mPT inhibitors nortriptyline and promethazine13,17 are found to protect cultured neurons against OGD-induced death. In addition, such mPT inhibitors are protective of mice with MCAO-induced ischemic injury. However, their studies suggest mPT prefer regulating necrotic but not apoptotic cell death. An earlier study also draws a similar conclusion by using a cell line that overexpresses cyclophilin D.26 Our data show that in the cellular model of cerebral ischemia, OGD induces apoptosis to a greater extent than necrosis and nortriptyline dramatically reduces such apoptotic cell death. The inconsistency of these observations can be explained in the following ways: (1) mPT plays different roles depending on the cell type and specific inducing stimuli. Compared with the other stresses, OGD activate apoptotic pathways that more likely resemble the events after acute injury in vivo. One way to clarify this is to perform an experiment in which PCNs from cyclophilin D-knockout mice are subjected to OGD. (2) mPT induction involves multiple components: adenine nucleotide translocase, voltage-dependent anion transporter, and cyclophilin D.27 Pharmacological inhibitors of mPT may function at multiple components rather than one, which is targeted by genetic method. Therefore, it could not be ruled out that mPT play a role, either primary or secondary, in modulating apoptosis.
Summary
The present study demonstrates that nortriptyline is significantly neuroprotective in both cellular and animal models of cerebral ischemia. Its mechanism of action is likely by inhibiting mPT-mediated mitochondrial events and downstream cell death pathways.
Acknowledgments
We thank Ethan Shimony for editorial assistance.
Sources of Funding This work was supported by grants from the NIH/NINDS (R.M.F./NS039324, NS041635; X.W.), the Huntington’s Disease Society of America/Coalition for the Cure (R.M.F.), Hereditary Disease Foundation (X.W., B.S.K.), High Q, Cure Huntington’s Disease Initiative (B.S.K.); NSF of China (W.Z.).
Footnotes
Disclosures None.
References
- 1.Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267:C313–339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- 2.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 3.Kristal BS, Dubinsky JM. Mitochondrial permeability transition in the central nervous system: induction by calcium cycling-dependent and -independent pathways. J Neurochem. 1997;69:524–538. doi: 10.1046/j.1471-4159.1997.69020524.x. [DOI] [PubMed] [Google Scholar]
- 4.Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem. 1994;269:16638–16642. [PubMed] [Google Scholar]
- 5.Kantrow SP, Piantadosi CA. Release of cytochrome c from liver mitochondria during permeability transition. Biochem Biophys Res Commun. 1997;232:669–671. doi: 10.1006/bbrc.1997.6353. [DOI] [PubMed] [Google Scholar]
- 6.Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu du C, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- 7.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petit PX, Goubern M, Diolez P, Susin SA, Zamzami N, Kroemer G. Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: the impact of irreversible permeability transition. FEBS Lett. 1998;426:111–116. doi: 10.1016/s0014-5793(98)00318-4. [DOI] [PubMed] [Google Scholar]
- 9.Friberg H, Wieloch T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie. 2002;84:241–250. doi: 10.1016/s0300-9084(02)01381-0. [DOI] [PubMed] [Google Scholar]
- 10.Zhang WH, Wang X, Narayanan M, Zhang Y, Huo C, Reed JC, Friedlander RM. Fundamental role of the rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proc Natl Acad Sci U S A. 2003;100:16012–16017. doi: 10.1073/pnas.2534856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, Chiarugi A, Thomas SS, Kohane DS, Korsmeyer SJ, Moskowitz MA. Bid mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci U S A. 2001;98:15318–15323. doi: 10.1073/pnas.261323298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kristal BS, Brown AM. Apoptogenic ganglioside gd3 directly induces the mitochondrial permeability transition. J Biol Chem. 1999;274:23169–23175. doi: 10.1074/jbc.274.33.23169. [DOI] [PubMed] [Google Scholar]
- 13.Stavrovskaya IG, Narayanan MV, Zhang W, Krasnikov BF, Heemskerk J, Young SS, Blass JP, Brown AM, Beal MF, Friedlander RM, Kristal BS. Clinically approved heterocyclics act on a mitochondrial target and reduce stroke-induced pathology. J Exp Med. 2004;200:211–222. doi: 10.1084/jem.20032053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krasnikov BF, Kuzminova AE, Zorov DB. The ca2+-induced pore opening in mitochondria energized by succinate-ferricyanide electron transport. FEBS Lett. 1997;419:137–140. doi: 10.1016/s0014-5793(97)01450-6. [DOI] [PubMed] [Google Scholar]
- 15.Guegan C, Vila M, Rosoklija G, Hays AP, Przedborski S. Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci. 2001;21:6569–6576. doi: 10.1523/JNEUROSCI.21-17-06569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G, Fishman MC, Greenberg AH, Moskowitz MA, Yuan J. Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narayanan MV, Zhang W, Stavrovskaya IG, Kristal BS, Friedlander RM. Promethazine: a novel application as a neuroprotectant that reduces ischemia-mediated injury by inhibiting mitochondrial dysfunction. Clin Neurosurg. 2004;51:102–107. [PubMed] [Google Scholar]
- 18.Matsumoto S, Friberg H, Ferrand-Drake M, Wieloch T. Blockade of the mitochondrial permeability transition pore diminishes infarct size in the rat after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1999;19:736–741. doi: 10.1097/00004647-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- 20.Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Cyclosporin a delays mitochondrial depolarization induced by n-methyl-d-aspartate in cortical neurons: evidence of the mitochondrial permeability transition. Neuroscience. 1996;75:993–997. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- 21.Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blomgren K, Zhu C, Hallin U, Hagberg H. Mitochondria and ischemic reperfusion damage in the adult and in the developing brain. Biochem Biophys Res Commun. 2003;304:551–559. doi: 10.1016/s0006-291x(03)00628-4. [DOI] [PubMed] [Google Scholar]
- 23.Chang LK, Johnson EM., Jr Cyclosporin a inhibits caspase-independent death of NGF-deprived sympathetic neurons: a potential role for mitochondrial permeability transition. J Cell Biol. 2002;157:771–781. doi: 10.1083/jcb.200112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci U S A. 1997;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu S, Li M, Figueroa BE, Liu A, Stavrovskaya IG, Pasinelli P, Beal MF, Brown RH, Jr, Kristal BS, Ferrante RJ, Friedlander RM. Prophylactic creatine administration mediates neuroprotection in cerebral ischemia in mice. J Neurosci. 2004;24:5909–5912. doi: 10.1523/JNEUROSCI.1278-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Johnson N, Capano M, Edwards M, Crompton M. Cyclophilin-d promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis. Biochem J. 2004;383:101–109. doi: 10.1042/BJ20040669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halestrap A. Biochemistry: a pore way to die. Nature. 2005;434:578–579. doi: 10.1038/434578a. [DOI] [PubMed] [Google Scholar]
- 28.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 29.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 30.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 31.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]