

Abstract
Efficient apoptotic cell clearance is critical for maintenance of tissue homeostasis, and to control the immune responses mediated by phagocytes. Little is known about the molecules that contribute “eat me” signals on the apoptotic cell surface. C1q, the recognition unit of the C1 complex of complement, also senses altered structures from self and is a major actor of immune tolerance. HeLa cells were rendered apoptotic by UV-B treatment and a variety of cellular and molecular approaches were used to investigate the nature of the target(s) recognized by C1q. Using surface plasmon resonance, C1q binding was shown to occur at early stages of apoptosis and to involve recognition of a cell membrane component. C1q binding and phosphatidylserine (PS) exposure, as measured by annexin V labeling, proceeded concomitantly, and annexin V inhibited C1q binding in a dose-dependent manner. As shown by cosedimentation, surface plasmon resonance, and x-ray crystallographic analyses, C1q recognized PS specifically and avidly (KD = 3.7-7 × 10-8 M), through multiple interactions between its globular domain and the phosphoserine group of PS. Confocal microscopy revealed that the majority of the C1q molecules were distributed in membrane patches where they colocalized with PS. In summary, PS is one of the C1q ligands on apoptotic cells, and C1q-PS interaction takes place at early stages of apoptosis, in newly organized membrane patches. Given its versatile recognition properties, these data suggest that C1q has the unique ability to sense different markers which collectively would provide strong eat me signals, thereby allowing efficient apoptotic cell removal.
Proper recognition and clearance of apoptotic cells is critical for many biological processes including development, tissue remodeling, and maintenance of homeostasis. The rapid removal of unwanted self-cells by professional or nonprofessional phagocytes prevents the release of their intracellular contents and is essential to reduce inappropriate inflammation and avoid autoimmune disorders. Nevertheless, apoptotic cell clearance is not neutral in immunological terms. In fact, apoptotic cell recognition critically regulates immune responses, particularly when achieved by professional phagocytes such as macrophages or immature dendritic cells (DCs).3 Thus, macrophages engulfing apoptotic cells secrete anti-inflammatory cytokines (1-3) and DCs that have captured apoptotic cells induce tolerance by silencing autoreactive T cells or by inducing T cell tolerance to self-Ags (4, 5).
The uptake of apoptotic cells involves a wide variety of phagocyte receptors and soluble bridging molecules (6) which bind these cells in response to “eat me” signals. Several receptors have been proposed to mediate recognition and engulfment of apoptotic bodies, but little is known about the molecules or patterns that contribute eat me signals on the apoptotic cell surface. To date, the best characterized molecule known to allow discrimination of apoptotic cells from normal ones and promote their clearance is phosphatidylserine (PS), which becomes exposed on the surface of cells undergoing programmed cell death. A candidate phagocyte receptor for PS (PSR) identified by phage display has been proposed (7, 8), but this hypothesis is challenged by studies using PSR-deficient animals (9-11). More recent studies (12) have demonstrated that calreticulin, a well-characterized protein chaperone, acts as an eat me signal on the surface of apoptotic cells, whereas this signal is overridden on viable cells by other “don't eat me” signals involving CD47.
The role of the bridging molecules in apoptotic cell recognition also awaits precise characterization. Among these molecules, the implication of C1q, the recognition subunit of the first complement component, is increasingly recognized. Its involvement in apoptotic cell recognition and clearance in vivo has been elegantly demonstrated by knockout experiments (13, 14), and C1q deficiency has been clearly shown to be associated with systemic lupus erythematosus and other autoimmune diseases (15, 16). The traditional portrait of C1q as a molecule able to trigger complement activation in an Ab-dependent manner has been largely reconsidered (17) following the discovery that C1q has the ability to recognize altered structures from self such as β-amyloid fibrils (18), the pathological form of the prion protein (19, 20), and apoptotic cells (21). Other recent data reveal a specific role of C1q in the biology of DCs. Thus, immature DCs are an important C1q source, whereas maturation abrogates C1q production (22). In contrast, opsonization with C1q targets apoptotic cells to DCs (23). These findings, together with previous observations, provide strong indication that C1q is a key player in the maintenance of immune tolerance.
C1q is a 460-kDa hexameric protein comprising six heterotrimeric collagen-like triple-helical fibers, each prolonged by a C-terminal globular region (GR) which supports most, if not all, of the C1 recognition activities (17, 24, 25). Using immunofluorescence microscopy, it was shown that the C1q GR specifically binds to the apoptotic cell surface (26). In vivo studies have established that apoptotic cell clearance is dependent on C1q in two ways, with or without activation of the classical complement pathway (14), but the mechanisms by which C1q recognizes “unwanted” self-cells remain elusive. The C1q-binding site(s) on the apoptotic cell membrane is (are) still poorly documented, due for a large part to the continuous modifications that take place on the surface of cells undergoing apoptosis. In a recent study (27), it was proposed that C1q senses nucleic acids rapidly exposed on the apoptotic cell surface, thereby triggering complement activation.
In view of the crucial role of C1q-mediated phagocytosis in the maintenance of immune tolerance, we undertook to identify the apoptotic cell component(s) recognized by C1q at the early stages of apoptosis, when efficient engulfment prevents release of intracellular components in the surrounding medium. This was achieved using a variety of molecular and cellular techniques. Altogether, our data demonstrate that PS, a classical marker of apoptosis, is a major ligand recognized by C1q on the apoptotic cell surface. This finding, together with the known binding versatility of C1q, sheds new light on the physiological role of C1q and provides insights into the recognition mechanism of this major actor in the clearance of unwanted self-cells and control of immune tolerance.
Materials and Methods
Proteins and lipids
C1q was purified from human serum and its GR was prepared as described previously (18, 28). Annexin V, PS, and phospho-L-serine were obtained from Sigma-Aldrich. Other phospholipids, including 06:0 PS were obtained from Avanti Polar Lipids. Purified calreticulin was provided by G. Houen (Statens Serum Institut, Copenhagen, Denmark). Antiserum against human C1q was obtained in our laboratory (29).
Cell culture and induction of apoptosis
HeLa cells (CCL-2; American Type Culture Collection (ATCC)) were cultivated in Glutamax DMEM medium (Invitrogen Life Technologies) supplemented with 10% (v/v) FCS, 2.5 U/ml penicillin, and 2.5 μg/ml streptomycin (Invitrogen Life Technologies). Cells (3 × 106) were grown in sterile dishes overnight to 60-80% confluence and exposed to 1000 mJ/cm2 UV-B irradiation at 312 nm in fresh DMEM medium. For some experiments, as indicated in the text, apoptosis was induced using 0.25 μM staurosporine (Sigma-Aldrich). The conditions for induction of apoptosis by UV-B and staurosporine were designed to yield comparable annexin V-positive cell numbers at each time point (respectively, 25 ± 4% and 50 ± 5% at 4 and 20 h posttreatment). Both types of conditions were optimal to render HeLa cells apoptotic as determined by preliminary studies. Cells were then incubated under 5% CO2 at 37°C for different periods.
Flow cytometry
Cells were harvested using trypsin-EDTA (Invitrogen Life Technologies) at varying times postirradiation. To analyze the whole apoptotic cell population, nonadherent cells present in the culture medium were added to harvested cells. Recognition of apoptotic cells by the C1q GR was assessed from binding of biotinylated GR. Cells (4 × 106/ml) were suspended in PBS (pH 7.4; Invitrogen Life Technologies) containing 0.3% BSA, incubated for 1 h on ice with 25 μg/ml GR biotinylated with the ECL protein biotinylation system (Pharmacia Biotech), washed twice, resuspended in PBS, and incubated on ice for 30 min with streptavidin-R-PE (BD Pharmingen/BD Biosciences). Cells were then washed twice, resuspended in PBS, and rapidly analyzed with a FACScan flow cytometer using CellQuest software (BD Biosciences). PS exposure and necrosis were assessed using an apoptosis detection kit (Oncogene VWR International). Briefly, HeLa cells (106 cells/ml) were incubated for 15 min with 1.5 μg/ml Annexin VFITC and then analyzed immediately by flow cytometry in the presence of 0.6 μg/ml propidium iodide (PI). PI-positive cells were regarded as necrotic permeabilized cells.
Cosedimentation analyses
Liposomes containing PS, phosphatidylcholine (PC), phosphatidylethanolamine, or a 1:1 PC:PS mixture (each 100 μg in 100 μl of PBS (pH 7.4)) were obtained by treatment for 20 min in a bath sonicator (30). The GRs (10 μg in 100 μl of PBS (pH 7.4)) were incubated with each liposome for 30 min at 22°C. Samples were centrifuged at 300,000 × g for 30 min at 4°C, allowing separation of the supernatant from the lipid-containing pellet. The GR contents of the supernatant and pellet fractions were determined by means of a 10% SDS-PAGE analysis followed by Coomassie blue staining.
Surface plasmon resonance (SPR) spectroscopy
Analyses were conducted on a BIAcore 3000 or BIAcore X instrument.
Analyses on phospholipid-coated surfaces
To prepare phospholipid micelles, aliquots of PS, PC, and phosphatidylethanolamine dissolved in chloroform were transferred into glass tubes, and a thin phospholipid film was formed on the wall by rotating the tube while evaporating chloroform under a stream of argon. Dried lipids were suspended in PBS (pH 7.4) to give a final concentration of 300 μg/ml and were then sonicated for 15 min just before preparation of the surface. The lipid layer on the HPA sensor chip (BIAcore) was then formed using the standard procedure recommended by BIAcore. The phospholipid-coated chips were blocked with 0.1 mg/ml BSA for 5 min. The specific binding signal shown was obtained by subtracting the background signal, obtained by injection of the protein sample over a surface saturated with BSA. C1q binding was routinely measured over 1500 resonance units of immobilized phospholipids at a flow rate of 10 μl/min in the running buffer (PBS (pH 7.4)). Complete analyte dissociation was achieved by injection of 10 μl of 12 mM EDTA.
Analyses on C1q- or phosphoserine-coated surfaces
The running buffer for C1q and phosphoserine immobilization was 145 mM NaCl, 5 mM EDTA, 10 mM HEPES (pH 7.4). C1q and phosphoserine were diluted to 40 μg/ml in 10 mM sodium acetate (pH 4.0) and to 16 mg/ml in 10 mM formate (pH 3.0), respectively, and immobilized onto a CM5 sensor chip (BIAcore) using the BIAcore amine coupling kit. Binding of 06:0 PS to immobilized C1q and of C1q to immobilized phosphoserine was measured at a flow rate of 20 μl/min in PBS containing 0.005% surfactant P20 (pH 7.4). Surfaces were regenerated by injection of 10 μl of 10 mM NaOH. The specific binding signal shown was obtained by subtracting the background signal, routinely obtained by injection of the sample over an activated-deactivated surface. All data were analyzed by global fitting to a 1:1 Langmuir binding model of both the association and dissociation phases for several concentrations simultaneously, using the BIAevaluation 3.2 software (BIAcore). In each case, the data presented were obtained with a statistic χ2 value <2. The apparent equilibrium dissociation constants (KD) were calculated from the ratio of the dissociation and association rate constants (koff/kon). Experiments performed in the presence of 1 mM CaCl2 or 1 mM EDTA provided no evidence for a requirement of Ca2+ for the interactions.
Analyses using viable or apoptotic cells
HeLa cells were suspended at 2.5 × 105 cells/ml in 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 25 mM HEPES (pH 7.4), containing 0.005% surfactant P20, and passed over a C1q-coated surface at a flow rate of 10 μl/min. The surface was regenerated by injection of 10 mM NaOH. The specific binding signal shown was obtained by subtracting the background signal obtained by injection of the cell sample over an activated-deactivated surface. For inhibition experiments, untreated control cells as well as apoptotic cells 2 h postirradiation were incubated with increasing concentrations of annexin V for 20 min at room temperature before injection over immobilized C1q.
Crystallization and data collection
C1q GR crystals suitable for diffraction were obtained as described previously (24). Briefly, the protein was concentrated in 250 mM NaCl, 2% glycerol, 50 mM Tris-HCl (pH 7.6), containing 100 mM nondetergent sulfo-betain 195 as a solubilizing agent. Crystals were obtained by the hanging-drop vapor-diffusion method, with the well solution containing 30-41% PEG 4000, 20 mM CaCl2, 10 mM 2-ME, 100 mM Tris-HCl (pH 7.0 or 7.4). Microseeding was used to obtain more reproducible native crystals. Cocrystallization trials with 06:0 PS were first conducted under vapor diffusion conditions close to those used to generate native GR crystals, using C1q GR solutions preincubated overnight with 10 or 20 mM 06:0 PS. Crystals were obtained under these conditions, but they proved to be native upon crystallographic analysis. Cocrystallization trials were also performed using commercial crystallization kits and the robot available at the European Molecular Biology Laboratory Grenoble outstation. The most successful soaking experiments were conducted using prolonged (≥2 days) soaking into a solution only containing 42% PEG 4000, 10 mM CaCl2, 100 mM Tris-HCl (pH 7.4), and a phosphoserine concentration of 12.5 mM, slightly above its upper solubility limit in this type of solution. Soaking experiments were not performed using 06:0 PS because of its heavy precipitation under similar conditions.
Diffraction data were collected on the European Synchrotron Radiation Facility beam lines ID14eh2, ID23eh2, and ID29. Reflection data were processed with the XDS package (31). Crystals grew in the monoclinic P1 space group (a = 48.09 Å, b = 48.07Å, c = 84.7Å, α = 91.34°, β = 93.34°, γ = 113.68°), with two C1q GR trimers per asymmetric unit.
Structure determination and refinement
The structure was solved by automated molecular replacement using AMoRe (32) or Phaser (33) using the native C1q trimer structure as a search model. Refinement was done using Refmac5 (34) alternately with inspection of electron density maps and model correction using Coot (35). Five percent of the reflections were not included in the refinement to monitor Rfree. The additional electron density observed in one trimer was better interpreted as two alternative conformations of the phosphoserine ligand (see Fig. 6B). The extra electron density seen in the second trimer was modeled as two alternative conformations of Arg111, likely indicating weak binding of the ligand at this position. The final crystallographic and refinement statistics are listed in Table II.
FIGURE 6.

X-ray structure of a phosphoserine-C1q GR complex. A, Overall view of the interaction. Subunits A-C are colored blue, green, and cyan, respectively, and are shown in ribbon representation. The ligand is shown in stick representation. The Ca2+ ion bound to the GR is represented as a golden sphere. In subunit C, the two surface loops involved in ligand stabilization are colored dark blue. N indicates the N-terminal end of the subunits, connecting the GR to the preceding collagen-like triple helix. According to the current C1q model (17), the B subunit marks the external face of each GR in the whole C1q molecule and the target surface is expected to lie roughly at the bottom of the GR structure. B, Detailed view of the interactions between subunit C and phosphoserine. The two alternative conformations of phosphoserine are shown. Electron densities (2mFo-Fc) are contoured at the 1σ level. C, Comparative detailed view of the phosphoserine-binding region in the native GR structure, illustrating the extended conformation of the side chain of Arg111. D, Overall model of the C1q molecule (17) illustrating the positioning of the phosphoserine-binding site. Arg111 and Arg98 are colored yellow and red, respectively. This figure was designed using Pymol and Grasp (62).
Table II.
Crystallographic and refinement statistics
| C1q GR- Phosphoserine Complex | Native C1q GR | |||||
|---|---|---|---|---|---|---|
| Unit cell (Å) | 48.09 | 48.07 | 84.70 | 48.15 | 48.24 | 87.79 |
| (°) | 91.338 | 93.340 | 113.678 | 92.454 | 92.646 | 113.532 |
| European Synchrotron Radiation Facility beamline | ID14-eh2 | ID29 | ||||
| Measured reflections | 75,256 | 107,754 | ||||
| Unique reflections | 38,993 | 50,005 | ||||
| Resolution (Å)a | 2.05 (-2.1) | 1.95 (-2.0) | ||||
| Completeness (%)a | 89.7 (77.2) | 95 (84.3) | ||||
| Rsyma,b | 4.2 (9) | 4.2 (8.4) | ||||
| 〈I〉/〈σ(I)〉a | 13.5 (7.9) | 16 (9.2) | ||||
| Refinement | ||||||
| Resolution range (Å) | 20-2.05 | 15-1.95 | ||||
| Reflections | 37,012 | 44,958 | ||||
| Rwork | 17.7 | 21.2 | ||||
| Rfree | 23.8 | 25.4 | ||||
| Protein atoms | 6,412 | 6231 | ||||
| Calcium atoms | 2 | 2 | ||||
| Solvent atoms | 364 | 193 | ||||
| Ligand atoms | 20 | 0 | ||||
| Rmsd bonds (Å) | 0.007 | 0.008 | ||||
| Rmsd angles (°) | 1.02 | 1.46 | ||||
Values for the outermost resolution shell are in parentheses.
Rsym = {ΣhklΣi|Ii(hkl) - 〈I(hkl)〉|/ΣhklΣiIi(hkl)} × 100, where Ii is the i-th measurement of reflection I(hkl).
Confocal laser scanning microscopy
HeLa cells were incubated with Annexin V-FluoProbes Alexa 488 according to the manufacturer's protocol (Interchim) to reveal PS exposure, and with 10 μg/ml C1q in 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 1 mg/ml BSA, 25 mM HEPES (pH 7.4) for 1 h at 4°C. Cells were then washed in the HEPES buffer and fixed for 5 min with 4% paraformaldehyde. C1q was detected by indirect immunofluorescence using an anti-C1q polyclonal Ab diluted 1/2000 in the HEPES buffer. Bound Abs were visualized with cyanine 3-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories) diluted 1/1000 in the HEPES buffer. DNA was then stained with 1 μg/ml Hoechst dye (Sigma-Aldrich) and cells were mounted on glass slides with 25 mg/ml DABCO (Sigma-Aldrich) in a PBS:glycerol 1:9 (v/v) solution. Cells were photographed using a laser confocal fluorescence microscope (Leica). To assess spatial localizations within the cell and at the surface, serial optical sections were taken at 0.7-μm intervals throughout the thickness of all cells examined. The proportion of labeled cells was estimated by counting an average of 150 cells on several observation fields at a 400-fold magnification.
Results
C1q recognizes HeLa cells at early stages of apoptosis
Previous studies based on indirect immunofluorescence experiments have shown that C1q binds to apoptotic cells through its GRs 4-6 h after apoptosis (26). To investigate in more detail the ability of C1q to recognize cells at the early stages of apoptosis, HeLa cells were rendered apoptotic by UV-B irradiation and C1q binding was analyzed by real-time SPR spectroscopy, using immobilized C1q and cells as fluid-phase ligands. A control experiment was performed initially using viable cells, revealing a significant increase in the binding signal (Fig. 1). This indicated that cells were captured to some extent by immobilized C1q, in agreement with previous studies reporting C1q-mediated adhesion of endothelial cells (36). A 2.5-fold increase in the binding was observed when cells were treated with UV-B and then passed over the sensor chip 2 h after irradiation, indicating that apoptosis induced a marked increase in C1q recognition. Increasing the postirradiation period to 4 and 6 h progressively reduced the binding signal to values that nevertheless remained higher than those observed for intact cells. As the SPR signal reflects mass variations at the surface of the sensor chip, a plausible hypothesis is that this decrease was an indirect consequence of a loss of mass occurring during progression of apoptosis. The binding signal remained remarkably stable during the dissociation phase, indicative of a high affinity interaction between C1q and apoptotic cells.
FIGURE 1.

SPR analysis of apoptotic cell capture on a C1q-coated surface. Viable or UV-B-irradiated HeLa cells were harvested using trypsin-EDTA, suspended at 2.5 × 105 cells/ml in 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, and 25 mM HEPES (pH 7.4) containing 0.005% surfactant P20, and then passed over a C1q-coated sensor chip at a flow rate of 10 μl/min. Analyses were performed on viable control cells, and on cells harvested 2, 4, and 6 h after UV-B irradiation, as indicated. Association and dissociation were monitored for 120 s, as indicated.
Similar experiments were next performed to test capture of apoptotic cells on a C1q GR-coated surface. This provided evidence for interaction (data not shown), but the binding signal was too weak to allow further analysis, likely reflecting the fact that, due to its monomeric character, the isolated GR was less efficient in mediating cell recognition than the whole hexameric C1q protein. Consequently, the ability of the GR to recognize apoptotic cells was investigated by flow cytometry using biotinylated GR and streptavidin-R-PE labeling (Fig. 2). Again, a significant binding to untreated HeLa cells was detected (Fig. 2A), in keeping with our preceding observations with intact C1q (Fig. 1). Nevertheless, compared with untreated cells, UV-B treated cells exhibited more pronounced GR binding as reflected by the increased fluorescence values of the main cell population at 4 h postirradiation (Fig. 2B). In addition, the apoptosis process generated cells with higher fluorescence values, and this population increased progressively up to 20 h. Analysis of forward (FSC) and side scattering (SSC) properties of the cells at 4 h postirradiation showed more spread FSC and SSC values with a low proportion of cells of small size (corresponding to the lower left part of the gated population). At 20 h, the proportion of these cells increased, consistent with the morphological changes known to occur during apoptosis (37).
FIGURE 2.

Flow cytometry analysis of the interaction between the C1q GR and apoptotic HeLa cells at various periods after UV-B irradiation or treatment with staurosporine. Cells were incubated with biotinylated GR molecules and binding was monitored by flow cytometry after addition of streptavidin-PE. Untreated cells (A), and apoptotic cells 4 and 20 h after treatment with UV-B (B) or staurosporine (C), were analyzed. The upper five panels show dot plots of FSC and SSC of the cell populations. The boxes define the population analyzed for GR binding (bold lines) on the lower five panels. In these panels, dotted lines represent the fluorescence background determined using streptavidin-PE alone. UV-B and staurosporine induction of apoptosis was designed to yield similar annexin V-positive cell numbers at each time point (see Materials and Methods).
For comparative purposes, the latter experiment was performed in parallel with cells treated with the widely used apoptosis-inducing agent staurosporine (38). Again, the early apoptotic cell population exhibited a more marked GR binding and this increased when increasing the cell culture period (Fig. 2C). The cell populations, analyzed on FSC and SSC dot plots, were similar to those observed after UV-B treatment.
The apoptotic state of the UV-B-treated cells was also assessed by double Annexin VFITC and PI labeling. As illustrated in Fig. 3, the number of annexin V-reactive cells (i.e., of cells exposing PS) started increasing at 2 h, when cell membranes had not yet undergone permeabilization as indicated by the absence of PI labeling. Permeabilization did not increase significantly before 6 h, indicating that at this time cells had not undergone significant necrosis. Similar results were obtained after treatment with staurosporine (data not shown). At 20 h postinduction, ~50% of the cells were PI positive. Incidentally, this indicated that the higher GR binding observed 20 h after cells treatment was likely due in part to cell permeabilization.
FIGURE 3.
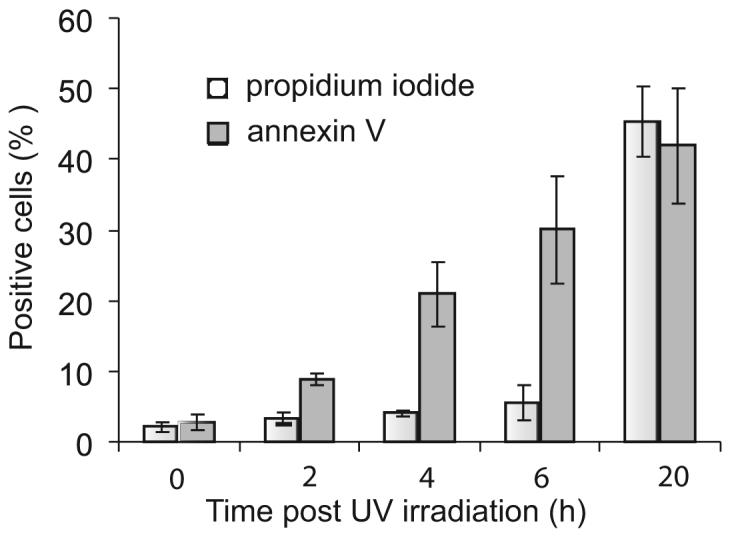
C1q GR binding to apoptotic cells is concomitant with PS exposure. Time course analysis of PS exposure and cell permeabilization during apoptosis. PS exposure and cell permeabilization were analyzed by double annexin V/PI labeling as described in Materials and Methods. The data shown represent the mean value ± SD of three independent experiments.
To assess the specificity of GR binding to apoptotic cells, untreated and UV-B irradiated cells were double-labeled with biotinylated GR and a fluorescent cell-permeable inhibitor of activated caspases (Poly Caspases Detection kit; Immunochemistry Technologies). Control cells were negative for caspase activation and displayed low GR binding, in keeping with the data shown in Fig. 2. In contrast, at 4 h postirradiation, 88.5% of UV-B irradiated cells bound GR, and ~20% were positive for both caspase activation and GR binding. Relative to untreated cells, caspase-positive cells exhibited a higher increase in the mean fluorescence value compared with the caspase-negative population (data not shown). These observations provided support to the specificity of GR binding to apoptotic cells.
Taken together, these experiments indicated that: 1) C1q was able to recognize apoptotic cells at a very early stage; 2) C1q likely recognized (a) cell membrane component(s), as interaction could be monitored by SPR in a configuration where C1q was immobilized and hence not expected to have access to inner cell components; 3) on the average cell population, as judged from GR binding and annexin V labeling, the increase in C1q binding was concomitant with PS exposure on the cell surface.
The C1q globular region associates with PS-containing liposomes
The above observations prompted us to investigate the ability of C1q to bind to PS and other membrane phospholipids. For this purpose, the C1q GRs were initially incubated with lipid vesicles composed of PS, phosphatidylethanolamine, or PC, and interaction was assessed by a cosedimentation assay, from the relative amount of GRs associated with the vesicles in the pellet after ultracentrifugation. As shown in Fig. 4, a significant proportion of the GRs was detected in the pellet fraction when incubated with vesicles containing PS alone or a PS/PC mixture. In contrast, no significant binding to PC- or phosphatidylethanolamine-containing vesicles was observed, because in both cases the whole GR population was found in the supernatants. This provided strong evidence for the ability of the C1q GRs to bind specifically to PS.
FIGURE 4.

Analysis by cosedimentation of the interaction between the C1q GR and PS-containing liposomes. The C1q GR was allowed to interact with liposomes containing PS, PC, phosphatidylethanolamine, or a 1:1 PC:PS mixture. After ultracentrifugation, the pellet and supernatant fractions were separated and their relative GR content was assessed by SDS-PAGE analysis as described in Materials and Methods. Molecular mass markers are indicated on the right. P, Pellet; S, supernatant. The data shown are representative of three independent experiments.
SPR analysis of C1q/PS interaction demonstrates high affinity
Further characterization of the C1q/PS interaction was achieved by SPR, using initially phospholipid surfaces assembled on HPA sensor chips and the intact C1q molecule or its GR as soluble analytes. The use of HPA hydrophobic surfaces allows formation of a flat rigid phospholipid monolayer with the polar groups oriented toward the fluid phase (39, 40). Preliminary control experiments showed that neither C1q nor its GR bound to a significant extent to an unmodified HPA surface saturated with BSA. We next tested the ability of the GRs to interact with phospholipid surfaces containing either PS, PC, or phosphatidylethanolamine. As demonstrated in Fig. 5A, the GRs readily bound to PS-containing monolayers, but no significant binding to PC- or phosphatidylethanolamine-coated surfaces was detected, providing further evidence of the C1q-binding specificity for PS. The kinetic parameters of PS recognition were determined by recording sensorgrams at varying GR (Fig. 5B) and C1q (Fig. 5C) concentrations. The association (kon) and dissociation (koff) rate constants, and the resulting KD were determined in both instances (Table I). Whereas koff values were similar in both cases, C1q exhibited a significantly higher kon value than its GR. As a result, the KD value of the interaction was ~7-fold higher for the GR compared with intact C1q, again accounting for a decreased binding avidity due to the monomeric structure of the GR.
FIGURE 5.

SPR analysis of the interaction between C1q or its GR and PS or PS derivatives. A and B, Binding of the C1q GR to immobilized PS. C, Binding of intact C1q to immobilized PS. D, Binding of 06:0 PS to immobilized C1q. E, Binding of C1q to immobilized phosphoserine. All interactions were measured in the running buffer at a flow rate of 10 μl/min. Association and dissociation curves were each recorded for 120 s. The concentrations of soluble ligands were as follows: GR: 0.75 μM (A) and 0.32, 0.75, 1.5, and 3 μM (B); C1q: 6, 12, 25, 70, and 100 nM (C) and 1.12, 3.25, 7.5, 15, and 30 nM (E); 06:0 PS: 0.15, 0.3, 0.6, and 0.9 μM (D). All other conditions are described in Materials and Methods.
Table I.
Kinetic and dissociation constants for the interaction ofC1q or its GR and PS and its derivativesa
| Soluble Analyte/Immobilized Ligand | Kon(M-1 × s-1) | Koff(s-1) | kD(M) |
|---|---|---|---|
| C1q/PSb | 2.9 × 104 | 2.1 × 10-3 | 7 × 10-8 |
| C1q GR/PSb | 3.5 × 103 | 1.8 × 10-3 | 5 × 10-7 |
| 06:0 PS/C1qc | 13.6 | 2.4 × 10-3 | 1.7 × 10-4 |
| C1q/phosphoserinec | 8.4 × 104 | 3.1 × 10-3 | 3.7 × 10-8 |
Interactions were measured on either HPAb or CM5c sensorchips, as described in Materials and Methods. The data shown correspond to a representative series of binding studies performed on the same sensor chip. In each case, similar results were reproduced from at least three independent experiments, using different phospholipid and protein preparations, and different sensor chips.
Further investigations of the C1q-PS interaction were conducted using a commercial water-soluble derivative (06:0 PS) comprising the polar moiety of PS connected to two six-carbon saturated hydrocarbon chains. In this case, C1q was coated on a CM5 sensor chip and 06:0 PS was used as a soluble ligand. As illustrated in Fig. 5D, SPR analysis at varying 06:0 PS concentrations demonstrated a dose-dependent interaction with immobilized C1q. Interestingly, the KD of the interaction was ~2400-fold higher than that determined for the interaction between immobilized PS and soluble C1q, due essentially to a dramatic decrease in the kon value (Table I). A plausible explanation for this decrease lies in the fact that C1q was immobilized in the latter case, and hence lost its binding avidity. In addition, it appears likely that C1q was chemically immobilized to the surface in a variety of configurations, not all optimal for ligand binding, further contributing to the marked decrease in kon.
To gain insights into the determinant(s) of the PS molecule involved in C1q recognition, its phosphoserine moiety was immobilized covalently on a CM5 sensor chip, and allowed to bind soluble C1q. As illustrated in Fig. 5E, C1q bound efficiently to phosphoserine, with a KD of ~3.7 × 10-8 M, comparable to the value determined for the interaction with immobilized PS (Table I). It became clear, therefore, that most, if not all, of the C1q/PS interaction occurred through the phosphoserine group.
X-ray structure of a phosphoserine-C1q GR complex
X-ray crystallography analyses were conducted with the aim to observe interaction between the C1q GR and PS-derived ligands at the atomic level. Two main strategies were used for this purpose: cocrystallization of the C1q GR with the ligand and soaking of native C1q GR crystals into ligand-containing solutions. In each case, x-ray diffraction data were collected, and structures were solved and refined to 2-Å resolution (see Materials and Methods). The crystallographic statistics are provided in Table II.
Compared with the native GR structure, no additional electron density was observed when the protein was crystallized in the presence of 06:0 PS (Fig. 6C), or after soaking crystals in the presence of phosphoserine for 2 h. In contrast, an extra density corresponding to the ligand was clearly seen after prolonged soaking in a phosphoserine solution (Fig. 6B). Crystals could only be obtained under conditions close to those used initially to crystallize the native C1q GR (24), and all other attempts using commercial screening kits proved unsuccessful. As depicted in Fig. 6A, phosphoserine is stabilized mainly by residues from two surface loops (108-111 and 126 -129) of subunit C of the C1q GR. The ligand is observed in two alternative conformations (Fig. 6B), and in both cases the phosphoester group is stabilized by hydrogen bonds with the main chain carbonyls of Ser126 and Thr127 and by electrostatic interaction with Arg111. In one conformation, the serine nitrogen atom is H-bonded to the carbonyl group of Leu109. According to our current model of the C1q molecule (17), this site is located at the inner face of the C1q cone and oriented toward the target surface (Fig. 6, A and D). Interestingly, the side chain of Arg111, which is involved in crystal contacts in the native structure, is markedly displaced toward the inside of the protein in the phosphoserine-GR complex (Fig. 6, B and C). Thus, phosphoserine is also partly stabilized by interactions with a surface loop (158-162) of subunit A from a neighboring molecule. Although the phosphoserine molecule is clearly attracted toward the observed position in the crystal, its orientation is strongly constrained by the crystal packing and might therefore be slightly different from the one achieved under physiological conditions. This hypothesis is consistent with the observation that, at variance with our SPR-binding experiments, the observed C1q GR-phosphoserine complex involves minimal interaction with the serine moiety of phosphoserine. In this respect, the neighboring area located between Arg111 and Arg98, which is not accessible in the crystal lattice, appears as a plausible alternative binding site. Accommodation of the phosphoserine group at this position would bring it slightly closer to the underlying target surface (Fig. 6D).
The observed structure clearly excludes a binding of phosphoserine through the Ca2+-binding site located at the top of the C1q GR, as the latter is fully accessible in the crystal lattice and shows no extra electron density. Consistent with this finding, similar binding curves were obtained by SPR in the presence of either Ca2+ ions or EDTA (data not shown). The binding site determined here is therefore clearly different from other known PS-binding structures such as annexin V (41) and protein kinase C (42), which both involve Ca2+-bridging mechanisms.
Annexin V inhibits apoptotic cell recognition by C1q
Taken together, our previous observations provided strong support for an implication of PS in the recognition of apoptotic cells by C1q. To test the relevance of this hypothesis at the cellular level, we investigated whether annexin V, an established ligand of the PS polar head group (41, 43), had the ability to compete for apoptotic cell recognition by C1q. Due to technical constraints, linked in particular to the fact that annexin V binds reversibly to PS, this could not be monitored using flow cytometry. In contrast, SPR appeared as a method of choice for this purpose, because it allowed us to monitor C1q binding in real time, immediately after incubation of the cells in the presence of annexin V. As illustrated in Fig. 7, both untreated and early apoptotic HeLa cells were allowed in parallel to bind immobilized C1q in the presence of increasing concentrations of soluble annexin V. Whereas annexin V had no significant effect on the capture of viable cells by C1q, it inhibited recognition of apoptotic cells in a dose-dependent manner, with nearly 50% inhibition at a concentration of 1.5 μM. This result provided direct evidence of the implication of PS as a major target for C1q on the apoptotic cell surface.
FIGURE 7.

Annexin V inhibits apoptotic cell recognition by C1q. Apoptotic HeLa cells were harvested 2 h after UV-B irradiation and incubated at 2.5 × 105 cells/ml in the presence of increasing concentrations of annexin V for 20 min at room temperature in 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, and 25 mM HEPES (pH 7.4) containing 0.005% surfactant P20. Apoptotic and control cell samples were then passed over a C1q-coated sensor chip as described in Materials and Methods and cell capture was measured after association for 2 min as described in the legend to Fig. 1. Results are expressed relative to the capture of either viable cells or apoptotic cells by C1q in the absence of annexin V. The data shown represent the mean value ± SD of three independent experiments.
C1q mostly clusters in patches containing PS on the apoptotic cell surface
Confocal laser microscopy was next used to investigate the distribution of C1q and PS on apoptotic HeLa cells. Double immunofluorescence labeling of normal, early apoptotic (2 h postirradiation), and late apoptotic (20 h postirradiation) unfixed cells was performed using annexin V and a polyclonal anti-C1q antiserum (Fig. 8). Nuclei were visualized by Hoechst labeling. The proportion of cells stained by annexin V and/or by C1q was evaluated by counting an average of 150 cells. Fluorescence images confirmed that 85 ± 5% of untreated cells displayed a low-level C1q labeling (Fig. 8, A, C, and D). In contrast, as expected, no significant PS exposure was detected on these cells (Fig. 8B). In contrast, within the early apoptotic cell population, 16 ± 3% displayed a strong PS exposure (Fig. 8, E and F) and 81 ± 3% were C1q positive, with a significantly higher average fluorescence intensity compared with control cells (Fig. 8, E and G). Superimposition of C1q and annexin V labeling (Fig. 8H) reveals that the C1q molecules were generally clustered in membrane patches where they colocalized with PS. This was true for all annexin V-positive cells, but reflected somewhat different individual situations, as illustrated in the representative cell samples shown in Fig. 8M. On cell 1, PS is clustered in discrete patches where C1q colocalizes. Cell 2, likely corresponding to a more advanced apoptosis stage, shows a homogeneous annexin V labeling covering most of the plasma membrane and again most, if not all of the C1q molecules are clustered in patches also containing PS.
FIGURE 8.

PS and C1q colocalize within membrane patches on apoptotic cells. Cells were submitted to a double immunofluorescent labeling for PS (green) and C1q (red) as described in Materials and Methods. Nuclei were labeled with Hoechst (blue). In overall views (A, E, and I), white arrows indicate selected cells shown in focused views. A-D, Untreated control HeLa cells. E-H, Early apoptotic cells (2 h post-UV-irradiation). I-L, Late apoptotic cells (20 h post-UV irradiation). M, A group of two cells (1, 2) representative of the whole apoptotic cell population 4 h postirradiation, is shown. Scale bars represent 20 μm. In control experiments, no labeling was detected when cells were stained with primary and secondary Ab without preincubation with C1q.
The late apoptotic cell population was quite heterogeneous (Fig. 8I), all cells being labeled by C1q and annexin V, but to varying extents (Fig. 8, I-L). As a result of membrane permeabilization, annexin V staining diffused inside the cell. Again, the majority of the C1q molecules colocalized in membrane patches with PS (Fig. 8L). Interestingly, despite membrane permeabilization, very few C1q molecules were detected inside the cells.
These data, together with our previous observation that annexin V inhibits apoptotic cell recognition by C1q, provided clear evidence of a direct C1q-PS interaction occurring in membrane areas likely corresponding to newly organized patches.
Discussion
This study provides the first experimental evidence that C1q, an established recognition protein of the innate immune system, recognizes PS exposed at the surface of apoptotic cells. This conclusion is based on the following concordant observations: 1) C1q binds to apoptotic cells at early stages of apoptosis, well before cell permeabilization, a property that is crucial in terms of maintenance of immune tolerance. 2) C1q binding and PS exposure proceed concomitantly, and annexin V inhibits C1q binding in a dose-dependent manner. 3) As shown by cosedimentation and SPR analyses, C1q recognizes PS specifically and avidly (KD = 3.7-7 × 10-8 M), through multiple interactions between its GRs and the phosphoserine head of PS. 4) The majority of the C1q molecules are remarkably distributed in membrane patches where they colocalize with PS. This latter observation validates the observed C1q/PS interaction at the cellular level.
The ability of C1q to recognize PS is further substantiated at the atomic level by the x-ray crystallographic analysis of a phosphoserine-C1q GR complex, revealing that phosphoserine is bound to subunit C of the C1q GR, through a site located inside the C1q cone and oriented toward the target surface (Fig. 6, A and D). C1q is known for its ability to sense an amazing variety of immune and nonimmune targets (25). Based on the C1q GR structure (24) and on site-directed mutagenesis studies (44), two major recognition sites have been identified, at the apex of the C1q head (C-reactive protein, IgM) and in the equatorial region of subunit B (IgG). The present study indicates that PS binding occurs at a distinct site, and hence has possibly little interference on binding to the above ligands. These findings fully support the current concept that the C1q recognition versatility arises from the existence of multiple binding sites on its heterotrimeric globular domain (24, 44).
A number of studies have now clearly established that PS exposure at the outer leaflet of the cell membrane is a general feature of apoptotic cells that allows their elimination and elicits suppressive pathways that inhibit the inflammatory response induced by phagocytes. PS is critical for the uptake process and is required for phagocyte engulfment (45-48). The view that PS serves as an eat me signal has gained more support following observations that PS-containing vesicles and phospho-L-serine partially block phagocytosis (49) and that annexin V impairs uptake of apoptotic cells by macrophages (50). In addition, engulfment of apoptotic cells involves the redistribution of membrane PS on both the phagocyte and the target cell (51). A presumptive PSR at the phagocyte surface has been identified by phage display screening (7), but its role has been challenged recently by two independent studies based on PSR gene knockout experiments (9, 11) and by the observation that PSR is a nuclear protein (10, 52). Although disruption of the PSR gene in mice was shown to have different indirect effects on apoptotic cell phagocytosis, it was also demonstrated that this molecule is not generally involved in the specific innate recognition or uptake of apoptotic cells (53). These recent data leave open the question of the recognition mechanism(s) of PS by phagocytes, and particularly of the identity of the bridging molecule(s) and receptor(s) involved in this process.
Although C1q is traditionally viewed as a molecule able to trigger activation of the classical complement pathway in response to direct or indirect pathogen recognition, there is increasing evidence that this multipotent molecule plays a crucial role in the detection of altered self structures, as demonstrated in the case of amyloid fibrils, the pathologic form of the prion protein, and apoptotic cells (18, 19, 21). Unequivocally, C1q deficiency induces major defaults in apoptotic cell uptake and consequently in the control of the inflammatory process (13, 15, 54). The recognized role of C1q in the maintenance of immune tolerance has been recently reinforced by the discovery that it is involved in the biology of dendritic cells (22). These considerations, together with our own finding that C1q binds with high avidity to PS exposed at the surface of early apoptotic cells, strongly support the proposal that C1q may represent a major PS recognition molecule involved in the engulfment of apoptotic cells. C1q possesses a receptor (calreticulin) at the surface of most mammalian cells (55, 56) and may therefore obviously serve as a bridging molecule between PS exposed at the apoptotic cell surface and the phagocyte. The fact that macrophages and dendritic cells synthesize and secrete C1q (57-59) strengthens this hypothesis.
New insights into the nature of the eat me signals exposed at the surface of apoptotic cells arise from the recent finding that, in addition to PS, calreticulin acts as a second general recognition ligand and triggers phagocytosis through binding and activation of LDL-receptor-related protein (or CD91) on the engulfing cell (12). This process involves changes on the apoptotic cell surface that create an environment where don't eat me signals are inactivated, whereas eat me signals, including calreticulin and phosphatidylserine but most probably also other as yet unidentified molecules, congregate together and signal for removal. Calreticulin is a chaperone that is classically known for its ability to act as a receptor for the collagenous tail of C1q (56). It has been shown that ingestion of apoptotic cells via a C1q-dependent mechanism involves stimulation of calreticulin/CD91 (60), and it was proposed that C1q recognizes apoptotic cells via its GRs and is in turn recruited by the phagocyte through interaction between its collagen-like tail and calreticulin. The finding that calreticulin provides a second eat me signal on the apoptotic cell surface (12) leads us to hypothesize that this protein may in fact play a dual role, not only as a receptor for the C1q collagenous tail on the phagocyte, but also as an additional eat me ligand of the C1q GR on the apoptotic cell. The latter proposal appears consistent, because both calreticulin and C1q colocalize with PS on the apoptotic cell surface (12; this study). To test this hypothesis, we have conducted preliminary analyses by SPR, and indeed these indicate that both the GR and the collagenous tail of C1q bind calreticulin immobilized on a sensor chip with high affinity (P. Tacnet-Delorme, G. Houen, H. Païdassi, G. J. Arlaud, and P. Frachet, unpublished data).
Previous studies by Navratil et al. (26) have provided strong evidence that the C1q collagenous tail is not involved in the interaction with apoptotic blebs. In the same way, our own studies demonstrate that the C1q GR alone is able to sense apoptotic cells. It appears unlikely, therefore, that the interaction between C1q and early apoptotic cells investigated in the present study involves a binding of the collagen moiety of C1q to calreticulin, at least to a significant extent. Whether calreticulin, in addition to PS, is indeed a ligand of the C1q GR on the apoptotic cell surface remains to be demonstrated at the cellular level. Nevertheless, there is increasing evidence that identification of apoptotic cells involves sensing of multiple eat me signals, possibly congregated within the same membrane patches. If this hypothesis is correct, then C1q may be expected to play a central role in apoptotic cell recognition, owing to its multimeric structure, and the multipotent character of its recognition properties, which arise for a large part from the heterotrimeric structure of its recognition domain (17, 25). Indeed, to our knowledge, this latter feature is uncommon among innate immunity recognition proteins which, with a few exceptions such as lung surfactant proteins (61), are highly specific molecules recognizing a single type of ligand. Thus, C1q combines particular structural and functional features that make it an ideal candidate to sense and collect different molecular motifs, including PS, on the apoptotic cell surface. Taken individually, these motifs would possibly not be sufficient to trigger efficient recognition and removal, but together would constitute a strong eat me signal. This particular ability of C1q to sense multiple signals, together with the fact that it recognizes apoptotic cells at early stages, provides a basis for its established implication as a major actor of immune tolerance.
Acknowledgments
We are grateful to Françoise Lacroix and Didier Grunwald for their help with confocal microscopy.
Footnotes
The atomic coordinates presented in this article have been deposited in the Protein Data Bank with the accession codes 2jG8 and 2jG9.
Abbreviations used in this paper: DC, dendritic cell; PS, phosphatidylserine; GR, globular region; PC, phosphatidylcholine; SPR, surface plasmon resonance; FSC, forward scattering; SSC, side scattering; PI, propidium iodide.
Publisher's Disclaimer: The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Disclosures The authors have no financial conflict of interest.
References
- 1.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 2.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J. Clin. Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 4.Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 2000;191:411–416. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol. 2002;23:445–449. doi: 10.1016/s1471-4906(02)02281-0. [DOI] [PubMed] [Google Scholar]
- 6.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 7.Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RA, Henson PM. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- 8.Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, Ridley AJ, Fadok VA, Henson PM. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J. Cell Biol. 2001;155:649–659. doi: 10.1083/jcb.200108080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williamson P, Schlegel RA. Hide and seek: the secret identity of the phosphatidylserine receptor. J. Biol. 2004;3:14. doi: 10.1186/jbiol14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cikala M, Alexandrova O, David CN, Proschel M, Stiening B, Cramer P, Bottger A. The phosphatidylserine receptor from Hydra is a nuclear protein with potential Fe(II) dependent oxygenase activity. BMC Cell Biol. 2004;5:26. doi: 10.1186/1471-2121-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bose J, Gruber AD, Helming L, Schiebe S, Wegener I, Hafner M, Beales M, Kontgen F, Lengeling A. The phosphatidylserine receptor has essential functions during embryogenesis but not in apoptotic cell removal. J. Biol. 2004;3:15. doi: 10.1186/jbiol10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 13.Botto M. C1q knock-out mice for the study of complement deficiency in autoimmune disease. Exp. Clin. Immunogenet. 1998;15:231–234. doi: 10.1159/000019076. [DOI] [PubMed] [Google Scholar]
- 14.Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, Savill JS, Henson PM, Botto M, Walport MJ. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J. Exp. Med. 2000;192:359–366. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carroll MC. The lupus paradox. Nat. Genet. 1998;19:3–4. doi: 10.1038/ng0598-3. [DOI] [PubMed] [Google Scholar]
- 16.Tsao BP. Genetic susceptibility to lupus nephritis. Lupus. 1998;7:585–590. doi: 10.1191/096120398678920695. [DOI] [PubMed] [Google Scholar]
- 17.Gaboriaud C, Thielens NM, Gregory LA, Rossi V, Fontecilla-Camps JC, Arlaud GJ. Structure and activation of the C1 complex of complement: unraveling the puzzle. Trends Immunol. 2004;25:368–373. doi: 10.1016/j.it.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Tacnet-Delorme P, Chevallier S, Arlaud GJ. β-Amyloid fibrils activate the C1 complex of complement under physiological conditions: evidence for a binding site for A β on the C1q globular regions. J. Immunol. 2001;167:6374–6381. doi: 10.4049/jimmunol.167.11.6374. [DOI] [PubMed] [Google Scholar]
- 19.Blanquet-Grossard F, Thielens NM, Vendrely C, Jamin M, Arlaud GJ. Complement protein C1q recognizes a conformationally modified form of the prion protein. Biochemistry. 2005;44:4349–4356. doi: 10.1021/bi047370a. [DOI] [PubMed] [Google Scholar]
- 20.Klein MA, Kaeser PS, Schwarz P, Weyd H, Xenarios I, Zinkernagel RM, Carroll MC, Verbeek JS, Botto M, Walport MJ, et al. Complement facilitates early prion pathogenesis. Nat. Med. 2001;7:488–492. doi: 10.1038/86567. [DOI] [PubMed] [Google Scholar]
- 21.Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J. Immunol. 1997;158:4525–4528. [PubMed] [Google Scholar]
- 22.Castellano G, Woltman AM, Nauta AJ, Roos A, Trouw LA, Seelen MA, Schena FP, Daha MR, van Kooten C. Maturation of dendritic cells abrogates C1q production in vivo and in vitro. Blood. 2004;103:3813–3820. doi: 10.1182/blood-2003-09-3046. [DOI] [PubMed] [Google Scholar]
- 23.Nauta AJ, Castellano G, Xu W, Woltman AM, Borrias MC, Daha MR, van Kooten C, Roos A. Opsonization with C1q and mannose-binding lectin targets apoptotic cells to dendritic cells. J. Immunol. 2004;173:3044–3050. doi: 10.4049/jimmunol.173.5.3044. [DOI] [PubMed] [Google Scholar]
- 24.Gaboriaud C, Juanhuix J, Gruez A, Lacroix M, Darnault C, Pignol D, Verger D, Fontecilla-Camps JC, Arlaud GJ. The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J. Biol. Chem. 2003;278:46974–46982. doi: 10.1074/jbc.M307764200. [DOI] [PubMed] [Google Scholar]
- 25.Kishore U, Gaboriaud C, Waters P, Shrive AK, Greenhough TJ, Reid KB, Sim RB, Arlaud GJ. C1q and tumor necrosis factor superfamily: modularity and versatility. Trends Immunol. 2004;25:551–561. doi: 10.1016/j.it.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Navratil JS, Watkins SC, Wisnieski JJ, Ahearn JM. The globular heads of C1q specifically recognize surface blebs of apoptotic vascular endothelial cells. J. Immunol. 2001;166:3231–3239. doi: 10.4049/jimmunol.166.5.3231. [DOI] [PubMed] [Google Scholar]
- 27.Elward K, Griffiths M, Mizuno M, Harris CL, Neal JW, Morgan BP, Gasque P. CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. J. Biol. Chem. 2005;280:36342–36354. doi: 10.1074/jbc.M506579200. [DOI] [PubMed] [Google Scholar]
- 28.Arlaud GJ, Sim RB, Duplaa AM, Colomb MG. Differential elution of Clq, Clr and Cls from human Cl bound to immune aggregates: use in the rapid purification of Cl subcomponents. Mol. Immunol. 1979;16:445–450. doi: 10.1016/0161-5890(79)90069-5. [DOI] [PubMed] [Google Scholar]
- 29.Villiers MB, Reboul A, Thielens NM, Colomb MG. Purification and characterization of C4-binding protein from human serum. FEBS Lett. 1981;132:49–54. doi: 10.1016/0014-5793(81)80425-5. [DOI] [PubMed] [Google Scholar]
- 30.Ishii M, Fujita S, Yamada M, Hosaka Y, Kurachi Y. Phosphatidylinositol 3,4,5-trisphosphate and Ca2+/calmodulin competitively bind to the regulators of G-protein-signalling (RGS) domain of RGS4 and reciprocally regulate its action. Biochem. J. 2005;385:65–73. doi: 10.1042/BJ20040404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993;26:795–800. [Google Scholar]
- 32.Navaza J. Implementation of molecular replacement in AMoRe. Acta Crystallogr. D Biol. Crystallogr. 2001;57:1367–1372. doi: 10.1107/s0907444901012422. [DOI] [PubMed] [Google Scholar]
- 33.Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta. Crystallogr. D Biol. Crystallogr. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macro-molecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Feng X, Tonnesen MG, Peerschke EI, Ghebrehiwet B. Cooperation of C1q receptors and integrins in C1q-mediated endothelial cell adhesion and spreading. J. Immunol. 2002;168:2441–2448. doi: 10.4049/jimmunol.168.5.2441. [DOI] [PubMed] [Google Scholar]
- 37.Hagenhofer M, Germaier H, Hohenadl C, Rohwer P, Kalden JR, Herrmann M. UV-B irradiated cell lines execute programmed cell death in various forms. Apoptosis. 1998;3:123–132. doi: 10.1023/a:1009601109509. [DOI] [PubMed] [Google Scholar]
- 38.Couldwell WT, Hinton DR, He S, Chen TC, Sebat I, Weiss MH, Law RE. Protein kinase C inhibitors induce apoptosis in human malignant glioma cell lines. FEBS Lett. 1994;345:43–46. doi: 10.1016/0014-5793(94)00415-3. [DOI] [PubMed] [Google Scholar]
- 39.Kalb E, Frey S, Tamm LK. Formation of supported planar bilayers by fusion of vesicles to supported phospholipid monolayers. Biochim. Biophys. Acta. 1992;1103:307–316. doi: 10.1016/0005-2736(92)90101-q. [DOI] [PubMed] [Google Scholar]
- 40.Saenko E, Sarafanov A, Ananyeva N, Behre E, Shima M, Schwinn H, Josic D. Comparison of the properties of phospholipid surfaces formed on HPA and L1 biosensor chips for the binding of the coagulation factor VIII. J. Chromatogr. A. 2001;921:49–56. doi: 10.1016/s0021-9673(01)00601-x. [DOI] [PubMed] [Google Scholar]
- 41.Swairjo MA, Concha NO, Kaetzel MA, Dedman JR, Seaton BA. Ca2+-bridging mechanism and phospholipid head group recognition in the membrane-binding protein annexin V. Nat. Struct. Biol. 1995;2:968–974. doi: 10.1038/nsb1195-968. [DOI] [PubMed] [Google Scholar]
- 42.Verdaguer N, Corbalan-Garcia S, Ochoa WF, Fita I, Gomez-Fernandez JC. Ca2+ bridges the C2 membrane-binding domain of protein kinase Cα directly to phosphatidylserine. EMBO J. 1999;18:6329–6338. doi: 10.1093/emboj/18.22.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huber R, Romisch J, Paques EP. The crystal and molecular structure of human annexin V, an anticoagulant protein that binds to calcium and membranes. EMBO J. 1990;9:3867–3874. doi: 10.1002/j.1460-2075.1990.tb07605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roumenina LT, Ruseva MM, Zlatarova A, Ghai R, Kolev M, Olova N, Gadjeva M, Agrawal A, Bottazzi B, Mantovani A, et al. Interaction of C1q with IgG1, C-reactive protein and pentraxin 3: mutational studies using recombinant globular head modules of human C1q A, B, and C chains. Biochemistry. 2006;45:4093–4104. doi: 10.1021/bi052646f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Callahan MK, Williamson P, Schlegel RA. Surface expression of phosphatidylserine on macrophages is required for phagocytosis of apoptotic thymocytes. Cell Death Differ. 2000;7:645–653. doi: 10.1038/sj.cdd.4400690. [DOI] [PubMed] [Google Scholar]
- 46.Fadok VA, Bratton DL, Frasch SC, Warner ML, Henson PM. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 1998;5:551–562. doi: 10.1038/sj.cdd.4400404. [DOI] [PubMed] [Google Scholar]
- 47.Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibro-blasts. J. Biol. Chem. 2001;276:1071–1077. doi: 10.1074/jbc.M003649200. [DOI] [PubMed] [Google Scholar]
- 48.Krahling S, Callahan MK, Williamson P, Schlegel RA. Exposure of phosphatidylserine is a general feature in the phagocytosis of apoptotic lymphocytes by macrophages. Cell Death Differ. 1999;6:183–189. doi: 10.1038/sj.cdd.4400473. [DOI] [PubMed] [Google Scholar]
- 49.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 50.Stach CM, Turnay X, Voll RE, Kern PM, Kolowos W, Beyer TD, Kalden JR, Herrmann M. Treatment with annexin V increases immunogenicity of apoptotic human T-cells in BALB/c mice. Cell Death Differ. 2000;7:911–915. doi: 10.1038/sj.cdd.4400715. [DOI] [PubMed] [Google Scholar]
- 51.Marguet D, Luciani MF, Moynault A, Williamson P, Chimini G. Engulfment of apoptotic cells involves the redistribution of membrane phosphatidylserine on phagocyte and prey. Nat. Cell Biol. 1999;1:454–456. doi: 10.1038/15690. [DOI] [PubMed] [Google Scholar]
- 52.Cui P, Qin B, Liu N, Pan G, Pei D. Nuclear localization of the phosphatidylserine receptor protein via multiple nuclear localization signals. Exp. Cell Res. 2004;293:154–163. doi: 10.1016/j.yexcr.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 53.Mitchell JE, Cvetanovic M, Tibrewal N, Patel V, Colamonici OR, Li MO, Flavell RA, Levine JS, Birge RB, Ucker DS. The presumptive phosphatidylserine receptor is dispensable for innate anti-inflammatory recognition and clearance of apoptotic cells. J. Biol. Chem. 2006;281:5718–5725. doi: 10.1074/jbc.M509775200. [DOI] [PubMed] [Google Scholar]
- 54.Botto M, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiology. 2002;205:395–406. doi: 10.1078/0171-2985-00141. [DOI] [PubMed] [Google Scholar]
- 55.Stuart GR, Lynch NJ, Lu J, Geick A, Moffatt BE, Sim RB, Schwaeble WJ. Localisation of the C1q binding site within C1q receptor/calreticulin. FEBS Lett. 1996;397:245–249. doi: 10.1016/s0014-5793(96)01156-8. [DOI] [PubMed] [Google Scholar]
- 56.Stuart GR, Lynch NJ, Day AJ, Schwaeble WJ, Sim RB. The C1q and collectin binding site within C1q receptor (cell surface calreticulin) Immunopharmacology. 1997;38:73–80. doi: 10.1016/s0162-3109(97)00076-3. [DOI] [PubMed] [Google Scholar]
- 57.Kaul M, Loos M. Expression of membrane C1q in human monocyte-derived macrophages is developmentally regulated and enhanced by interferon-γ. FEBS Lett. 2001;500:91–98. doi: 10.1016/s0014-5793(01)02592-3. [DOI] [PubMed] [Google Scholar]
- 58.Petry F, Botto M, Holtappels R, Walport MJ, Loos M. Reconstitution of the complement function in C1q-deficient (C1qa-/-) mice with wild-type bone marrow cells. J. Immunol. 2001;167:4033–4037. doi: 10.4049/jimmunol.167.7.4033. [DOI] [PubMed] [Google Scholar]
- 59.Vegh Z, Goyarts EC, Rozengarten K, Mazumder A, Ghebrehiwet B. Maturation-dependent expression of C1q binding proteins on the cell surface of human monocyte-derived dendritic cells. Int. Immunopharmacol. 2003;3:39–51. doi: 10.1016/s1567-5769(02)00211-4. [DOI] [PubMed] [Google Scholar]
- 60.Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, Henson PM. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 2001;194:781–795. doi: 10.1084/jem.194.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ogasawara Y, Kuroki Y, Akino T. Pulmonary surfactant protein D specifically binds to phosphatidylinositol. J. Biol. Chem. 1992;267:21244–21249. [PubMed] [Google Scholar]
- 62.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]