

Abstract
Ionized nucleobases are required for folding, conformational switching or catalysis in a number of functional RNAs. A common strategy to study these sites employs nucleoside analogs with perturbed pKa, but the interpretation of these studies is often complicated by the chemical modification introduced, in particular modifications that add, remove, or translocate hydrogen bonding groups in addition to perturbing pKa values. In the present study we present a series of fluorine substituted adenosine analogs that produce large changes in N1 pKa values with minimal structural perturbation. These analogs include fluorine for hydrogen substitutions in the adenine ring of adenosine and 7-deaza-adenosine with resulting N1 pKa values spanning more than four pKa units. To demonstrate the utility of these analogs we have conducted a nucleotide analog interference mapping (NAIM) study on a self-ligating construct of the Varkud Satellite (VS) ribozyme. We find that each of the analogs is readily incorporated by T7 RNA polymerase and produce fully active transcripts when substituted at the majority of sites. Strong interferences are observed for three sites known to be critical for VS ribozyme function, most notably A756. Substitutions at A756 lead to slight enhancements in activity for elevated pKa analogs, and dramatic interferences in activity for reduced pKa analogs, supporting the proposed catalytic role for this base. The structural similarity of these analogs, combined with their even incorporation and selective interference, provides an improved method for identifying sites of adenosine protonation in a variety of systems.
Keywords: adenosine analog, nucleoside, fluorine, pKa, Varkud Satellite, ribozyme, interference
Introduction
Ribonucleic acids are increasingly found to be involved in a wide variety of cellular functions outside their traditionally accepted roles in protein translation, where they act as information carriers (mRNA), decoding elements (tRNA) or ribosomal constituents (rRNA). These functions include RNA splicing1, tRNA maturation2, plasmid replication3, and gene regulation, either through metabolite binding4 or via a number of RNA mediated gene silencing pathways5. Many of these functions require RNA sequences capable of adopting well-defined folds, and in some cases these folds produce active sites capable of catalysis, most commonly the breakdown or formation of phosphodiester bonds, either at a defined site within the structured RNA itself or in some external substrate RNA sequence. These RNA based enzymes, or ribozymes, were originally thought to exist exclusively as metalloenzymes, employing positioned divalent cations in their active sites to catalyze phosphodiester transfer reactions6. However, a number of nucleolytic ribozymes, which produce free 5′-hydroxyl and 2′,3′-cyclic phosphates as cleavage products, have been shown to be functional in the absence of divalent metals7. These results in combination with pH profiles of reaction rate and an increasing number of crystal structures showing appropriate geometries suggest some ribozymes may be capable general acid/base catalysis, with the nucleobases themselves acting as the site of proton transfer8-10. In these systems the active site nucleobases are predicted to perform a role analogous to that of histidines in protein ribonucleases11.
General acid/base mechanisms for ribozyme catalyzed reactions require substantial pKa perturbations. The RNA functional groups with unperturbed pKa values closest to neutrality are the nitrogens of nucleobase heterocycles, namely N3 of cytidine (pKa = 4.2), N1 of adenosine (pKa = 3.5), N1 of guanosine (pKa = 9.2) and N3 of uridine (pKa = 9.2)10. Because ribozymes are folded polyanions the stabilization of the protonated forms of bases, leading to elevation in pKa values, is expected to be favored. This stabilization makes cytidine and adenosine attractive candidates for sites of proton transfer. Elevated pKa values for these bases have been observed with spectroscopic techniques in a number of systems12-17, and calculations suggest these shifts arise from a combination of non-canonical base pairing interactions and focused electrostatic potentials arising from the phosphate backbone18.
While spectroscopic techniques provide an important tool for identifying sites of pKa perturbation they provide no information on the functional consequence of protonation at these sites. This is of particular concern when truncated or stabilized versions of the system are required, as is often the case in NMR studies, particularly those that investigate individual domains of the full ribozyme15,17. An alternative approach is to incorporate nucleotide analogs with perturbed pKa and to assay the effect of analog substitution on catalysis. One challenge of this approach is the design of analogs that produce large changes in pKa while maintaining other structural features, most notably hydrogen bonding patterns. Analogs must then be incorporated into the functional RNA in a manner that allows for a site by site evaluation of the effect on function. The most straightforward method of incorporation is to introduce analogs site specifically via oligonucleotide synthesis and to measure the effect on observed rates directly. The limitations of this strategy include the stability of the desired analog to RNA synthesis conditions and the relatively short RNA sequences accessible by direct synthesis. Although longer RNA sequences can be reassembled through RNA ligations or alternative methods the need to repeat this process for each site of interest often makes a complete analysis impractical.
Nucleotide analog interference mapping (NAIM) provides a semi-quantitative alternative to site specific incorporation. The NAIM method utilizes 5′-O-(1-thio)nucleotide triphosphates with an additional chemical modification in the ribose or nucleobase. Unlike synthetic methods, the analogs are incorporated by in vitro transcription, allowing any functional RNA transcript of virtually any length to be investigated. NAIM studies consist of four basic steps (for reviews of the NAIM method see19,20). i) Transcripts of the functional RNA are prepared where one of the four bases (A, U, G, or C) is substituted at low frequency with a nucleotide analog. In this step the ratio of nucleotide triphosphate and analog 5′-O-(1-thio)nucleotide triphosphate is adjusted such that any individual transcript contains approximately one analog substitution at a random site. ii) The active members of the transcribed pool are separated from the inactive members, most commonly by a change of electrophoretic mobility or a transfer of a radioisotope. iii) The phosphorothioate linkage introduced at sites of analog substitution is selectively cleaved by treatment with iodine. iv) Sites of analog interference are identified by separating the iodine cleaved RNAs by gel electrophoresis and quantitating relative band intensities by autoradiography. NAIM studies have successfully identified RNA functional groups required for catalysis, folding or ligand binding in a number of systems21-25.
Identification of nucleobase protonation sites by NAIM requires a series of analogs with perturbed pKa. Previous studies investigating adenosine protonation have used analogs in one of two categories, those that make carbon/nitrogen substitutions in the adenine ring, including 3-deaza-adenosine (c3A), 7-deaza-adenosine (7dA), Formycin (FormA), and 8-aza-adenosine (n8A), and those that add, remove, or translocate the exocyclic amine, including 2,6-diaminopurine (DAP), purine (Pur), and 2-aminopurine (2AP)21,23. Several of these analogs are incorporated with relatively poor efficiency by T7 RNA polymerase (c3A, n8A, Pur, 2AP)20, others alter standard base pairing interactions (Pur, 2AP) or exist in multiple tautomeric forms (FormA)26. To improve the identification of protonated adenosines in functional RNAs we set out to design a new series of structurally similar adenosine analogs that retain hydrogen bonding patterns on the Watson-Crick face of the base, are readily incorporated by T7 polymerase, and which produced large shifts in N1 pKa values.
The analogs presented in this study make fluorine for hydrogen substitutions in adenosine or 7-deaza-adenosine (Figure 1). This series was chosen because fluorine for hydrogen substitutions produce large pKa shifts in nitrogen heterocycles with relatively modest structural perturbation27. Fluorine substitutions were introduced into adenosine at the 2 position (2F-A) and 7-deaza-adenosine at the 2 or 7 position (2F-7dA and 7F-7dA, Figure 1). These structurally similar analogs have N1 pKa values spanning more than four pKa units and are readily incorporated by T7 RNA polymerase (see below). Importantly, all five members of the series retain the exocyclic amine for standard base pairing interactions.
Figure 1.

pKa perturbed adenosine analogs.
To demonstrate the utility of this new series of adenosine analogs we conducted a NAIM study on a ligating form of the Varkud Satellite (VS) ribozyme21,28 (Figure 2). We chose to study a ligating form of the VS ribozyme because less information is currently available for the VS catalyzed reaction in the direction of ligation than in the direction of cleavage. The ligation of a radiolabeled substrate also provides a straightforward functional selection, which simplifies the NAIM experiment. The role of A756 is of particular interest since this nucleotide has been proposed as a site of proton transfer in the cleavage reaction29-31. A previous NAIM study on this construct using the analogs Pur, n8A, FormA and 7dA observed an interference pattern consistent with A756 protonation, but these analogs also produced a number of interferences at other sites that were less easily interpreted21. We set out to confirm the importance of A756 protonation with a distinct class of pKa perturbed analogs, and to see if the structural similarity of these analogs would produce a more selective and interpretable interference pattern.
Figure 2.

Predicted secondary structure of the VSE ribozyme (VS RNA NT 599-791 containing point mutants A782U and U785C). Helices are numbered I-VII, with the site of ligation marked with the arrow, and the tertiary “kissing” interaction shown with the dashed line. The unligated VSE RNA (NT 621-791) was transcribed with analogs 7a - 7e randomly incorporated, and the substrate RNA (NT 599-620) was prepared with a 2′,3′-cyclic phosphate terminus. The NAIM analysis assayed the effect of analog substitution from A645-A767. Sites of interference with at least one analog are denoted with filled triangles.
Results
Synthesis
7-fluoro-7-deaza-adenosine was synthesized essentially as described by Migawa32 (Scheme 1). Selective electrophilic fluorination of 6-chloro-7-deazapurine with the reagent Selectfluor afforded 1, which was coupled to 1-O-Acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose to produce the protected nucleoside 2. Simultaneous deprotection and aromatic substitution in methanolic ammonia provided 3 in addition to the methoxide substituted product in a ratio of 2:1. A similar coupling strategy with 6-chloro-7-deazaguanine did not produce the desired protected nucleoside, starting either from the unprotected or acetyl protected base. We turned instead to a nucleobase-anion glycosylation strategy33,34 to obtain 4, which was converted to 5 with aqueous ammonia (Scheme 2). Diazotation of 5 in the presence of HF produced 2-fluro-7-deaza-adenosine, 6, in low yield (the efficiency of this transformation is variable and dependent on heterocycle substitution patterns35-37). 2-fluoro-adenosine is available from commercial sources. 5′-O-(1-thio)nucleotide triphosphates were obtained from the corresponding nucleosides by the method of Frey38 (Scheme 3) or from commercial sources (analogs 7a,7b).
Scheme 1.

a) Selectfluor, CH3CN, acetic acid, 70°C 15 hr. b) BSA, CH3CN, 1-O-Acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose, TMSOTf, 80°C1 hr. c) NH3/MeOH, 73°C 18 hr.
Scheme 2.

a) 5′-O-tertbutyldimethylsilyl-2,3-O-isopropylidene-D-ribose, THF, CCl4,P(NMe2)3,−72°C 2 hr. b) 6-chloro-7-deazaguanine, CH3CN, KOH, TDA-1, product of a) r.t. 20 hr. c) TFA, r.t. 50 min. d) NH3(aq), 75°C 22 hr. e) HF/pyr, KNO2, 0°C 15 min.
Scheme 3.

a) PSCl3, TEP, r.t. 1 hr. b) TBAP, TEP, r.t. 1 hr.
pKa Values of Analogs
To determine the N1 pKa values of the fluoro-adenosine analogs we recorded the 19F NMR signal of analog nucleosides as a function of pH in aqueous solutions. The 19F NMR titration of 7F-7dA (Figure 3A) produces a titration curve that fits well to a single ionization model (Figure 3B). This fit yields a pKa value of 4.5, which lies between that of A (pKa = 3.5) and 7dA (pKa = 5.2) as expected. 19F NMR titrations were also performed for 2F-A and 2F-7dA. Neither of these analogs showed substantial frequency shifts within easily buffered pH ranges. However, titrations conducted with aqueous solutions of sulfuric acid provided completed titration curves for both 2F-A and 2F-7dA by 20 wt% H2SO4, with large 19F frequency shifts (+4.7 ppm for 2F-A and −10.2 ppm for 2F-7dA). A combination of 19F, 15N and 13C NMR data confirmed that the site of protonation for 7F-7dA and 2F-7dA is N1, while the most basic site of 2F-A is N7 (see Supporting Information). Because 2F-A and 2F-7dA are protonated at different sites we report an upper limit for N1 pKa values for these analogs of 1.0 (Figure 1).
Figure 3.

19F NMR titration of 7F-7dA. (A) 19F NMR spectra referenced to internal fluorobenzene in 10% D2O. (B) 19F NMR frequency as a function of pH. A fit to a single ionization model (dashed line) provides a pKa value of 4.5. pH was maintained with 100 mM phosphate (◆), citrate (●) or Tris (■) buffers.
Incorporation of Analogs by T7 RNA Polymerase
To test the ability of T7 RNA polymerase to incorporate analogs 7c - 7e we performed transcriptions with variable concentrations of these analogs and compared the incorporation level to transcriptions containing 0.05 mM 5′-O-(1-thio)adenosine triphosphates, 7a. Transcriptions using EarI cut pHHVS as template and variable concentrations of individual analogs were purified, 5′-end labeled, and iodine cleaved (see Experimental Section). The products of iodine cleavage were separated by gel electrophoresis and incorporation levels were determined by quantitating band intensities (data not shown). These results provided the analog concentrations required to match the incorporation levels seen with 7a (Table 1). The analog concentrations in Table 1 were used in all subsequent transcriptions for production of VSE RNA. It has been previously shown that 7dA is incorporated with the same efficiency as A19, and we find 7F-7dA is similarly efficient, while fluorine substitutions at the 2 position of adenosine (2F-A) or ***7-deaza-adenosine (2F-7dA) require slightly higher analog concentrations. In addition to high incorporation efficiency, NAIM experiments require relatively even incorporation at all coded sites, and minimal misincorporation. The intensity of iodine cleavage bands from unselected pools of unligated VSE RNA shows even incorporation with no evidence of misincorporation (Figures 4B,D), allowing the entire series to be used for NAIM studies.
Table 1.
Analog concentrations required for 5% incorporation level.
| Analog | Analog Conc. (mM)a |
ATP Conc. (mM) |
|---|---|---|
| A | .05 | 1.0 |
| 7dA | .05 | 1.0 |
| 7F-7dA | .05 | 1.0 |
| 2F-7dA | .20 | 1.0 |
| 2F-A | .20 | 1.0 |
Concentrations are of Sp diasteriomers of 5′-O-(1-thio)nucleotide triphosphates. A and 7dA were added to reactions as pure Sp diasteriomers, while remaining analogs were added as 50:50 mixtures of diasteriomers. See Experimental Section for transcription details.
Figure 4.

(A,C) Sequencing gel of NAIM ligation selections run for sufficient times to resolve short (A) or long (C) iodine cleavage products. Ligation products were treated with iodine (lanes 6-10) or without iodine (lanes 1-5) prior to being loaded onto the gel. The analog incorporated for each selection is shown above lanes and nucleotide positions are shown to the right. Sites of modified activity are marked as follows; strong interference (κ > 4) filled triangle, modest interference (4 > κ > 2) open triangle, enhancement (1/κ > 2) filled circle. (B,D) Sequencing gel of 5′-end labeled unligated VSE RNAs labeled as above, run to control for level of analog incorporation.
VSE Ribozyme Interference Mapping
The functional consequence of analog substitution at individual sites of the VSE ribozyme was investigated by performing a ligation selection (see Experimental Section). Five unligated VSE RNA transcripts (NT 621-791, Figure 2), each containing one of the analogs 7a - 7e randomly incorporated at ∼ 5% incorporation level were incubated with a 5′-end labeled substrate (NT 599-620, Figure 2). This substrate RNA was constructed with a 2′,3′-cyclic phosphate at its 3′ terminus, as required for VSE ligation. Ligation reactions were allowed to proceed to ∼ 10% completion, then run on PAGE sequencing gels with or without iodine cleavage. Because only functional VSE transcripts are capable of ligating the substrate, the band intensities from the iodine cleaved selection report on the ability of an analog to promote the ligation reaction when incorporated at a particular site. An increased band intensity relative to an unselected control corresponds to an enhancement in activity, while a decrease in band intensity corresponds to interference at that site.
NAIM sequencing gels typically resolved 36 of the 48 VSE adenosine positions, including all positions in the core of the ribozyme (Figures 4A,C). Only three of these sites, A698, A712 and A756, show interference with any analog. The strongest of these interferences are easily identified from the sequencing gels directly by comparing band intensities from the VSE ligation selection experiment (Figures 4A,C) to the band intensities from 5′-end labeled unselected controls (Figures 4B,D). Interferences were further quantitated by drawing line profiles through the lanes, measuring the area of each band, and calculating interference (κ) or enhancement (1/κ) values for each position (see Experimental Section). A plot of average interference and enhancement values for each analog at each site shows a selective and reproducible interference pattern for A698, A712 and A756, with very little effect at other sites (Figures 5). None of the sites of interference is paired in the predicted secondary structure of the VS ribozyme and each has been shown to be critical to function by mutagenesis studies39-41.
Figure 5.
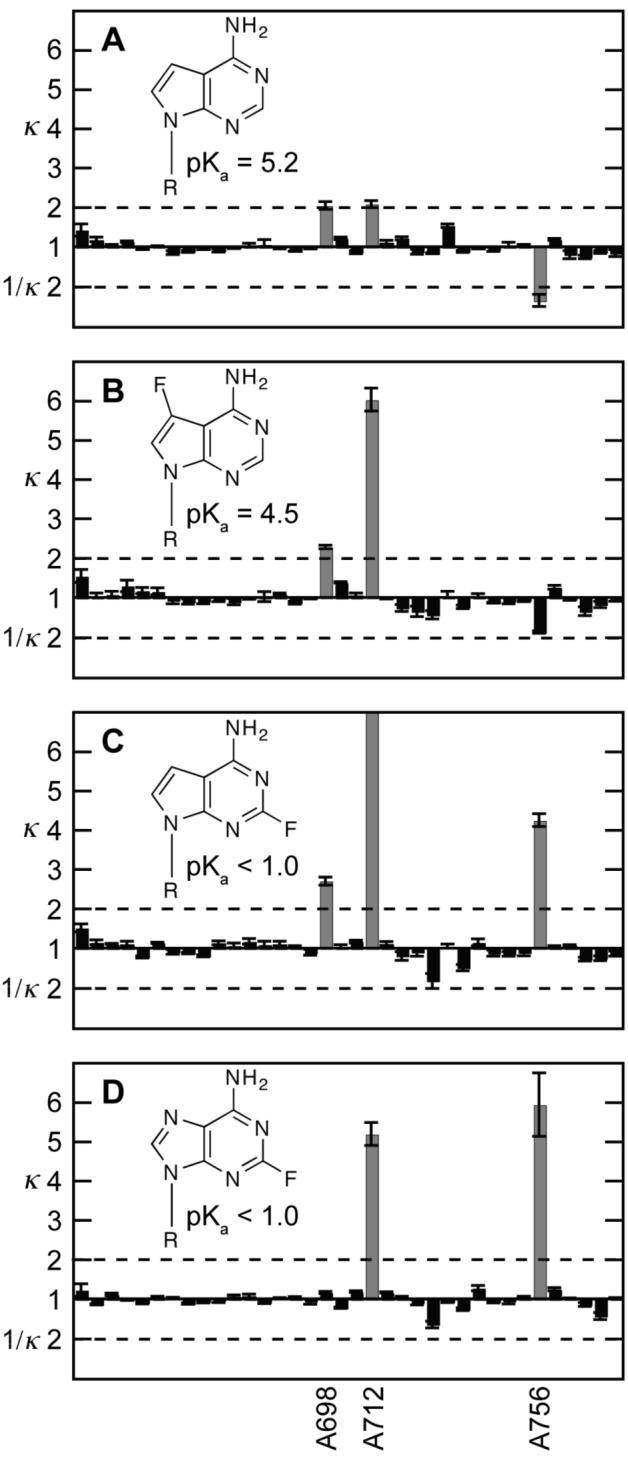
Calculated interference (κ) or enhancement (1/κ) values for adenosine analogs 7dA (A), 7F-7dA (B), 2F-7dA (C) and 2F-A (D). Values are provided for adenosine sites A645 (left) to A767 (right) and represent average of four measurements with standard deviation error bars. Interferences or enhancements greater than 2 (dashed line) are considered significant and shaded grey.
Discussion
Attributes of Fluoro-Adenosine Analogs
Analogs 7a - 7e produce the largest span of N1 pKa values of any adenosine series employed in NAIM studies to date, with the N1 pKa values of 2F-A and 2F-7dA at least a full unit below that of any previously reported analog21,23. Despite this dramatic range of pKa values the minimal structural perturbation introduced by fluorine substitution allows this series to be introduced by transcription more readily and evenly than previous low pKa analogs, while producing a more selective interference pattern. This finding is consistent with the observation that the normal triphosphate of 2F-A can be readily incorporated by in vitro transcription, and that RNA duplexes containing this modification are only modestly destabilized42. A NAIM approach is particularly attractive for 2F-A since fluorine is readily displaced from this analog under standard oligonucleotide synthesis deprotection conditions, making its introduction by chemical synthesis challenging43. In contrast 7F-7dA is stable under nucleophilic substitution conditions (Scheme 1), and could be incorporated site specifically via chemical synthesis. The large 19F frequency shift observed between protonated and neutral forms (Figure 3) suggests this analog may be useful as a spectroscopic reporter of adenosine protonation in folded RNA, as has been demonstrated for 5-fluoro-uracil44,45.
NAIM Results and Proposed VS Ribozyme Mechanism
Since the original report of VS ribozyme cleavage activity46 a large body of kinetic and low resolution structural data has been collected. Extensive mutagenesis throughout the core of the ribozyme41,47, as well as in the substrate of a trans cleaving construct30 have identified two nucleotides critical for cleavage activity, G638 and A756. Although A756 is far from the cleavage site in primary sequence or secondary structure, UV cross-linking experiments place it near the scissile bond in the folded ribozyme48, supporting its potential catalytic role. A number of base modifications have been introduced at A756 by reassembling a three piece construct, with significant reductions in activity found for modifications that alter the Watson-Crick face of the base49. Introduction of an imidazole nucleotide at A756 also supports cleavage, though at a greatly reduced rate, suggesting the ability to conduct proton transfer may be the key feature of this catalytic residue50. These data, combined with the pH dependent rates observed for the fastest known constructs30,31, and the lack of a divalent metal requirement7 have led to a proposed general acid-base mechanism for VS ribozyme catalysis, with G638 and A756 as the sites of proton transfer30.
We conducted a NAIM study to test the proposed general acid-base mechanism of the VS ribozyme in the direction of ligation, with particular interest in the interference pattern at A756. If A756 is a site of proton transfer in the reaction then reduced pKa analogs would be expected to reduce activity when substituted at A756, either because of their reduced N1 nitrogen basicity (A756 as general base) or because of their reduced probability of being protonated at neutral pH (A756 as general acid)51. Because some chemical modification is require to perturb pKa values, the effect of modifications unrelated to pKa shifts is a concern. For this reason the close structural similarity between analogs 7a - 7e aids greatly in the interpretation of their interference patterns.
The effect of analog substitution at A756 (Figures 5A-D) ranges from significant enhancement (7dA) to strong interference (2F-7dA and 2F-A). The enhancement observed for 7dA, a pKa elevated base, is reduced when the 7-fluoro substitution is introduced, presumably because 7F-7dA moves the N1 pKa back toward that of A. The enhancement observed for 7dA is unlikely to originate from the loss of N7 because 2F-7dA shows strong interference. For the pKa reduced analogs the interference observed for 2F-A is greater than that of 2F-7dA, indicative of a pKa effect since 2F-A is expected to have the lower N1 pKa (see Supporting Information). A756 is the only site that shows an interference pattern directly related to N1 pKa, and this pattern of interference suggests protonation at this site is required for VSE function.
In addition to the pKa dependent interference observed at A756, analogs 7b - 7e also produced interferences at two important structural positions, A698 and A712. The interference pattern observed at both sites support proposed uridine turns, defined by the consensus sequence U-N-R (where N is any nucleotide and R is a purine). The structure of canonical uridine turns includes a key hydrogen bond between the 2′-hydroxyl of the U and the N7 of the purine52. Consistent with the proposed U696-G697-A698 uridine turn interference at A698 results from the loss of N7 (7dA, 7F-7dA, 2F-7dA) and is independent of N1 pKa, as demonstrated by the lack of interference for 2F-A (Figure 5). This pattern supports a long-range pseudoknot between helices I and V built in part from a uridine turn involving A69839. These interferences also confirm the functional relevance of the uridine turn observed in the NMR structure of VS nucleotides 693-70253.
The interference pattern at A712 is less clearly interpretable, in part because significant interference has been observed with nearly every analog tested to date21. A uridine turn has also been proposed for residues U710-G711-A712 based on mutagenesis and chemical modification protection40, and the interferences observed for analogs lacking N7 is consistent with that prediction. However, in contrast to A698, which can be replaced with G in the context of the U631C/A698G double mutant, the A712G mutant is inactive, suggesting an additional interaction with this base or very tight packing at this position. Although the origin of base selectivity at A712 in the native sequence is unclear it is known that this site is not directly involved in catalysis since residues 710-712 can be replaced with hairpins of various length and sequence with modest decreases in rate40.
The current model for VS ribozyme catalysis employs A756 as a general acid or general base, with some other functional group, possibly G638, as the complementary site of proton transfer30 (Figure 6). The interference pattern observed with the fluoro-adenosine analogs of this study strongly supports a functional role of A756 protonation, as this is the only site in the ribozyme that shows a pKa dependent interference pattern. However, the interference pattern itself cannot distinguish between a potential general acid or general base role for A756, nor does it preclude a structural role for the protonated form of A75651. One approach to resolve this ambiguity is structural studies. Although there are no atomic structures yet available for the VS ribozyme it has been suggested30 that the VS may position functional groups in a manner similar to the Hairpin ribozyme, which places a G at the 2′-oxygen and an A near the 5′-hydroxyl54. In this scenario A756 would act as the general base in the ligation reaction (Figure 6B), and interferences caused by low pKa analogs such as 2F-A would arise from the analog's reduced N1 basicity. This dependence on active site adenosine N1 pKa would be analogous to that observed in the Hairpin ribozyme, where rescue of the A38 abasic substitution shows a strong dependence on exogenous nucleobase pKa55.
Figure 6.

Two potential catalytic roles for A756 protonation. In a general acid/base mechanism A756 could act either as the general acid (A) or general base (B) in the ligation reaction, with another ionizable group, HA, as the complementary site of proton transfer.
Conclusions
The fluoro-adenosine analogs presented in this study provide an improved series to determine sites of adenosine protonation by NAIM analysis. These analogs have N1 pKa values spanning more than four pKa units while maintaining a normal Watson-Crick hydrogen bonding face and introducing minimal steric perturbation. Each of the analogs is evenly incorporated by T7 RNA polymerase with good fidelity at relatively low analog concentrations. A NAIM analysis carried out on a ligating form of the VS ribozyme led to three sites of interference, and a single site, A756, whose interference pattern is consistent with functional protonation. The small number of interferences observed demonstrates the ability of this new series to produce extremely selective interference patterns. Because the entire series is related by minor chemical modifications, interferences not originating from N1 pKa perturbations are readily controlled for. In addition to its use in activity studies we anticipate 7F-7dA may find use as a spectroscopic probe of pKa values because of its moderate pKa, large 19F NMR frequency shift upon protonation, and ease of incorporation.
Experimental Section
Synthesis
Reagents: 2-fluoro-adenosine (SynQuest), 6-chloro-7-deazapurine, 6-chloro-7-deazaguanine, and 5′-O-tert-butyldimethylsilyl-2,3-O-isopropylidene-D-ribose (Toronto Research Chemicals), 1-O-Acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose (Acros Organics), NMR solvents (Cambridge Isotopes), all remaining reagents and solvents (Sigma-Aldrich). Instrumentation: 1H NMR (Bruker, 400 or 500 MHz), 19F NMR and 31P NMR (Bruker, 400 MHz), MS (Waters, Micromass ZQ), high pressure bomb (Parr Instruments, bomb number 4745), FPLC (Amersham, AKTAprime), anion exchange column (GE Healthcare, HiTrap Q HP).
4-chloro-5-fluoro-7H-pyrrolo[2,3-d]pyrimidine (1)
6-chloro-7-deazapurine (258 mg, 1.7 mmol) was dried by co-evaporation of pyridine (5 mL × 3) and placed under vacuum overnight. Selectfluor (890 mg, 2.5 mmol) was added followed by anhydrous acetonitrile (12.5 mL) and acetic acid (2.5 mL). The solution was refluxed at 70°C under Ar for 15 hours and cooled to r.t. Solvent was removed under reduced pressure and coevaporated with toluene (5 mL × 2) to leave a brown solid. A suspension of this solid in 1:1 DCM:EtOAc (10 mL) was filtered through 1 cm silica, which was washed with 1:1 DCM:EtOAc (150 mL). Evaporation of washes left a yellow solid that was purified on a silica column with DCM:EtOAc (4:1) to give 125 mg (43 % yield) of 1 as white solid. Rf = 0.32 (1:1 DCM:EtOAc). 1H NMR (DMSO-d6) δ 12.47 (br s, 1H), 8.61 (s, 1H), 7.71 (d, 1H, J = 2.2 Hz); 19F NMR (DMSO-d6)δ −170.75 (d, J = 2.2 Hz). MS expected (M + H)+ = 172.0, observed 171.9 (M + H)+.
4-Chloro-5-fluoro-7-(2,3,5,-tri-O-benzoyl-β-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine (2)
In separate r.b. flasks 4-chloro-5-fluoro-7H-pyrrolo[2,3-d]pyrimidine (1) (125 mg, 0.73 mmol) and 1-O-Acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose (400 mg, 0.79 mmol) were coevaporated with acetonitrile and dried under vacuum overnight. To a suspension of 4-chloro-5-fluoro-7H-pyrrolo[2,3-d]pyrimidine in anhydrous acetonitrile (3.0 mL) was added N,O-Bis(trimethylsilyl)acetamide (0.21 mL, 0.82 mmol), to form a clear slightly purple solution. 1-O-Acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose was dissolved in anhydrous acetonitrile (1.6 mL) with mild heating and added to the reaction, followed by trimethylsily trifluoromethanesulfonate (0.14 mL, 0.79 mmol) dropwise to produce a clear yellow solution. The reaction was stirred at r.t. for 15 min, then refluxed at 80°C for 1 hour. The orange/brown reaction was cooled to r.t., diluted with EtOAc (25 mL) and washed with sat. NaHCO3 (25mL) andsat. NaCl (25 mL), then dried over MgSO4 and concentrated to give a yellow oil. Purification by silica column with 10-25% EtOAc in hexanes gave 2 (150 mg, 33% yield) as a yellow/white foam. Rf = 0.33 (30% EtOAc in hexanes). 1H NMR (CDCl3) δ 8.57 (s, 1H), 8.10 (d, 2H, J = 7.1 Hz), 7.98 (d, 2H, J = 7.1 Hz), 7.89(d, 2H, J = 7.1 Hz), 7.60-7.32 (m, 9H), 7.15 (d, 1H, J = 2.5 Hz), 6.68 (d, 1H, J = 2.8 Hz), 6.11 (m, 1H), 6.05 (m, 1H), 4.85 (m, 1H), 4.76 (m, 1H), 4.65 (m, 1H). 19F NMR (CDCl3) δ −165.80 (s). MS expected (M + H)+ = 616.1, observed 616.3 (M + H)+, 638.1 (M + Na)+.
7-fluoro-7-deaza-adenosine (3)
4-Chloro-5-fluoro-7-(2,3,5,-tri-O-benzoyl-β-D-ribofuranosyl) pyrrolo[2,3-d]pyrimidine (2) (150 mg, 0.24 mmol) was dried under vacuum overnight in a r.b., then cooled to 0°C under Ar. A 7N solution of ammonia in MeOH (10.0 mL) was added and the solution was stirred at 0°C until a clear solution formed. This solution was transferred to the teflon insert of a stainless steel bomb and heated to 73°C for 18 hours. After cooling the solvent was removed to leave a yellow solid that was coevaporated with MeOH (20 mL × 3). Purification by silica column with 5-15% MeOH in DCM gave 3 (27 mg, 38% yield) as a white solid. Rf = 0.23 (15% MeOH in DCM). 1H NMR (DMSO-d6) δ 8.05 (s, 1H), 7.34 (d, 1H, J = 1.9 Hz), 7.00 (br s, 2H), 6.05 (m, 1H) 5.35 (d, 1H, J = 5.8 Hz), 5.14 (m, 2H), 4.27 (m, 1H), 4.04 (m, 1H), 3.85 (m, 1H), 3.61-3.47 (m, 2H). 19F NMR (DMSO-d6) δ −167.73 (s). MS expected (M + H)+ = 285.1, observed 285.1 (M + H)+, 307.1 (M + Na)+.
4-chloro-7-(β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidin-2-amine (4)
Compound 4 was synthesized in three steps by the method of Seela33,34,56. a) Synthesis of 5′-O-tert-butyldimethylsilyl-2,3-O-isopropylidene-D-ribose chloride: 5′-O-tert-butyldimethylsilyl-2,3-O-isopropylidene-D-ribose (1.36 g, 4.47 mmol) was dried under vacuum overnight and placed under Ar. Anhydrous THF (14.0 mL) and CCl4 (0.52 mL) were added and the solution was cooled to −72°C. To the cooled solution tris(dimethylamino)phosphane (0.83 mL, 4.57 mmol) was added dropwise over 15 min and stirred for 2 hours with brief periods of slight warming to prevent gel formation. The reaction was concentrated to 4.5 mL and used immediately without purification in following step. b) Nucleobase-anion glycosylation: In separate flasks 6-chloro-7-deazaguanine (733 mg, 4.35 mmol) and powdered potassium hydroxide (760 mg, 13.5 mmol) were dried under vacuum overnight. Anhydrous acetonitrile (36.0 mL) was added to the potassium hydroxide, followed by tris[2-(2-methoxyethoxy)ethyl]amine (0.028 mL) and stirred for 10 min at r.t. 6-chloro-7-deazaguanine was added and stirred 10 min at r.t. to produce a clear yellow solution. To this solution the crude THF solution of 5′-O-tert-butyldimethylsilyl-2,3-O-isopropylidene-D-ribose chloride from the previous step was added, and stirred at r.t for 20 hours, changing in color from light pink to dark brown. Insoluble material was filtered off and the concentrated filtrate was purified on a silica column with 20% EtOAc in hexanes to provide the protected nucleoside (Rf = 0.24, 20% EtOAc in hexanes). c) Nucleoside deprotection: To protected nucleoside of previous step was added 90% aqueous trifluoroacetic acid (3.7 mL) and stirred at r.t for 50 min. The reaction was concentrated under reduced pressure and coevaporated with MeOH (10 mL × 5). Purification by silica column with 10% MeOH in DCM gave 4 (350 mg, 27% overall yield) as a white solid. Rf = 0.23 (10% MeOH in DCM). 1H NMR (DMSO-d6) δ 7.37 (d, 1H, J = 3.8 Hz), 6.69 (br s, 1H), 6.36 (d, 1H, J = 3.8 Hz), 5.97 (d, 1H, J = 6.3 Hz), 5.28 (d, 1H, J = 6.2 Hz), 5.08 (d, 1H, J = 4.6 Hz), 4.97 (t, 1H, J = 5.4 Hz), 4.29 (m, 1H), 4.04 (m, 1H), 3.83 (m, 1H), 3.57-3.50 (m, 2H). MS expected (M + H)+ =301.1, observed 301.1 (M + H)+, 323.2 (M + Na)+.
7-(β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidin-2,4-diamine (5)
4-chloro-7-(β-Dribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidin-2-amine (4) (250 mg, 0.83 mmol) was transferred to the teflon insert of a steel bomb and 30% aqueous ammonia (15 mL) was added. The reaction was heated to 75°C for 22 hours, cooled, and evaporated to dryness. Purification by silica column with 40% MeOH in DCM gave 5 (211 mg, 90% yield) as a white solid. Rf = 0.49 (40% MeOH in DCM). 1H NMR (DMSO-d6) δ 6.87 (d, 1H, J = 3.6 Hz), 6.57 (br s, 2H), 6.35 (d, 1H, J = 3.6 Hz), 5.82 (d, 1H, J = 6.4 Hz), 5.49 (br s, 2H), 5.33 (br s, 1H), 5.16 (d, 1H, J = 6.3 Hz), 5.00 (d, 1H, J = 4.5 Hz), 4.32 (m, 1H), 4.10 (m, 1H), 4.02 (m, 1H), 3.81 (m, 1H), 3.57-3.46 (m, 2H). MS expected (M + H)+ =282.1, observed 282.1 (M + H)+, 304.0 (M + Na)+.
2-fluoro-7-deaza-adenosine (6)
7-(β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidin-2,4-diamine (5) (211 mg, 0.75 mmol) was transferred to a polypropylene tube with anhydrous pyridine (0.4 mL). To stirred suspension cooled to 0°C, 70% hydrogen fluoride in pyridine (0.67 mL) was added. Potassium nitrite (80 mg, 0.94 mmol) dissolved in water (0.16 mL) was then added slowly to the stirred reaction, leading to evolution of gas and a green color. After 15 min at 0°C the reaction was poured into a stirred suspension of calcium carbonate (1.7g) in water (3.0 mL). The slurry was stirred at 0°C for 2 hours, warmed to r.t. and stirred for 18 hours, then filtered to give a red/brown solution. The insoluble material was washed with water (2.0 mL) and 1:1 water:EtOH (2.0 mL × 2), concentrated and coevaporated with toluene (10 mL × 3). Purification by silica column with 20% MeOH in DCM gave 6 (23 mg, 11% yield) as a white solid. Rf = 0.34 (20% MeOH in DCM). 1H NMR (DMSO-d6) δ 7.54(brs,2H), 7.31 (d, 1H,J = 3.7 Hz), 6.60 (d, 1H, J = 3.7 Hz), 5.87 (d, 1H, J = 6.2 Hz), 5.27 (d, 1H, J = 6.5 Hz), 5.10 (d, 1H, J = 4.9 Hz), 4.99 (t, 1H, J = 5.5Hz), 4.31 (m, 1H), 4.04 (m, 1H), 3.85 (m, 1H), 3.63-3.46 (m, 2H). MS expected (M + H)+ =285.1, observed 285.1 (M + H)+, 307.0 (M + Na)+. 19F NMR (DMSO-d6) δ −53.84 (s).
5′-O-(1-thio)nucleotide triphosphates (7c - 7e)
Representative procedure: The nucleoside (0.0095 mmol) was dried by co-evaporation with pyridine (1.0 mL × 3), put under high vacuum overnight, dissolved in triethyl phosphate (0.5 mL) with gentle heating and cooled to r.t under Ar. To this solution trioctylamine (0.062 mL, 0.014 mmol) was added, followed by thiophosphoryl chloride (0.014 mL, 0.014 mmol) and the reaction was stirred at r.t. for 1 hour. Dried tributylammonium pyrophosphate (250 mg, 0.055 mmol) was dissolved in triethyl phosphate (2.0 mL) with heating, cooled, added to reaction and stirred for 1 hour at r.t. To the stirred reaction triethyl amine (0.8 mL) was added, precipitating a white solid that was collected by centrifugation. The precipitate was dissolved in triethyl amine bicarbonate buffer (TEAB, 5.0 mL, 50 mM pH = 8.0), and incubated at room temperature for 12 hours. The crude product, containing mono, di, tri, tetra , and penta phosphates, was filtered and applied to a HiTrap Q HP anion exchange column and eluted with a linear gradient of TEAB (0.05 - 1.0 M). Purification of analog triphosphates by anion exchange column chromatography yielded a 50:50 mixture of Rp and Sp diasteriomers, as determined by 31P NMR. Triphosphate, cyclic triphosphate and (1-thio) cyclic triphosphate coeluted with analog triphosphates in some preparations, and were observed in the mass-spectra and 31P NMR spectra of these samples. No effort was made to eliminate these impurities as they did not interfere with subsequent transcriptions. (7c) 31P NMR (10% D2O) δ 42.69 (m, 1P), −6.55 (d, 1P, J = 20.2 Hz), −23.07 to −23.42 (m, 1P). 19F NMR (10% D2O) δ −166.16 (d, J = 42.9 Hz). MS expected (M)− = 538.9, observed 539.4 (M)−. (7d) 31P NMR (10% D2O) δ 42.61 (m, 1P), −7.35 (d, 1P, J = 19.8 Hz), −23.45 to −23.80 (m, 1P). 19F NMR (10% D2O) δ −55.78 (s). MS expected (M)− = 538.9, observed 539.1 (M)−. (7e) 31P NMR (10% D2O) δ 42.71 (m), −6.67 (d, 1P, J = 19.8 Hz), −23.16 to −23.51 (m, 1P). 19F NMR (10% D2O) δ −52.92 (s). MS expected (M)− = 539.9, observed 540.0 (M)−.
19F NMR titrations
Nucleosides were dissolved in 10% D2O solutions to a final concentrations of approximately 1.0 mM. The pH of each solution was set by either 100 mM buffer (from pH = 2.0 to 8.0) or by wt% H2SO4 solutions (Hammett acidity function57 H0 = −1.2 to 2.0). Fluorobenzene was included in each sample as an internal reference. 19F NMR spectra were obtained and peak positions fit to the following single ionization model to yield pKa values and frequency shifts:
where Fobs is the observed frequency, FA is the frequency of the neutral nucleoside, and FHA is the frequency of the protonated nucleoside.
Extinction Coefficients
Extinction coefficients for analogs 7c - 7e were estimated by obtaining UV-vis spectra of solutions of known concentration of the corresponding nucleosides in 100 mM phosphate buffer (pH = 7.0) (see Supporting Information) and yielded the following absorption maxima and extinction coefficients; 7F-7dA (278 nm, ε = 7,700 M−1cm−1), 2F-7dA (269 nm, ε = 9,200 M−1cm−1), 2F-A (262 nm, ε = 13,400 M−1cm−1). These extinction coefficients were used to calculate analog concentrations for subsequent transcription reactions.
VSE RNA Transcription
Unligated VSE RNA (NT 621-791) was obtained by transcription of EarI cut pHHVS plasmid. This RNA contains two mutations to the native VS sequence, A782U and U785C that increase ligation activity28. The pHHVS plasmid21 contains a Hammerhead ribozyme upstream of the VSE sequence and an engineered EarI site at U791. Hammerhead processing during transcription produces a uniform 5′-hydroxyl at A621, which acts as the nucleophile in subsequent ligation selection. VSE transcriptions (100 uL) contained 40 mM Tris buffer (pH = 8.0), 10 mM MgCl2, 2 mM spermidine, 5 mM DTT, 0.005 % Triton X-100, 1 mM of each NTP, analogs 7a - 7e to reach concentrations in Table 1, 2 ug of EarI cut pHHVS, 1 unit inorganic pyrophosphatase (Sigma) and T7 RNA polymerase to a final concentration of 0.1 mg/mL. Transcriptions were incubated for 2 hours at 37°C, which led to nearly complete Hammerhead processing following transcription. VSE RNA was purified by 8% denaturing PAGE and eluted at r.t. for 30 min with 10 mM Tris buffer (pH = 8.0), 0.1 mM EDTA, 250 mM NaCl, 2% w/w SDS. Elutions were PCA extracted and concentrated by ethanol precipitation, resuspended in T10E0.1 (Tris buffer (pH = 8.0), 0.1 mM EDTA) and stored at −20°C. For 5′-end labeled controls unligated VSE RNAs were labeled with 5 mCi [γ-32P] ATP (Perkin Elmer) with T4 polynucleotide kinase (New England Biolabs), and purified as above. Labeled samples were split into two aliquots, treated with iodine (1/10 volume of 100 mM iodine in ethanol) or ethanol alone and run on an 8% denaturing PAGE sequencing gels.
Ligation Substrate
The 22 NT substrate used for ligation selections corresponds to VS nucleotides 599-620. To produce the substrate with the required 2′,3′-cyclic phosphate terminus a trans-cleaving VS ribozyme (denoted Rz58) was used. First a 41 NT oligo corresponding to VS nucleotides 599-639 was synthesized (Dharmacon) and labeled with 5 mCi [γ-32P] ATP (Perkin Elmer) using T4 polynucleotide kinase (New England Biolabs). This oligo contains the stem I helix recognized by the trans-cleaving Rz ribozyme. Following the labeling reaction the solution was heated to 75°C for 20 min to deactivate the kinase and cooled to 50°C. Trans-cleavage buffer and Rz RNA were added to give the following final concentrations; 100 mM Tris Buffer (pH = 7.5), 25 mM KCl, 20 mM MgCl2, 2 mM spermidine, 4 mM DTT, 2 μM Rz RNA, and 2 μM labeled 41 NT oligo. After slow cooling to 30°C the trans-cleavage reaction was incubated at 30°C for 1.5 hours (this procedure typical provided >75% cleavage product). Residual [γ-32P] ATP was removed with size exclusion columns (Microspin G-50, GE Healthcare), and the 22NT substrate was purified by 20% non-denaturing PAGE, eluted into T10E0.1, PCA extracted and concentrated by ethanol precipitation.
NAIM Ligation Selections
Individual ligation reactions were performed with labeled 22NT substrate and unlabeled VSE RNAs. Each of the VSE RNAs contained one of the analogs 7a - 7e randomly incorporated at adenosine sites. VSE RNAs were incubated in 2x ligation buffer at 50°C for 15 min and slow cooled to 30°C over 30 min. 22NT substrate was heated to 30°C and added to reactions to give the following final concentrations in a volume of 10 μL; 50 mM Tris buffer (pH = 7.5), 5 mM MgCl2 , 25 mM KCl, 0.4 μM VSE RNA, 0.05 μM 22NT substrate. Reactions were incubated at 30°C for 30-45 minutes which was sufficient time to produce 10-15% ligation. Reactions were quenched with 10 μL formamide loading buffer and split into two aliquots. Iodine (1.0 μL of 100 mM in ethanol) was added to one aliquot to cleave phosphorothioate linkages, while the other aliquot was treated with ethanol alone to control for non-specific cleavage. Cleaved and uncleaved samples were run on 8% denaturing PAGE sequencing gels. Because only active members of the VSE RNA pool become radiolabeled separation of active and inactive members was unnecessary.
Interference Quantitation
Ligation selection gels and 5′-end labeled control gels were dried and exposed to phosphorimager screens. Interferences or enhancements in activity are recognized as decreases or increases in band intensity in the ligation selection gel (Figures 4A,C) relative to the unligated 5′-end labeled controls (Figures 4B,D). Because the phosphorothioate substitution can itself lead to changes in activity a control for this substitution is needed, namely the parental analog 7a which introduces the backbone modification without nucleobase substitutions. If significant non-specific degradation is observed the relative band intensities no longer report on sites of analog incorporation. Iodine free controls were run on the same gels to check for this possibility (Figures 4A-D, lanes 1-5). To quantitate sites of interference line profiles were drawn through ligation selection and control lanes and peak areas calculated. From these peak areas an interference value, κ, is calculated20:
where each member of the fraction is the peak area for a particular site in the ligation or 5′-end labeled gel, NαS is the parental analog 7a, and δαS is one of the pKa perturbed analogs (7b - 7e). A normalization factor (NF) is calculated from the ratios to account for uneven loading of the lanes20. Interference values calculated in this way normalize both for the extent of analog incorporation at a particular site, and for the effect the phosphorothioate substitution at that site. Interference values between 0.5 and 2.0 are typically viewed as insignificant, while those below 0.5 are often expressed as 1/κ enhancements. For large interference values increasingly weak bands must be quantitated in the background of minor non-specific degradation. Interference values above 6.0 are considered indistinguishable for this reason. Figure 5A-D provides the average of four interference values calculated for each of the analogs of this study. Because large iodine cleavage products were insufficiently resolved in some gels interference values are reported for sites A645-A767.
Supplementary Material
Acknowledgements
We thank M.T. Migawa for guidance in the synthesis of 7F-7dA. This work was supported by NSF grant MCB-0544255 and an NIH NRSA fellowship to I.T.S (GM078764).
Footnotes
Supporting Information Available. Extinction spectra of compounds 7c - 7e; 19F titrations for 7F-7dA, 2F-7dA and 2F-A; 1H-15N HMBC spectra of A, 7F-7dA and 2F-A in protonated and unprotonated form; 13C chemical shifts for 7F-7dA, 2F-7dA and 2F-A in protonated and unprotonated form; Complete ref 35. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Cell. 1982;31:147–157. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 2.Guerriertakada C, Gardiner K, Marsh T, Pace N, Altman S. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 3.Kennell JC, Saville BJ, Mohr S, Kuiper MTR, Sabourin JR, Collins RA, Lambowitz AM. Genes Dev. 1995;9:294–303. doi: 10.1101/gad.9.3.294. [DOI] [PubMed] [Google Scholar]
- 4.Mandal M, Breaker RR. Nat. Rev. Mol. Cell Biol. 2004;5:451–463. doi: 10.1038/nrm1403. [DOI] [PubMed] [Google Scholar]
- 5.Hannon GJ. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 6.Pyle AM. Science. 1993;261:709–714. doi: 10.1126/science.7688142. [DOI] [PubMed] [Google Scholar]
- 7.Murray JB, Seyhan AA, Walter NG, Burke JM, Scott WG. Chem. Biol. 1998;5:587–595. doi: 10.1016/s1074-5521(98)90116-8. [DOI] [PubMed] [Google Scholar]
- 8.Das SR, Piccirilli JA. Nat. Chem. Biol. 2005;1:45–52. doi: 10.1038/nchembio703. [DOI] [PubMed] [Google Scholar]
- 9.Nakano S, Chadalavada DM, Bevilacqua PC. Science. 2000;287:1493–1497. doi: 10.1126/science.287.5457.1493. [DOI] [PubMed] [Google Scholar]
- 10.Bevilacqua PC, Brown TS, Nakano S, Yajima R. Biopolymers. 2004;73:90–109. doi: 10.1002/bip.10519. [DOI] [PubMed] [Google Scholar]
- 11.Raines RT. Chem. Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 12.Gong B, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR. J. Am. Chem. Soc. 2007;129:13335–13342. doi: 10.1021/ja0743893. [DOI] [PubMed] [Google Scholar]
- 13.Moody EM, Brown TS, Bevilacqua PC. J. Am. Chem. Soc. 2004;126:10200–10201. doi: 10.1021/ja047362h. [DOI] [PubMed] [Google Scholar]
- 14.Legault P, Pardi A. J. Am. Chem. Soc. 1997;119:6621–6628. [Google Scholar]
- 15.Ravindranathan S, Butcher SE, Feigon J. Biochemistry. 2000;39:16026–16032. doi: 10.1021/bi001976r. [DOI] [PubMed] [Google Scholar]
- 16.Luptak A, Ferre-D'Amare AR, Zhou KH, Zilm KW, Doudna JA. J. Am. Chem. Soc. 2001;123:8447–8452. doi: 10.1021/ja016091x. [DOI] [PubMed] [Google Scholar]
- 17.Flinders J, Dieckmann T. J. Mol. Biol. 2001;308:665–679. doi: 10.1006/jmbi.2001.4627. [DOI] [PubMed] [Google Scholar]
- 18.Tang CL, Alexov E, Pyle AM, Honig B. J. Mol. Biol. 2007;366:1475–1496. doi: 10.1016/j.jmb.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Ryder SP, Ortoleva-Donnelly L, Kosek AB, Strobel SA. Methods Enzymol. 2000;317:92–109. doi: 10.1016/s0076-6879(00)17008-9. [DOI] [PubMed] [Google Scholar]
- 20.Cochrane JC, Strobel SA. Current Protocols in Nucleic Acid Chemistry. Vol. 2. Wiley; New York: 2004. [DOI] [PubMed] [Google Scholar]
- 21.Jones FD, Strobel SA. Biochemistry. 2003;42:4265–4276. doi: 10.1021/bi020707t. [DOI] [PubMed] [Google Scholar]
- 22.Oyelere AK, Kardon JR, Strobel SA. Biochemistry. 2002;41:3667–3675. doi: 10.1021/bi011816v. [DOI] [PubMed] [Google Scholar]
- 23.Ryder SP, Oyelere AK, Padilla JL, Klostermeier D, Millar DP, Strobel SA. RNA. 2001;7:1454–1463. [PMC free article] [PubMed] [Google Scholar]
- 24.Szewczak AA, Ortoleva-Donnelly L, Zivarts MV, Oyelere AK, Kazantsev AV, Strobel SA. Proc. Natl. Acad. Sci. U. S. A. 1999;96:11183–11188. doi: 10.1073/pnas.96.20.11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwon MY, Strobel SA. RNA. 2008;14:25–34. doi: 10.1261/rna.771608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kierdaszuk B, Modrak-Wojcik A, Wierzchowski J, Shugar D. Biochim. Biophys. Acta, Protein Struct. M. 2000;1476:109–128. doi: 10.1016/s0167-4838(99)00225-3. [DOI] [PubMed] [Google Scholar]
- 27.Perrin DD. Dissociation Constants of Organic Bases in Aqueous Solution. Butterworths; London: 1965. [Google Scholar]
- 28.Jones FD, Ryder SP, Strobel SA. Nucleic Acids Res. 2001;29:5115–5120. doi: 10.1093/nar/29.24.5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao ZY, McLeod A, Harusawa S, Araki L, Yamaguchi M, Kurihara T, Lilley DMJ. J. Am. Chem. Soc. 2005;127:5026–5027. doi: 10.1021/ja0502775. [DOI] [PubMed] [Google Scholar]
- 30.Wilson TJ, McLeod AC, Lilley DMJ. EMBO J. 2007;26:2489–2500. doi: 10.1038/sj.emboj.7601698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith MD, Collins RA. Proc.Natl.Acad.Sci.U.S.A. 2007;104:5818–5823. doi: 10.1073/pnas.0608864104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang XJ, Seth PP, Ranken R, Swayze EE, Migawa MT. Nucleos. Nucleot. Nucl. 2004;23:161–170. doi: 10.1081/ncn-120027825. [DOI] [PubMed] [Google Scholar]
- 33.Rosemeyer H, Seela F. Helv. Chim. Acta. 1988;71:1573–1585. [Google Scholar]
- 34.Seela F, Soulimane T, Mersmann K, Jurgens T. Helv. Chim. Acta. 1990;73:1879–1887. [Google Scholar]
- 35.Eldrup AB, et al. J. Med. Chem. 2004;47:5284–5297. doi: 10.1021/jm040068f. [DOI] [PubMed] [Google Scholar]
- 36.Liu MC, Luo MZ, Mozdziesz DE, Sartorelli AC. Nucleos. Nucleot. Nucl. 2005;24:45–62. doi: 10.1081/NCN-46784. [DOI] [PubMed] [Google Scholar]
- 37.Krolikiewicz K, Vorbruggen H. Nucleos. Nucleot. 1994;13:673–678. [Google Scholar]
- 38.Arabshahi A, Frey PA. Biochem. Biophys. Res. Commun. 1994;204:150–155. doi: 10.1006/bbrc.1994.2438. [DOI] [PubMed] [Google Scholar]
- 39.Rastogi T, Beattie TL, Olive JE, Collins RA. EMBO J. 1996;15:2820–2825. [PMC free article] [PubMed] [Google Scholar]
- 40.Sood VD, Collins RA. J. Mol. Biol. 2001;313:1013–1019. doi: 10.1006/jmbi.2001.5119. [DOI] [PubMed] [Google Scholar]
- 41.Lafontaine DA, Wilson TJ, Norman DG, Lilley DMJ. J. Mol. Biol. 2001;312:663–674. doi: 10.1006/jmbi.2001.4996. [DOI] [PubMed] [Google Scholar]
- 42.Scott LG, Geierstanger BH, Williamson JR, Hennig M. J. Am. Chem. Soc. 2004;126:11776–11777. doi: 10.1021/ja047556x. [DOI] [PubMed] [Google Scholar]
- 43.Peng XH, Li H, Seela F. Nucleic Acids Res. 2006;34:5987–6000. doi: 10.1093/nar/gkl719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sowers LC, Eritja R, Kaplan B, Goodman MF, Fazakerly GV. J. Biol. Chem. 1988;263:14794–14801. [PubMed] [Google Scholar]
- 45.Hardin CC, Gollnick P, Kallenbach NR, Cohn M, Horowitz J. Biochemistry. 1986;25:5699–5709. doi: 10.1021/bi00367a053. [DOI] [PubMed] [Google Scholar]
- 46.Saville BJ, Collins RA. Cell. 1990;61:685–696. doi: 10.1016/0092-8674(90)90480-3. [DOI] [PubMed] [Google Scholar]
- 47.Sood VD, Collins RA. J. Mol. Biol. 2002;320:443–454. doi: 10.1016/s0022-2836(02)00521-1. [DOI] [PubMed] [Google Scholar]
- 48.Hiley SL, Sood VD, Fan J, Collins RA. EMBO J. 2002;21:4691–4698. doi: 10.1093/emboj/cdf462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lafontaine DA, Wilson TJ, Zhao ZY, Lilley DMJ. J. Mol. Biol. 2002;323:23–34. doi: 10.1016/s0022-2836(02)00910-5. [DOI] [PubMed] [Google Scholar]
- 50.Lilley DMJ. Biol. Chem. 2007;388:699–704. doi: 10.1515/BC.2007.069. [DOI] [PubMed] [Google Scholar]
- 51.Bevilacqua PC. Biochemistry. 2003;42:2259–2265. doi: 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- 52.Quigley GJ, Rich A. Science. 1976;194:796–806. doi: 10.1126/science.790568. [DOI] [PubMed] [Google Scholar]
- 53.Campbell DO, Bouchard P, Desjardins G, Legault P. Biochemistry. 2006;45:10591–10605. doi: 10.1021/bi0607150. [DOI] [PubMed] [Google Scholar]
- 54.Salter J, Krucinska J, Alam S, Grum-Tokars V, Wedekind JE. Biochemistry. 2006;45:686–700. doi: 10.1021/bi051887k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuzmin YI, Da Costa CP, Cottrell JW, Fedor MJ. J. Mol. Biol. 2005;349:989–1010. doi: 10.1016/j.jmb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 56.Wilcox CS, Otoski RM. Tetrahedron Lett. 1986;27:1011–1014. [Google Scholar]
- 57.Albert A, Serjeant EP. The Determination of Ionization Constants. 3rd ed. Chapman and Hall; New York, NY: 1984. [Google Scholar]
- 58.Guo HCT, Collins RA. EMBO J. 1995;14:368–376. doi: 10.1002/j.1460-2075.1995.tb07011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.