

Abstract
Background
Prolonged breathing of nitric oxide reduces myocardial ischemia-reperfusion injury but the precise mechanisms responsible for the cardioprotective effects of inhaled nitric oxide are incompletely understood.
Methods
We investigated the fate of inhaled nitric oxide (80 parts per million) in mice, and quantified the formation of nitric oxide metabolites (NO-metabolites) in blood and tissues. We tested whether the accumulation of NO-metabolites correlated with the ability of inhaled nitric oxide to protect against cardiac ischemia-reperfusion injury.
Results
Mice absorbed nitric oxide in a nearly linear fashion (0.19±0.02 μmol/g·h). Breathing nitric oxide rapidly increased a broad spectrum of NO-metabolites. Levels of erythrocytic S-nitrosothiols, N-nitrosamines and nitrosyl-hemes exceeded nitrite within 30 sec of commencing nitric oxide inhalation. Marked increases of lung S-nitrosothiols and liver N-nitrosamines levels were measured, as well as elevated cardiac and brain NO-metabolite levels. Breathing hypoxic concentrations potentiated the ability of inhaled nitric oxide to increase cardiac NO-metabolite levels. Concentrations of each NO-metabolite, except nitrate, rapidly reached a plateau and were similar after 5 and 60 minutes. Studying a murine cardiac ischemia-reperfusion injury model, breathing nitric oxide for either 5 or 60 min before reperfusion decreased MI/AAR by 31 and 32%, respectively.
Conclusions
Breathing nitric oxide leads to the rapid accumulation of a variety of NO-metabolites in blood and tissues, contributing to the ability of brief periods of nitric oxide inhalation to provide cardioprotection against ischemia-reperfusion injury. The NO-metabolite concentrations achieved in a target tissue may be more important than the absolute amounts of nitric oxide absorbed.
Introduction
Inhaled nitric oxide is a selective pulmonary vasodilator that does not produce systemic hypotension when breathed at concentrations up to 80 parts per million (ppm). Inhaled nitric oxide is widely used to treat neonatal hypoxemia and acute pulmonary hypertension1. The selectivity of inhaled nitric oxide for the pulmonary vasculature is attributed to its high affinity for the heme moiety of hemoglobin and its rapid conversion, in the presence of oxygenated hemoglobin, to nitrate and met-hemoglobin. However, as early as 1993, it was appreciated that breathing nitric oxide had systemic effects and could prolong the bleeding time (rabbits and humans)2. Subsequent reports demonstrated that breathing nitric oxide could decrease neointima formation after carotid artery injury (rats)3, decrease thrombosis after thrombolysis (dogs)4, and reduce reperfusion injury after mesenteric artery ischemia (cats)5 or cardiac ischemia-reperfusion (mice6 and pigs7). Moreover, recent human studies show that inhaled nitric oxide decreased reperfusion-associated inflammatory responses in ischemic limbs8 and decreased hepatic injury after liver transplantation9.
Since nitric oxide has a short half-life in biological fluids, it is unlikely that nitric oxide molecules absorbed into the bloodstream during inhalation reach the periphery in an unmodified form10. A variety of nitric oxide adducts and nitric oxide derived metabolites have been identified in animal and human blood during nitric oxide inhalation. Formation of these nitric oxide products can be direct11 via binding to heme-containing proteins through the transition metal center forming nitrosyl-heme species (NO-heme)12 such as NO- hemoglobin 13. Alternatively, nitric oxide metabolites (NO-metabolites) may be generated indirectly14 via secondary reactions with oxygen that form nitrosating species, such as N2O3. These nitrosating species, in turn, react with thiols to form S-nitrosothiols (RSNO)15 or with amine groups to form N-nitrosamines (RNNO). In addition, breathing nitric oxide increases the blood levels of the end products of nitric oxide oxidation, nitrite and nitrate16. Despite early recognition that NO-metabolites were formed during inhalation of nitric oxide16, a quantitative evaluation of the array of NO-metabolites generated over time by breathing nitric oxide has not been reported.
To investigate the fate of inhaled nitric oxide, the first objective of this study was to determine the rate of nitric oxide absorption through inhalation and quantify the formation of NO-metabolites in blood and various body tissues. The second objective was to determine whether levels of NO-metabolites in blood or heart correlate with the duration of nitric oxide inhalation required to protect against cardiac ischemia-reperfusion injury. We report that nitric oxide inhalation leads to rapid accumulation of a broad spectrum of NO-metabolites in the blood and tissues, and that even a brief period of nitric oxide inhalation (<15min) produces elevation of NO-metabolites, and is associated with a reduction of cardiac ischemia-reperfusion injury.
Materials & Methods
Experimental animals
Male C57BL/6J mice fed a standard diet (RMH 3000, Prolab, PMI International, St. Louis, MO) were studied. All animal experimental protocols were approved by both the Subcommittee on Research Animal Care at Massachusetts General Hospital and the Institutional Animal Care and Use Committee at Boston University School of Medicine (Boston, Massachusetts).
Measurement of nitric oxide absorption
Mice (N=4) were placed in a chamber (PLY3211, Buxco Research Systems, Wilmington, NC) and exposed to air supplemented with 80 ppm nitric oxide (ppm NO) for 60 min. The quantity of nitric oxide absorption was calculated from the difference between inlet and outlet nitric oxide concentrations (ppm), multiplied by the gas flow rate (3 liters/min) and divided by the molar volume of nitric oxide at standard temperature and pressure (22.4 liters/mol). In order to account for the dilution of nitric oxide with ambient air when the chamber was opened to insert the mouse (3 sec), as well as the generation of nitrogen dioxide in the chamber, “sham absorption of nitric oxide” was measured under the same conditions without inserting a mouse (3.3±0.1 μmol/h, n=4). Nitric oxide absorbed (total absorption of nitric oxide, 7.9±0.6 μmol/ mouse·h, minus “sham absorption of nitric oxide”, 3.3 μmol), was divided by body weight and was expressed as μmol NO/g body weight per hour (Additional information regarding our detailed description of nitric oxide absorption measurements is available on the Anesthesiology Web site at http://www.anesthesiology.org. - Web Enhancement #1 Materials and Methods).
Whole body, tissue, blood, and urine sampling for measurement of NO-metabolites
For measurement of whole body NO-metabolite levels, mice breathed air without (n=5) or with (n=4) 80 ppm NO for one hour in the chamber, were subsequently anesthetized with diethyl ether, and euthanized by cervical dislocation. Rapid full-body homogenization was achieved with a Waring blender (Waring Products, Torrington, CT) using a mixture of frozen and chilled phosphate buffered saline (at 1:5, wt/vol) containing N-ethylmaleimide (10 mM) and EDTA (2.5 mM), and the resulting homogenate was immediately analyzed.
Additional mice were placed in the chamber and exposed to air or 80 ppm NO in air for 0.5, 5, 15, and 60 min (n=4-7 per time point). Following the exposure, mice were anesthetized with diethyl ether. Blood was withdrawn from the left ventricle (LV) and immediately centrifuged at 16,000 g for 3-5 min at room temperature (22°C) to separate erythrocytes from plasma. Erythrocytes were subjected to hypotonic lysis in water containing N-ethylmaleimide (10 mM) and EDTA (2.5 mM). To remove blood, tissues were perfused via the LV with room air-equilibrated phosphate buffered saline supplemented with N-ethylmaleimide (10 mM) and EDTA (2.5 mM) for 1 min. Brain, heart, liver, kidney, lung, and fat were harvested, homogenized, and subjected to immediate analysis. Urine was obtained via direct puncture of the bladder. Nitrite, nitrate, RSNO, RNNO, and NO-heme species were quantified in plasma, erythrocytes, tissues, and whole body homogenates. Due to volume limitations, urine analysis was restricted to nitrite and nitrate only.
To investigate the effect of reduced oxygen availability as occurring during ischemia on NO-metabolite levels in the heart, mice were placed in the chamber and breathed either 8% oxygen in nitrogen (Hypoxia, n=5) or 80 ppm NO in 8% oxygen for 60 min (Hypoxia+NO, n=5). Following the exposure, mice were anesthetized with diethyl ether, and cardiac NO-metabolite concentrations were measured immediately after the tissue perfusion with phosphate buffered saline containing N-ethylmaleimide (10 mM) and EDTA (2.5 mM) and homogenizing.
Quantitation of nitroso/nitrosyl species and oxidation products of nitric oxide
Methods for the detection of nitroso (RSNO and RNNO) and nitrosyl (NO-heme) compounds, as well as the oxidation products of nitric oxide (nitrite and nitrate), in blood and tissues have been detailed previously17. Quantification was achieved by group-specific denitrosation after injection of biological samples into either a triiodide-containing reaction mixture (nitroso species) or a potassium ferricyanide solution (nitrosyl species) constantly purged with nitrogen, and oxidation products of nitric oxide was measured with the aid of a gas phase chemiluminescence detector (CLD 77am sp, EcoPhysics, Ann Arbor, MI). Nitrate and nitrite concentrations were quantified by ion chromatography (ENO20 Analyzer, Eicom, San Diego, CA).
Myocardial ischemia-reperfusion injury
Mice were anesthetized by intraperitoneal administration of ketamine (120 mg/kg) and xylazine (5 mg/kg) and ventilated (MiniVent 845, Hugo Sachs Elektronik -Harvard Apparatus GmbH, March-Hugstetten, Germany) at FiO2 of 0.99-1.0. Myocardial ischemia was induced by ligation of the left coronary artery for 60 min, followed by reperfusion for 24 h6. Nitric oxide was administered during ischemia for 60 min, 5 min, or 0.5 min, just prior to reperfusion. During the surgical procedures, an FiO2 of 0.99-1.0 without or with 80 ppm NO was applied using two separate mechanical ventilators. After 24 h, the artery was religated, and either fluorescent microspheres (0.2 ml; 10 μm diameter, FluoSpheres; Invitrogen Corporation, Carlsbad, CA; for 60 min and 5 min nitric oxide inhalation study) were injected into the LV, or tissue marking dye (0.2 ml, TMD-BL; Triangle Biomedical Science Inc., Durham, NC; for 0.5 min nitric oxide inhalation study) was injected into the right carotid artery, to determine the area at risk (AAR). The heart was excised, and four consecutive 1 mm cardiac slices were stained with 2,3,5-triphenyltetrazolium chloride (1% wt/vol; Sigma-Aldrich, St. Louis, MO) for the measurement of myocardial infarction (MI) size. LV, AAR, and MI area were measured by computer-assisted planimetry (NIH Image J 1.34), and AAR/LV and MI/AAR ratios were calculated6.
Data acquisition and statistical analysis
All data are presented as mean ± standard error of the mean (mean±SEM). Data were analyzed using one-way analysis of variance with means comparison using Bonferroni test (Origin 7.0, OriginLab Corporation, Northampton, MA). To compare the changes of NO-metabolites in blood, tissues and urine, each time point was compared with the corresponding baseline value. For the comparison of cardiac NO-metabolite levels in mice breathing low oxygen concentrations, a separate one-way analysis of variance with a means comparison using the Bonferroni test was performed for the comparison of the four groups; i.e. baseline, breathing nitric oxide in air for 60 min, breathing 8% oxygen for 60 min, and breathing nitric oxide in 8% oxygen for 60 min. To analyze the effects of breathing nitric oxide on ischemia-reperfusion injury, analyses were performed for 60, 5, and 0.5 min inhalations of nitric oxide and compared to each control group. P values less than 0.05 were considered significant.
Results
Uptake of inhaled nitric oxide
To investigate the metabolic fate of inhaled nitric oxide, we first measured the absorption of nitric oxide from ambient gas during spontaneous ventilation in awake mice. The rate of nitric oxide uptake was nearly linear with time (0.19±0.02 μmol NO/g body weight·h, fig. 1).
Figure 1.

The absorption of nitric oxide by mice breathing 80 ppm nitric oxide (ppm NO) for 60 min. Mice breathed air supplemented with 80 ppm NO for 60 min in a vented chamber (n=4). Nitric oxide concentrations were recorded continuously at the chamber outlet during the first 30 sec, and thereafter, inlet and outlet were sampled every 15 sec. The difference between inlet and outlet concentrations multiplied by the gas flow rate and time provides an accurate measurement of total nitric oxide gas absorbed by each mouse. The line represents the mean absorption of nitric oxide, and SEM is shown at one minute intervals.
Inhaled nitric oxide is converted into longer-lived metabolites
To further characterize the pharmacokinetics of absorbed nitric oxide, we examined how much of the absorbed nitric oxide could be recovered as NO-metabolites. To measure the accumulation of NO-metabolites during nitric oxide inhalation (80 ppm, 1 h), mice were euthanized and homogenized, and individual NO-metabolites in whole body extracts were quantified. Nitric oxide inhalation led to an increase in the total body concentrations of all the NO-metabolites we examined (table 1). Nitrate concentrations increased 18-fold and represented 97% of the total NO-metabolites measured. Levels of NO-heme increased 13-fold, RSNO 8-fold, RNNO 5-fold, and nitrite 2-fold (table 1). Fifty-three percent (0.10±0.02 μmol/g) of the nitric oxide absorbed from the gas phase during inhalation for 1 h was recovered as NO-metabolites in the whole body extracts.
Table 1.
Whole body analysis of NO metabolites in mice breathing air or air supplemented with 80 ppm NO for 60 min.
| Control (n=5) | Inhaled nitric oxide (n=4) | |
|---|---|---|
| Nitrate | 5.6±1.4 | 98±22** |
| Nitrite | 0.96±0.17 | 2.2±0.5* |
| RSNO | 0.06±0.01 | 0.50±0.08* |
| RNNO | 0.05±0.00 | 0.27±0.05* |
| NO-heme | 0.02±0.00 | 0.26±0.06* |
Data are expressed as μM.
P<0.05,
P<0.01 differs vs. control.
Abbreviations: nitrosyl-heme species (NO-heme), N-nitrosamines (RNNO), S-nitrosothiols (RSNO).
Nitric oxide inhalation increases NO-metabolite concentrations in blood and tissues
To gain detailed insight into the dynamics of uptake, distribution, and secondary metabolism of the nitric oxide absorbed during inhalation, the concentrations of NO-metabolites were measured in blood (both erythrocytes and plasma, fig. 2) and tissues (heart, lung, brain, liver, kidney, and fat; fig. 3) of mice breathing air with or without 80 ppm NO for 0, 0.5, 5, 15, and 60 min (Additional information regarding each concentration, the number of animals studied, and the P-value are available on the Anesthesiology Web site at http://www.anesthesiology.org. - Web Enhancement #2-table 1 and Web Enhancement #3-table 2). Breathing air without nitric oxide for varying periods of time (5, 15, and 60 min) did not alter NO-metabolite concentrations in blood or any of the tissues we studied (data not shown).
Figure 2.
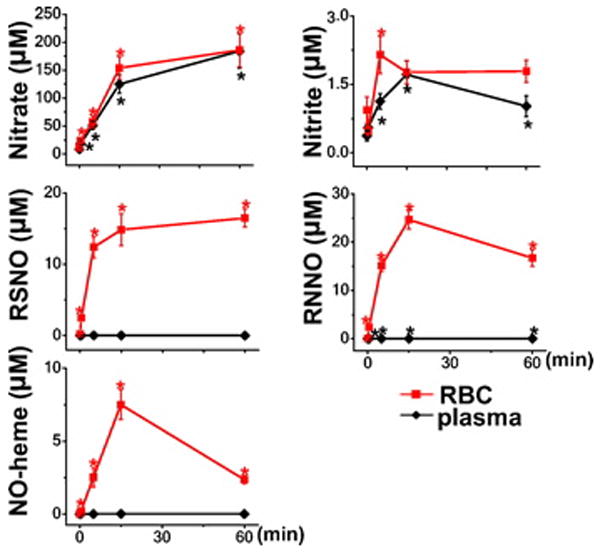
Distribution and kinetics of accumulation of NO-metabolites in mice breathing nitric oxide (plasma and erythrocytes). Concentrations of NO-metabolites were measured in blood of mice breathing air supplemented with nitric oxide for 0, 0.5, 5, 15, and 60 min (n=5-7). Abbreviations: erythrocytes (RBC), nitrosyl-heme species (NO-heme), N-nitrosamines (RNNO), S-nitrosothiols (RSNO).*P<0.05 vs. mice not breathing nitric oxide. Additional information regarding each concentration, the number of animals studied, and the P-value are available on the Anesthesiology Web site at http://www.anesthesiology.org. - Web Enhancement #2-Table 1.
Figure 3.
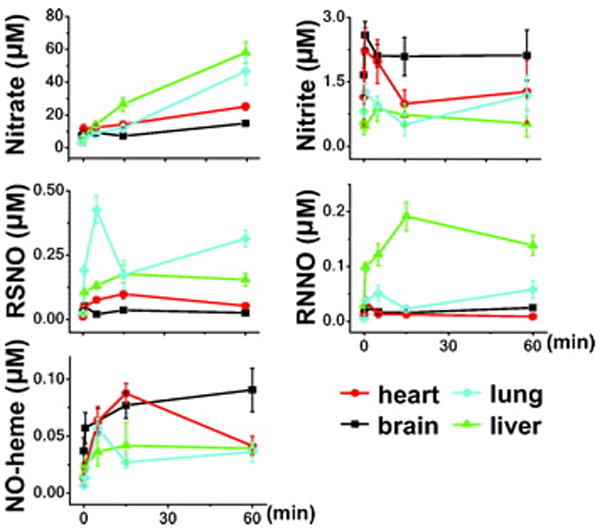
Distribution and kinetics of accumulation of NO-metabolites in mice breathing NO (heart, lung, brain and liver). Concentrations of NO-metabolites were measured in tissues of mice breathing air supplemented with nitric oxide for 0, 0.5, 5, 15, and 60 min (n=4-7). Abbreviations: nitrosyl-heme species (NO-heme), N-nitrosamines (RNNO), S-nitrosothiols (RSNO). Additional information regarding each concentration, the number of animals studied, and the P-value are available on the Anesthesiology Web site at http://www.anesthesiology.org. - Web Enhancement #3-Table 2.
During inhalation of nitric oxide, nitrate concentrations in plasma and erythrocytes increased linearly over the first 15 min and then tended to reach a plateau level (fig. 2). Nitrate concentrations were almost identical in plasma and erythrocytes at all time points. Nitrite concentrations in plasma reached a plateau within 15 min, whereas erythrocytic nitrite peaked as early as 5 min. Nitrite levels were lower than nitrate levels in plasma and erythrocytes by two orders of magnitude. The concentrations of RSNO, RNNO, and NO-heme increased markedly in erythrocytes (610-fold for RNNO, 535- fold for NO-heme, and 85-fold for RSNO; P<0.001 for all), which greatly exceeded the erythrocytic or plasma nitrite concentration. In contrast, breathing nitric oxide did not significantly increase RSNO or NO-heme concentrations in plasma, and plasma RNNO levels increased only 3-fold (P<0.001 vs. baseline). These results suggest that plasma nitrite, nitrate, and RNNO, as well as erythrocytic RSNO, RNNO, NO-heme, nitrite, and nitrate, may all contribute to the transport of bioavailable nitric oxide from the lung to the periphery.
In the heart, increased RSNO and NO-heme levels were detected at 0.5 and 5 min, respectively, and maximum levels were attained after 15 min of nitric oxide inhalation (9- and 7-fold increases, respectively, P<0.001 differs vs. baseline for both, fig. 3). Cardiac RNNO concentrations were maximal at 0.5 min (P<0.05 vs. baseline) and returned to baseline thereafter despite continued inhalation of nitric oxide (fig. 3). The concentration of nitrate increased as early as 0.5 min and remained elevated thereafter. In contrast, cardiac nitrite levels were not elevated significantly during the inhalation of nitric oxide.
To study the effect of hypoxia on the heart, cardiac NO-metabolite levels were measured in mice breathing low oxygen concentration, with and without 80 ppm NO. Cardiac nitrate levels markedly increased after awake mice breathed 8% oxygen for 60 min (Hypoxia; a 9-fold increase vs. baseline, P<0.05, fig. 4). After 80 ppm NO was breathed in 8% oxygen for 60 min (Hypoxia+NO), the levels of NO-heme, RSNO and RNNO were markedly elevated over those of Normoxia+NO (7-fold, 6-fold and 5-fold increases, respectively, P<0.05, fig. 4).
Figure 4.
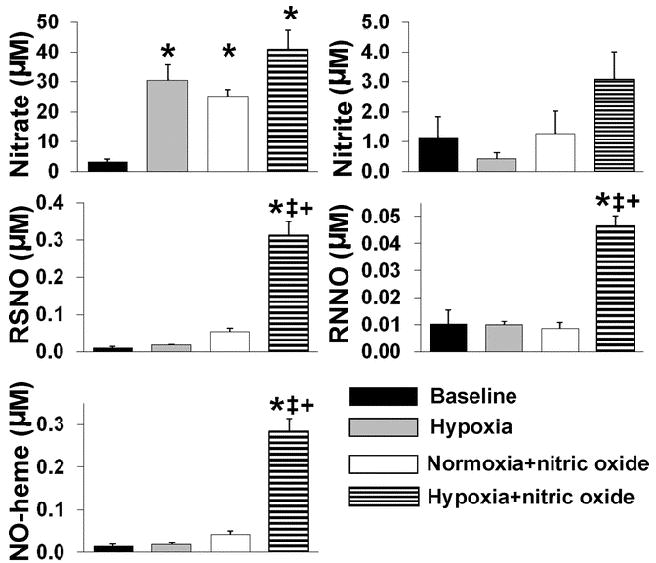
Effects of hypoxia on the cardiac levels of NO-metabolites. Concentrations of cardiac NO-metabolites were measured in cardiac tissue of mice breathing air (Baseline), 8% oxygen (Hypoxia), air supplemented with nitric oxide (Normoxia+nitric oxide), or 8% oxygen balance nitrogen supplemented with nitric oxide (Hypoxia+nitric oxide) for 60 min. *P<0.05 vs. Baseline, ‡P<0.05 vs. Hypoxia, +P<0.05 vs. Normoxia+nitric oxide.
Inhalation of nitric oxide led also to rapid increases in NO-metabolite concentrations in the lung, brain, and liver (fig. 3), but not in kidney and fat (Additional information regarding the concentrations of NO-metabolites in kidney and fat are available on the Anesthesiology Web site at http://www.anesthesiology.org. - Web Enhancement #3-table 2). The highest RSNO levels were achieved in the lung with peak concentrations attained within 5 min. During inhalation of nitric oxide, RNNO concentrations increased markedly in the liver and less so in the lung. In contrast, in the brain, inhalation of nitric oxide led to the accumulation of NO-heme, but not RSNO or RNNO. Breathing nitric oxide did not significantly increase nitrite levels in any of the tissues that we studied. Taken together, marked differences in NO-metabolite regulation exist between the blood, heart, and other tissues, suggesting that generation and/or metabolism of NO-metabolites is quite tissue-specific.
Urinary excretion of nitrite and nitrate
During nitric oxide inhalation, nitrite and nitrate began to accumulate in the urine as early as 0.5 min (data not shown). After 60 min of nitric oxide inhalation, concentrations of nitrite in the urine were 0.7±0.2 μM (P<0.01 differs vs. baseline level, fig. 5A) and were similar to those detected in plasma (1.0±0.3 μM). In contrast, in mice breathing nitric oxide for 60 min, urinary nitrate concentrations (3.5±0.5 mM) were 19-fold greater than those of plasma (P<0.0001 vs. baseline, fig. 5B). As an estimate of the quantity of absorbed nitric oxide that was excreted in the urine, the average concentration of nitrate after 60 min nitric oxide breathing (3.5 mM) was multiplied by the volume of urine collected (119±16 μl, n=9) at the same time point. We estimate that ~9% of the nitric oxide absorbed over one hour is excreted in the urine.
Figure 5.

Measurement of nitrite (A) and nitrate (B) in urine from mice breathing nitric oxide. Mice received air supplemented with nitric oxide for 0, 5, and 60 min (n=8, 7, and 9, respectively). *P<0.05 vs. mice not breathing nitric oxide.
Short-term inhalation of nitric oxide protects against myocardial ischemia-reperfusion injury
In a previous study of mice subjected to 60 min of cardiac ischemia and 24 h of reperfusion, we learned that continuous breathing of 80 ppm NO for 24 h decreased MI size as a fraction of myocardial area at risk (MI/AAR)6. In the current study, the observation that blood levels of nearly all NO-metabolites detected after breathing nitric oxide for 5 min were similar to those detected at 60 min (fig. 2), led us to determine whether a shorter duration of nitric oxide inhalation (≤ 60 min) could modify MI/AAR at 24 h after reperfusion. The overall 24 h mortality rate of mice in our study after ischemia-reperfusion was 8%. In mice breathing nitric oxide for 60 or 5 min before reperfusion, MI/AAR was decreased by 32% (P<0.05, fig. 6 panel A) and 31 % (P<0.05, fig. 6 panel B), respectively. In contrast, breathing nitric oxide for only 30 sec just before reperfusion did not alter the degree of cardiac ischemia-reperfusion injury (fig. 6 panel C).
Figure 6.
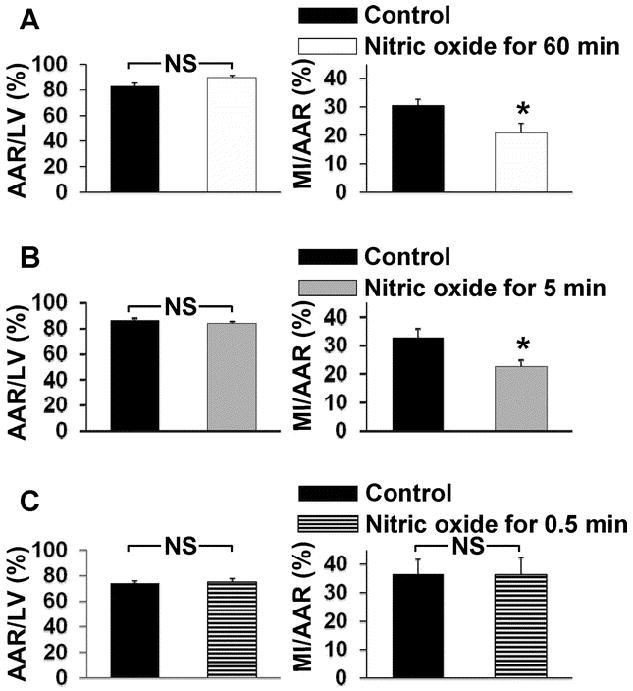
Inhalation of nitric oxide for short durations limits myocardial ischemia-reperfusion injury. All mice underwent left coronary artery occlusion for 60 min followed by 24 h of reperfusion. Mice received nitric oxide during ischemia for 60 min (n=10 and 9 for control and nitric oxide inhaled mice, respectively, Panel A), 5 min immediately before reperfusion (n=9 and 8 for control and nitric oxide inhaled mice, respectively, Panel B), or 0.5 min immediately before reperfusion (n=7 and 6 for control and nitric oxide inhaled mice, respectively, Panel C). Control mice did not receive nitric oxide. *P<0.05 vs. control. Abbreviations: area at risk (AAR), left ventricle (LV), myocardial infarction (MI).
Discussion
In the present study, we provide the first quantitative and temporal characterization of the levels of NO-metabolites that accumulate in the blood and peripheral tissues during nitric oxide inhalation. Moreover, we report that inhalation of nitric oxide for as little as 5 min before reperfusion can reduce infarct size in a murine model of myocardial ischemia-reperfusion injury, which supports the notion that increased levels of one or more NO-metabolite(s) in the blood contribute(s) to the cardioprotective effects of breathing nitric oxide.
It is increasingly appreciated that breathing nitric oxide can elicit a wide spectrum of physiological effects in peripheral tissues18; however, the mechanisms responsible for these salutary effects are incompletely understood. One possibility is that exposure of leukocytes and platelets to high nitric oxide concentrations as they transit the lung may inhibit their activation in peripheral tissues. On the other hand, multiple research groups have observed that inhalation of nitric oxide leads to the formation of NO-metabolites in the bloodstream12,16 and tissues19. To better understand the mechanisms responsible for the extrapulmonary effects of inhaled nitric oxide, we quantitatively assessed the fate of inhaled nitric oxide in whole body extracts, as well as in blood and representative tissues.
In mice breathing nitric oxide (80 ppm), the rate of nitric oxide absorption was essentially linear, and approximately 0.19 μmol/g body weight was absorbed within one hour. Of the gaseous nitric oxide absorbed, we estimate that about 9% (0.017 μmol/g body weight) was excreted in the urine. RSNO, RNNO, NO-heme, nitrite, and nitrate recovered from whole body extracts accounted for 53% (0.10 μmol/g body weight) of the absorbed nitric oxide. The fate of the absorbed nitric oxide that was not detected as NO-metabolites is currently unknown. Some of the absorbed nitric oxide may have been converted to metabolites not readily detected by the techniques we used (such as nitrotyrosine, nitrated fatty acids, and other nitrated species or stable C- or N-nitroso compounds). Alternatively, nitric oxide may have been reduced to nitrous oxide or nitrogen and exhaled. In whole body extracts, nitrate represented nearly 97% of the NO-metabolites accumulating during nitric oxide inhalation, consistent with previous studies showing that conversion of absorbed nitric oxide into nitrate represents the major metabolic pathway for inhaled nitric oxide 16,20.
Lecour and colleagues reported that breathing 100 or 200 ppm NO increased nitric oxide concentrations in peripheral tissues, as detected by electron spin resonance spectroscopy combined with a spin-trapping technique19. However, in contrast to our observations demonstrating that NO-heme levels increased as early as 5 min after the start of 80 ppm NO inhalation, Lecour and colleagues did not detect an increase in cardiac nitric oxide concentrations in rats breathing 100 ppm NO for 45 min. This discrepancy may be explained by the differing techniques for nitric oxide detection or trapping: our chemiluminescence-based technique is sufficiently sensitive to quantify the steady-state concentrations achieved in a given compartment at baseline while nitric oxide is bound to its natural ligands. In contrast, electron spin resonance spectroscopy-based techniques require the accumulation of nitric oxide over a period of time using a transition metal/thiol complex as the trapping agent. In the latter technique, nitric oxide is bound to an exogenous ligand rather than a natural ligand, and higher than normal tissue nitric oxide concentrations are achieved, facilitating detection. However, techniques that utilize exogenous nitric oxide trapping agents inevitably perturb endogenous equilibria involving nitric oxide and its metabolites, and the apparent nitric oxide concentrations achieved depend on the probe’s distribution and saturation characteristics, the stability of the nitric oxide complex formed, and the duration of nitric oxide accumulation.
In a recent study, Lang and colleagues reported that in patients undergoing liver transplantation, breathing 80 ppm NO increased plasma nitrate and nitrite concentrations, as well as erythrocytic nitrate and NO-heme levels, but did not increase erythrocytic RSNO and RNNO levels9. In contrast, we observed that breathing nitric oxide markedly increased erythrocytic RNNO and RSNO levels (600- and 80-fold, respectively). The reasons for this discrepancy are unclear. It is possible that differences in thiol reactivity between rodent and human hemoglobin contribute to differences in the concentrations of nitroso products formed21 However, Gladwin and colleagues also reported that breathing nitric oxide markedly increases RSNO levels (i.e. SNO-Hemoblogin), as well as plasma nitrate and met-hemoglobin, in healthy volunteers22.
During inhalation of nitric oxide, the rate of NO-metabolite accumulation differed depending on the tissue we studied. Increased levels of RSNO, RNNO and NO-heme were measured in the heart of mice breathing nitric oxide, and importantly, the concentrations achieved were similar to those detected in mice carrying a transgene which directs systemic expression of nitric oxide synthase 323. Of note, this strain of mice was shown to be protected from cardiac ischemia-reperfusion injury23. The accumulation of RSNO in the lung during inhalation of nitric oxide is consistent with the observations of Moya and colleagues24. In brain, only NO-heme levels increased after breathing nitric oxide for 15 min. The constancy of increased NO-heme levels in the brain despite continued nitric oxide inhalation may reflect decreased import or increased export of NO-metabolites and/or down-regulation of endogenous nitric oxide production. In comparison to the other tissues we studied, breathing nitric oxide led to the greatest accumulation of RNNO in the liver (a 6.6-fold increase by 15 min), and increased hepatic RSNO and NO-heme concentrations were observed. Differences in the distribution of NO-metabolites detected in these tissues strongly suggest that detection of these metabolites is not attributable to blood contaminating the tissues. Moreover, these findings suggest that the uptake, metabolism, and/or excretion of NO-metabolites are regulated in a tissue-specific manner.
In the murine model of cardiac ischemia-reperfusion injury, nitric oxide was administered only during the ischemia period, in which NO-metabolites can reach the ischemic tissue via the circulation only after subsequent coronary reperfusion. Using this model, breathing nitric oxide for 5 min (just prior to reperfusion) significantly decreased the cardiac injury. In contrast, breathing nitric oxide for 0.5 min just before reperfusion (a duration of breathing nitric oxide which resulted in NO-metabolite concentrations in blood that were consistently lower than those measured in mice breathing nitric oxide for 5 or 60 min) did not protect against cardiac ischemia-reperfusion injury. We acknowledge that, since the dead space and cardiac output differ between spontaneous breathing and mechanically-ventilated animals, the uptake and distribution of nitric oxide in awake mice may differ from that in mice undergoing a thoracotomy and transient coronary artery occlusion (during which nitric oxide was administered through the animal’s ventilator). Nevertheless, our findings correlated with the observation that brief periods of nitric oxide inhalation were capable of protecting against cardiac ischemia-reperfusion injury in mice. The substantial elevations of cardiac NO-metabolites, measured in mice breathing 8% oxygen supplemented with nitric oxide in this study suggests that myocardial ischemia is likely to alter the cardiac NO-metabolite levels produced by nitric oxide inhalation. To our knowledge, this is the first report of the hypoxia-related effects of inhaled nitric oxide on NO-metabolites in cardiac tissue.
Importantly, all of the NO-metabolites we found to be elevated in the blood, are capable of producing nitric oxide-related effects in the periphery. For example, RSNOs (e.g. SNO-hemoglobin and SNO-albumin) are known to dilate blood vessels25,26, and RNNOs can increase cyclic guanosine monophosphate concentrations27 and induce relaxation of bovine coronary arteries28. Especially in the ischemic myocardium, where tissue pH may fall below 5.5 after 30 min of ischemia, nitrite29 or nitrate30 may generate nitric oxide. Accordingly, although the half life of nitric oxide is short, blood NO-metabolites with longer lifetimes can deliver nitric oxide from the lung to distant organs and regenerate nitric oxide in the periphery. Moreover, it is also possible that the downstream effects of NO-metabolites may themselves be longer lasting.
Since any one NO-metabolite can be converted into many others, the determination of which single NO-metabolite confers cardioprotection, either solely or in concert with other NO-metabolites, remains a challenge. It has been previously reported that many NO-donors and NO-metabolites, including S-nitrosoglutathione and nitrite, can protect the heart from ischemia-reperfusion injury, perhaps via differing pathways. Sun et al. reported an attenuation of cardiac ischemia-reperfusion injury by S-nitrosoglutathione, which was associated with an increase of S-nitrosylation of the mitochondrial L-type Ca2+ channel31. Nitrite protected the heart when injected into the left ventricle at 5 min before the reperfusion32 and produced cardioprotection by attenuating mitochondrial respiration via inhibiting complex I in vitro33. Moreover, a recent study indicates that S-nitrosocysteine may protect the heart through nitric oxide-independent pathways34. Thus, while nitric oxide itself may be the active molecule in cardioprotection, RSNOs and other NO-metabolites seem to have a similar capability of protecting the heart.
Although our results and those of others suggest that inhaled nitric oxide can decrease cardiac ischemia-reperfusion injury in animal models, it is not known whether breathing nitric oxide will decrease MI size in patients suffering an acute coronary artery occlusion (ST-segment elevation MI). It is encouraging to note that breathing 80 ppm NO has shown to decrease ischemia-reperfusion injury in human studies8,9. If our observations in mice can be extrapolated to humans, the findings suggest the possibility that brief durations of nitric oxide inhalation may prove beneficial in patients at risk for cardiac ischemia-reperfusion injury.
In summary, we report that inhaled nitric oxide dynamically increases the levels of blood and tissue NO-metabolites and that the degree of accumulation of each NO-metabolite is quite tissue-specific. Moreover, brief periods of nitric oxide inhalation can reduce infarct size in a murine model of cardiac ischemia-reperfusion injury with similar efficacy as much longer periods of nitric oxide inhalation, suggesting that the concentrations of NO-metabolites achieved in the target tissue may be more important for protection than the absolute amounts of nitric oxide absorbed or delivered. The protective effects of breathing nitric oxide are likely to be attributable to NO-metabolites that are rapidly transported in a bioactive form via the blood from the lung to the heart.
Acknowledgments
The authors are grateful to Nathan S. Bryan, Ph.D. (Assistant Professor, Center for Cell Signaling, The Brown Foundation Institute of Molecular Medicine for the Prevention of Human Diseases, The University of Texas, Houston, Texas), Rong Liu, M.D. (Research Fellow, Division of Thoracic Surgery, Brigham and Women’s Hospital, Boston, Massachusetts), and Michael J. Raher, B.S. (Technician, Department of Anesthesia and Critical Care, Massachusetts General Hospital, Boston, Massachusetts) for their advice and skillful assistance, and Hui Zheng, Ph.D. (Assistant Professor of Medicine, Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts) for the statistical assistance.
Source of financial support for the work: This study was supported in part by National Institute of Health Grants (Bethesda, Maryland) HL42397 to W.M. Zapol, DA020644 to M. Feelisch, and HL70896 to K.D. Bloch, a multidisciplinary training in cardiovascular research grant HL07224 to B.O. Fernandez, and a sponsored research agreement between Massachusetts General Hospital and Ikaria (to K.D. Bloch).
Footnotes
Conflict of Interest Statement: The authors, Drs. Zapol and Bloch, have obtained patents relating to the use of inhaled nitric oxide. These patents are assigned to Massachusetts General Hospital (Boston, Massachusetts), which has licensed them to IKARIA (Clinton, New Jersey) and Linde Gas Therapeutics (Lidingo, Sweden). Dr. Zapol receives royalties and Dr. Bloch has received grants from IKARIA and Linde, which helped us to study inhaled nitric oxide.
Meetings at which the work has been presented: Annual Meetings of American Heart Association, November 7, 2007, Orlando, Florida. Abstract Oral Sessions (presentation number 1484)
Summary Statement: Breathing nitric oxide causes rapid accumulation of diverse nitric oxide metabolites in blood and tissues. This contributes to the ability of brief periods of nitric oxide inhalation to provide cardioprotection against murine cardiac ischemia-reperfusion injury.
Footnote statement describing the Web Enhancement: The additional information regarding this is available on the Anesthesiology Web site at http://www.anesthesiology.org. Detailed description of the nitric oxide absorption measurement, and blood and tissue analyses for each NO-metabolite are described in the Web Enhancement materials.
References
- 1.Griffiths MJ, Evans TW. Inhaled nitric oxide therapy in adults. N Engl J Med. 2005;353:2683–95. doi: 10.1056/NEJMra051884. [DOI] [PubMed] [Google Scholar]
- 2.Hogman M, Frostell C, Arnberg H, Hedenstierna G. Bleeding time prolongation and NO inhalation. Lancet. 1993;341:1664–5. doi: 10.1016/0140-6736(93)90802-n. [DOI] [PubMed] [Google Scholar]
- 3.Lee JS, Adrie C, Jacob HJ, Roberts JD, Jr, Zapol WM, Bloch KD. Chronic inhalation of nitric oxide inhibits neointimal formation after balloon-induced arterial injury. Circ Res. 1996;78:337–42. doi: 10.1161/01.res.78.2.337. [DOI] [PubMed] [Google Scholar]
- 4.Adrie C, Bloch KD, Moreno PR, Hurford WE, Guerrero JL, Holt R, Zapol WM, Gold HK, Semigran MJ. Inhaled nitric oxide increases coronary artery patency after thrombolysis. Circulation. 1996;94:1919–26. doi: 10.1161/01.cir.94.8.1919. [DOI] [PubMed] [Google Scholar]
- 5.Fox-Robichaud A, Payne D, Hasan SU, Ostrovsky L, Fairhead T, Reinhardt P, Kubes P. Inhaled NO as a viable antiadhesive therapy for ischemia/reperfusion injury of distal microvascular beds. J Clin Invest. 1998;101:2497–505. doi: 10.1172/JCI2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hataishi R, Rodrigues AC, Neilan TG, Morgan JG, Buys E, Shiva S, Tambouret R, Jassal DS, Raher MJ, Furutani E, Ichinose F, Gladwin MT, Rosenzweig A, Zapol WM, Picard MH, Bloch KD, Scherrer-Crosbie M. Inhaled nitric oxide decreases infarction size and improves left ventricular function in a murine model of myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;291:H379–84. doi: 10.1152/ajpheart.01172.2005. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, Huang Y, Pokreisz P, Vermeersch P, Marsboom G, Swinnen M, Verbeken E, Santos J, Pellens M, Gillijns H, Van de Werf F, Bloch KD, Janssens S. Nitric oxide inhalation improves microvascular flow and decreases infarction size after myocardial ischemia and reperfusion. J Am Coll Cardiol. 2007;50:808–17. doi: 10.1016/j.jacc.2007.04.069. [DOI] [PubMed] [Google Scholar]
- 8.Mathru M, Huda R, Solanki DR, Hays S, Lang JD. Inhaled nitric oxide attenuates reperfusion inflammatory responses in humans. Anesthesiology. 2007;106:275–82. doi: 10.1097/00000542-200702000-00015. [DOI] [PubMed] [Google Scholar]
- 9.Lang JD, Jr, Teng X, Chumley P, Crawford JH, Isbell TS, Chacko BK, Liu Y, Jhala N, Crowe DR, Smith AB, Cross RC, Frenette L, Kelley EE, Wilhite DW, Hall CR, Page GP, Fallon MB, Bynon JS, Eckhoff DE, Patel RP. Inhaled NO accelerates restoration of liver function in adults following orthotopic liver transplantation. J Clin Invest. 2007;117:2583–91. doi: 10.1172/JCI31892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelm M. Nitric oxide metabolism and breakdown. Biochim Biophys Acta. 1999;1411:273–89. doi: 10.1016/s0005-2728(99)00020-1. [DOI] [PubMed] [Google Scholar]
- 11.Wink DA, Mitchell JB. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med. 1998;25:434–56. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 12.Oda H, Kusumoto S, Nakajima T. Nitrosyl-hemoglobin formation in the blood of animals exposed to nitric oxide. Arch Environ Health. 1975;30:453–6. doi: 10.1080/00039896.1975.10666749. [DOI] [PubMed] [Google Scholar]
- 13.Gow AJ, Stamler JS. Reactions between nitric oxide and haemoglobin under physiological conditions. Nature. 1998;391:169–73. doi: 10.1038/34402. [DOI] [PubMed] [Google Scholar]
- 14.Ridnour LA, Thomas DD, Mancardi D, Espey MG, Miranda KM, Paolocci N, Feelisch M, Fukuto J, Wink DA. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol Chem. 2004;385:1–10. doi: 10.1515/BC.2004.001. [DOI] [PubMed] [Google Scholar]
- 15.Kharitonov VG, Sundquist AR, Sharma VS. Kinetics of nitrosation of thiols by nitric oxide in the presence of oxygen. J Biol Chem. 1995;270:28158–64. doi: 10.1074/jbc.270.47.28158. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida K, Kasama K, Kitabatake M, Okuda M, Imai M. Metabolic fate of nitric oxide. Int Arch Occup Environ Health. 1980;46:71–7. doi: 10.1007/BF00377461. [DOI] [PubMed] [Google Scholar]
- 17.Feelisch M, Rassaf T, Mnaimneh S, Singh N, Bryan NS, Jourd’Heuil D, Kelm M. Concomitant S-, N-, and heme-nitros(yl)ation in biological tissues and fluids: implications for the fate of NO in vivo. Faseb J. 2002;16:1775–85. doi: 10.1096/fj.02-0363com. [DOI] [PubMed] [Google Scholar]
- 18.McMahon TJ, Doctor A. Extrapulmonary effects of inhaled nitric oxide: role of reversible S-nitrosylation of erythrocytic hemoglobin. Proc Am Thorac Soc. 2006;3:153–60. doi: 10.1513/pats.200507-066BG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lecour S, Clermont G, du Toit E, Gilson L, Maupoil V, Lowe S, Dupuis P, Girard C, Rochette L. Evidence for the extrapulmonary localization of inhaled nitric oxide. Heart Dis. 2003;5:372–7. doi: 10.1097/01.hdx.0000098613.53486.08. [DOI] [PubMed] [Google Scholar]
- 20.Westfelt UN, Benthin G, Lundin S, Stenqvist O, Wennmalm A. Conversion of inhaled nitric oxide to nitrate in man. Br J Pharmacol. 1995;114:1621–4. doi: 10.1111/j.1476-5381.1995.tb14948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rassaf T, Bryan NS, Maloney RE, Specian V, Kelm M, Kalyanaraman B, Rodriguez J, Feelisch M. NO adducts in mammalian red blood cells: too much or too little? Nat Med. 2003;9:481–3. doi: 10.1038/nm0503-481. [DOI] [PubMed] [Google Scholar]
- 22.Gladwin MT, Ognibene FP, Pannell LK, Nichols JS, Pease-Fye ME, Shelhamer JH, Schechter AN. Relative role of heme nitrosylation and beta-cysteine 93 nitrosation in the transport and metabolism of nitric oxide by hemoglobin in the human circulation. Proc Natl Acad Sci U S A. 2000;97:9943–8. doi: 10.1073/pnas.180155397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elrod JW, Greer JJ, Bryan NS, Langston W, Szot JF, Gebregzlabher H, Janssens S, Feelisch M, Lefer DJ. Cardiomyocyte-specific overexpression of NO synthase-3 protects against myocardial ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2006;26:1517–23. doi: 10.1161/01.ATV.0000224324.52466.e6. [DOI] [PubMed] [Google Scholar]
- 24.Moya MP, Gow AJ, McMahon TJ, Toone EJ, Cheifetz IM, Goldberg RN, Stamler JS. S-nitrosothiol repletion by an inhaled gas regulates pulmonary function. Proc Natl Acad Sci U S A. 2001;98:5792–7. doi: 10.1073/pnas.091109498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stamler JS, Jaraki O, Osborne J, Simon DI, Keaney J, Vita J, Singel D, Valeri CR, Loscalzo J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci U S A. 1992;89:7674–7. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221–6. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 27.DeRubertis FR, Craven PA. Calcium-independent modulation of cyclic GMP and activation of guanylate cyclase by nitrosamines. Science. 1976;193:897–9. doi: 10.1126/science.7837. [DOI] [PubMed] [Google Scholar]
- 28.Gruetter CA, Barry BK, McNamara DB, Gruetter DY, Kadowitz PJ, Ignarro L. Relaxation of bovine coronary artery and activation of coronary arterial guanylate cyclase by nitric oxide, nitroprusside and a carcinogenic nitrosoamine. J Cyclic Nucleotide Res. 1979;5:211–24. [PubMed] [Google Scholar]
- 29.Zweier JL, Wang P, Samouilov A, Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nat Med. 1995;1:804–9. doi: 10.1038/nm0895-804. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Samouilov A, Liu X, Zweier JL. Characterization of the magnitude and kinetics of xanthine oxidase-catalyzed nitrate reduction: evaluation of its role in nitrite and nitric oxide generation in anoxic tissues. Biochemistry. 2003;42:1150–9. doi: 10.1021/bi026385a. [DOI] [PubMed] [Google Scholar]
- 31.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–63. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 32.Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, Gladwin MT, Lefer DJ. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–40. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, Wang X, MacArthur PH, Shoja A, Raghavachari N, Calvert JW, Brookes PS, Lefer DJ, Gladwin MT. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hogg N, Broniowska KA, Novalija J, Kettenhofen NJ, Novalija E. Role of S-nitrosothiol transport in the cardioprotective effects of S-nitrosocysteine in rat hearts. Free Rad Biol Med. 2007;43:1086–94. doi: 10.1016/j.freeradbiomed.2007.06.016. [DOI] [PubMed] [Google Scholar]