

Abstract
Many breast cancer cells typically exhibit lower expression of manganese superoxide dismutase (MnSOD) compared to the normal cells from which they arise. This decrease can often be attributed to a defect in the transcription of SOD2, the gene encoding MnSOD; however, the mechanism responsible for this change remains unclear. Here, we describe how altered histone modifications and a repressive chromatin structure constitute an epigenetic process to down regulate SOD2 in human breast carcinoma cell lines. Utilizing chromatin immunoprecipitation (ChIP) we observed decreased levels of dimethyl H3K4 and acetylated H3K9 at key regulatory elements of the SOD2 gene. Consistent with these results, we show that loss of these histone modifications creates a repressive chromatin structure at SOD2. Transcription factor ChIP experiments revealed that this repressive chromatin structure influences the binding of SP-1, AP-1, and NFκB to SOD2 regulatory cis-elements in vivo. Lastly, we show that treatment with the histone deacetylase inhibitors trichostatin A and sodium butyrate can reactivate SOD2 expression in breast cancer cell lines. Taken together, these results indicate that epigenetic silencing of SOD2 could be facilitated by changes in histone modifications and represent one mechanism leading to the altered expression of MnSOD observed in many breast cancers.
Keywords: methylation, chromatin, mammary, carcinoma, antioxidant
Introduction
Tumor cells are characterized by aberrant production of reactive oxygen species and an atypical redox state [1]. This has led to the hypothesis that reactive oxygen species may be causally involved in carcinogenesis. Changes in free radical biology in tumor cells can be linked to the altered expression pattern of antioxidant enzymes like MnSOD. Manganese superoxide dismutase (MnSOD) is a mitochondrial matrix enzyme encoded by the nuclear gene SOD2. Cancers originating from diverse tissue types often exhibit decreased MnSOD activity compared to the tissues from which they arise. At the same time, forced MnSOD over-expression in tumor derived cell lines attenuates their malignant phenotype and lowers their metastatic potential [2-6]. These observations suggest that MnSOD may function as a new type of tumor suppressor gene [7].
Breast cancer cells often have decreased levels of MnSOD compared to their normal counterparts, and this is generally attributed to a transcriptional defect of SOD2 [8]. SOD2 is regulated by several transcriptional regulatory elements controlled that contain binding sites for AP-2, EGR-1, AP-1 and SP-1, NFκB and FOXO3a [9-14]. While the cis-regulatory elements bound by these transcription factors are not commonly deleted or mutated in breast cancer [15], their accessibility to binding is required for them to play an active role in SOD2 transcription. The accessibility of DNA and transcriptional regulatory elements is actuated through epigenetic processes.
Epigenetic alterations are a means by which gene expression can be altered independent of changes in DNA sequence. In the past decade it has become abundantly clear that epigenetic changes play just as important a role as mutations in carcinogenesis. Cytosine methylation in CpG di-nucleotides is an epigenetic process that can silence gene expression. Hypermethylation of CpG di-nucleotides within the SOD2 basal promoter has been correlated with reduced levels of MnSOD in multiple myeloma and pancreatic carcinoma [16-18]. Similar studies from our group have revealed that DNA methylation reduces MnSOD levels in immortalized fibroblasts and breast cancer cells [19, 20]. However, this observation was not ubiquitous among all breast cancer cell lines with decreased expression of SOD2. For example, MB-231and T47D, two cell lines used in this study, are two examples of human breast carcinoma cell lines that have decreased MnSOD expression, but whose SOD2 promoters are unmethylated (Supplemental Figure 1 and data not shown). Here, we chose to investigate whether changes in the post-translational modification of histone tails could facilitate the decreased levels of MnSOD in human breast cancer cell lines.
The post-translational acetylation and methylation of lysine residues in histone tails is another epigenetic process that can regulate gene expression [21, 22]. Reduced levels of H3 histones methylated at lysine-4 and acetylated at lysine-9 within gene regulatory elements are indicative of gene silencing [23]. Previous studies have shown that MnSOD levels directly correlate with the acetylation status of histones present at SOD2 [10, 24]. Collectively these studies define the potential role for epigenetic regulation of SOD2.
We hypothesized that decreased levels of MnSOD in human breast cancer cell lines may be due, at least in part, to altered histone modifications and condensed chromatin structure at SOD2. In this study we identified decreased MnSOD protein and mRNA in four breast cancer cell lines compared to the immortalized, nontumorigenic breast epithelial cell line MCF-10A. Chromatin immunoprecipitation assays determined that the decreased expression of MnSOD in breast cancer cell lines correlated with hypoacetylation of H3K9 and hypomethylation of H3K4 at seven transcriptional regulatory elements within SOD2. Chromatin accessibility assays revealed that the chromatin structure of these seven regions is inaccessible compared to MCF-10A. Transcription factor chromatin immunoprecipitation uncovered decreased binding of NFκB, AP-1 and SP-1 at SOD2 in breast cancer cell lines. A causal role of histone hypoacetylation in repressing MnSOD levels was determined by activating SOD2 transcription using histone deacetylase inhibitors. Combined, these results suggest that MnSOD expression can be repressed in breast cancer by hypoacetylation and hypomethylation of histones, thus forming a closed chromatin structure at SOD2 that inhibits transcription factor function.
Methods and Materials
Tissue culture and drug treatments
The cell lines used in this study were obtained from the American Type Culture Collection (ATCC) and grown as previously described [20]. Cells were maintained at 37°C and 5% CO2 during all experiments. MB-231, T47D, MCF7 and MCF7-5C cells were either left untreated, or treated with 200 ng/ml of trichostatin A, or sodium butyrate dissolved in DNMSO.
Quantitative reverse transcription real-time PCR analysis
Real-time PCR analysis of SOD2 expression was carried out as previously described [20]. Analysis of SOD2 transcript levels was determined using 2 × SYBR real time master mix (Applied Biosystems) and primers specific to SOD2, or the control gene 18S. SYBR green flouresence was then monitored using an ABI 7000 Sequence Detection System (Applied Biosystems). Once reactions were complete, a threshold of amplification was selected to calculate the cycle threshold (Ct) for each sample using the ABI Sequence Detection software (Applied Biosystems). The Ct values from triplicate reactions were then averaged for SOD2 and 18S. Relative SOD2 expression between cell lines was then calculated by normalizing to the 18S control using the ΔΔCt method.
Western blot analysis
Total protein extracts were resolved by 4-20% SDS-polyacrylamide gels (Bio-Rad) and transferred to a nitrocellulose membrane for western analysis. After transfer was complete blots were stained with Ponceau S to ensure equal loading and transfer. Membranes were then probed with Anti-MnSOD primary antibody in PBS + 5% non-fat dry milk in PBS containing 0.1% TWEEN 20, followed by washing with PBS. Secondary anti-rabbit horseradish peroxidase antibody conjugate was then added. After subsequent washing, antigens were visualized using the ECL Plus kit (Amersham Pharmacia) and exposing the membranes to BioMax MR film (Eastman Kodak).
MnSOD activity assay
Total protein extracts were prepared as described under western blot analysis. The SOD activity gel assay was carried out as described [25].
Chromatin immunoprecipitation assay
The preparation of cells sonicated chromatin was carried out as described by Hitchler et al. 2006 [20]. Prepared chromatin was then mixed with antibodies specific to: di-methylH3K4, acetylated H3K9, NFκB p65, SP-1 (Upstate Biotech), AP-1 (Santa Cruz Biotechnology) and control nonspecific IgG (Sigma) and allowed to mix overnight at 4°C with agitation. Protein/Antibody/DNA complexes were then collected using 70 μl Protien A agarose (Upstate Biotech) followed by washing and elution according to the suggested protocol. The specificity of each antibody to pull down its respective antigen was then confirmed by western blot (Supplemental Figure 2) Protein/DNA crosslinks were then reversed using 200 mM NaCl at 65°C for 4 h and DNA purified from input chromatin and immunoprecipitation reactions using the Qiagen DNeasy Kit (Qiagen) according to manufacture’s suggested protocol. Purified DNA was then quantified by using a NanoDrop ND-1000 (NanoDrop). All samples were analyzed by SYBR green quantitative real time PCR with primers designed to GAPDH (GAPDHf: 5′-CGTCCTTGACTCCCTAGTGTCCT-3′, GAPDHr 5′-CCTACTTTCTCCCCGCTTTTTT-3′) and the seven regions of SOD2 (Fig. 2). Amplification of the target amplicon was monitored as a function of increased SYBR green fluorescence. An analysis threshold was set and the cycle threshold (Ct) computed for each sample. The fold enrichment of each target sequence was determined using the following formula (Fold enrichment=2(Ct of Ip’ed DNA-Ct of input)).
Fig. 2.
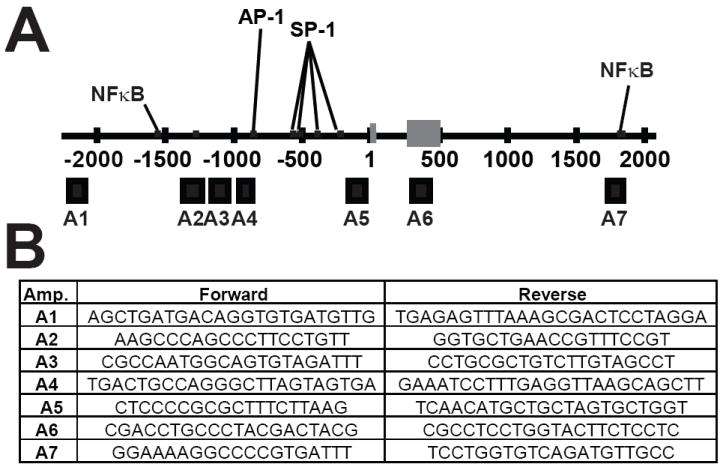
Locations of the seven real time PCR amplicons (A1-A7) used to analyze SOD2 by chromatin immunoprecipitation (ChIP) and chromatin accessibility. Binding sites for AP-1, NFκB and SP-1 within SOD2 are indicated by black lines. (A) The seven real time PCR amplicons used during histone and transcription factor ChIP as well as chromatin accessibility represented by black boxes below the line. Exon 1 of the SOD2 gene are designated by grey boxes. Numbering scheme was derived from build 17 of the human genome project. (B) Oligonucleotide primer sequences used to analyze the seven regions of SOD2 during chromatin immunoprecipitation and chromatin accessibility experiments.
Chromatin accessibility assay
Chromatin accessibility experiments were conducted as described by Rose et al. 2006 [26]. The accessibility indices of cis-regualtory regions were determined using the same quantitative real-time PCR primers for amplicons A1-A7 using reactions as described under ChIP experiments (Fig. 2). The accessibility index for each amplicon was determined by the following formula (Accessibility index=2((Ct Dnase treated) − (Ct Uncut))).
Statistical analysis
Statistical analysis of ChIp and Chromatin accessibility was determined using a one-way ANOVA analysis with Tukey’s multiple comparison post test. All analysis was done using GraphPad Prism Version 4.0 (GraphPad Software Inc).
Results
Breast cancer cell lines have decreased expression of MnSOD and SOD2 mRNA
To begin our study we analyzed MnSOD protein and SOD2 mRNA in cell lines that are devoid of aberrant DNA methylation (Supplemental Figure 1 and data not shown).
Quantitative RT-PCR demonstrated that MB-231, T47D, MCF7 and MCF7-5C breast cancer cell lines had significantly decreased SOD2 mRNA, and were approximately one-third of the steady state levels of SOD2 mRNA compared to nontumorigenic MCF-10A (Fig. 1A). To query how this change in SOD2 expression might influence free radical biology in these cell lines MnSOD protein levels and activity were also analyzed. MnSOD protein and activity was determined to be decreased in MB-231, T47D, and MCF7 breast cell lines (Fig. 1B). These results are consistent with our quantitative RT-PCR studies and confirm the previously reported observation that breast cancer cell lines generally have decreased MnSOD mRNA, protein, and activity [2].
Fig. 1.
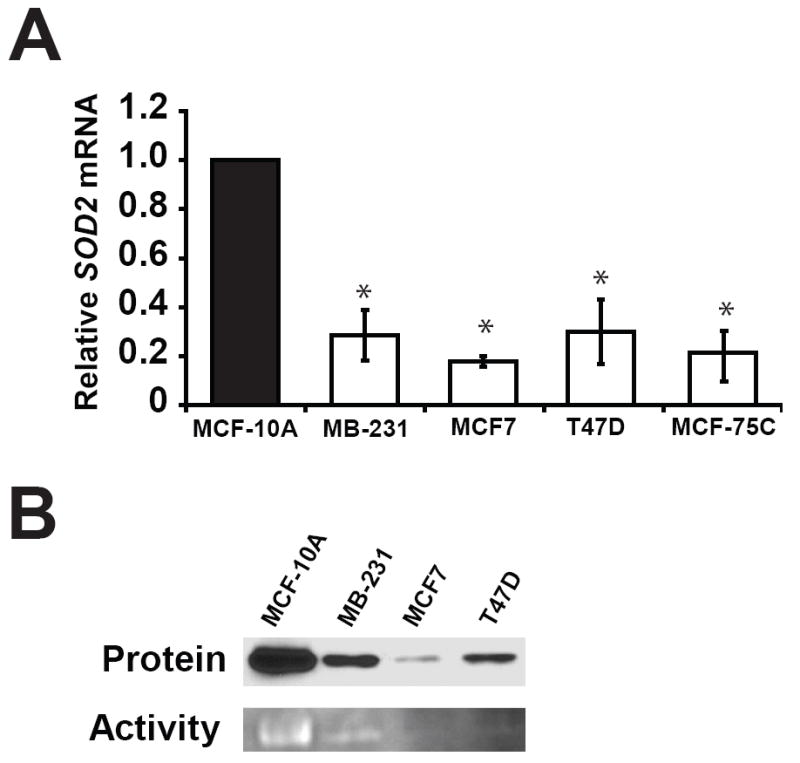
Levels of MnSOD mRNA, protein, and activity are lower in breast cancer cell lines than immortalized breast epithelial cells. (a) Relative SOD2 mRNA levels in breast cell lines determined by SYBR green real-time PCR. The steady state levels of SOD2 mRNA in the cell lines were normalized to 18S control, then compared to MCF-10A using the ΔΔCt method. Values shown are the average of three independent experiments ± standard error of mean (* p < 0.05). (b) The levels of MnSOD protein and activity in the model cell lines used during this study.
Breast cancer cell lines have a repressive histone code at SOD2
Different histone modifications at a gene can collectively form a “histone code” that can tightly regulate gene expression [21, 22]. Others have demonstrated that transcription of MnSOD directly correlates with its H3K9 acetylation after induction of SOD2 [10]. We sought to investigate whether the decreased levels of MnSOD in our breast cancer cell lines could be attributed to loss of two histone modifications associated with active transcription, H3K4 dimethylation and H3K9 acetylation, at SOD2 [23]. To meet this objective we chose to compare and contrast the levels of these two modifications at transcriptional regulatory elements within SOD2 between breast cancer cell lines and MCF-10A using chromatin immunoprecipitation (ChIP). Amplicon 1 (A1) is located in the extreme 5′ region of SOD2 and is devoid of known transcriptional regulatory elements. The other six regions were chosen because they have established roles in regulating SOD2 (Fig. 2). Enrichment of dimethyl H3K4 was significantly different between transcriptionally inactive A1 and the other six amplicons (A2-A7) in MCF-10A and suggests that H3K4 methylation may have a role in controlling SOD2 (Fig. 3). Similar analysis in MB-231, T47D, and MCF7 and MCF7-5C showed the level of dimethyl H3K4 enrichment was significantly different from MCF-10A at amplicons A2, A3, A4, and A7, but not at A1, A5, A6 or GAPDH (Fig. 3) (ANOVA p < 0.05, for individual statistical analysis see Supplemental Table S1). Similar quantitative ChIP analysis of H3K9 acetylation in MCF-10A again revealed that this active mark of transcription was statistically different at A1 compared to amplicons A2-A7. We were also able to determine that acetylated H3K9 enrichment was significantly different between respective amplicons in MCF-10A and MB-231, T47D, MCF7 and MCF7-5C breast cancer cell lines (Fig. 4). No enrichment of these seven amplicons was detected in control IgG immunoprecipitations (data not shown). Taken together, these tight associations give insight into the influence exerted by histone modifications on transcriptional regulatory elements of SOD2. Our analysis further suggests that SOD2 may be transcriptionally repressed in cancer cells by a repressive histone code.
Fig. 3.
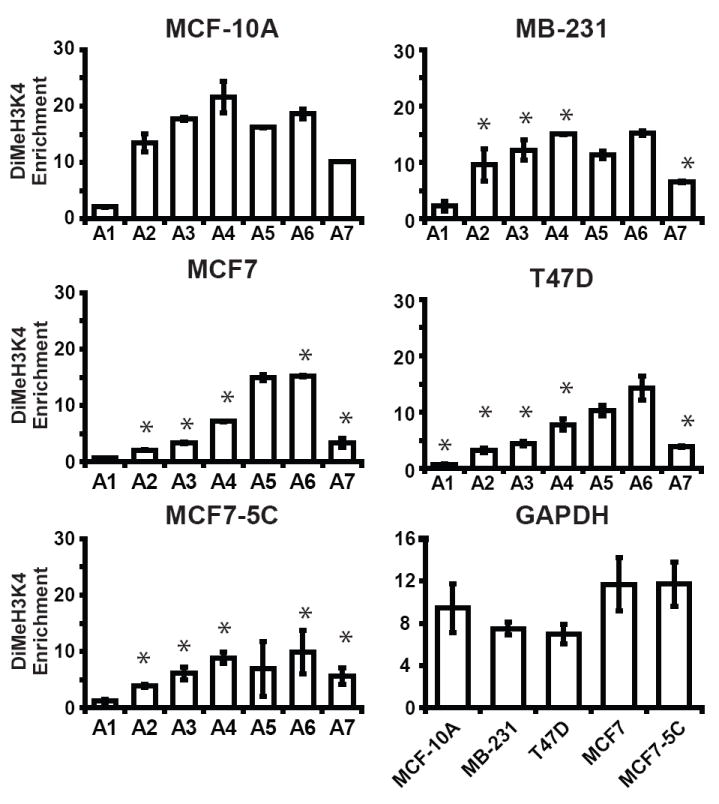
Breast cancer cells have decreased levels of dimethyl H3K4 at SOD2 compared to immortalized breast epithelial. Methylation of H3K4 histones at seven regions (A1-A7) of the SOD2 promoter and at GAPDH in breast cell lines was determined using chromatin immunoprecipitation (ChIP). Fold enrichments of dimethyl H3K4 at these regions were calculated as described in methods and materials. Values given are the averages of three independent experiments ± the standard error. (* p < 0.05 between MCF-10A and breast cancer cell lines at respective amplicons. For individual statistical analysis between MCF-10A and breast cancer cell lines see Supplemental Table S1).
Fig. 4.
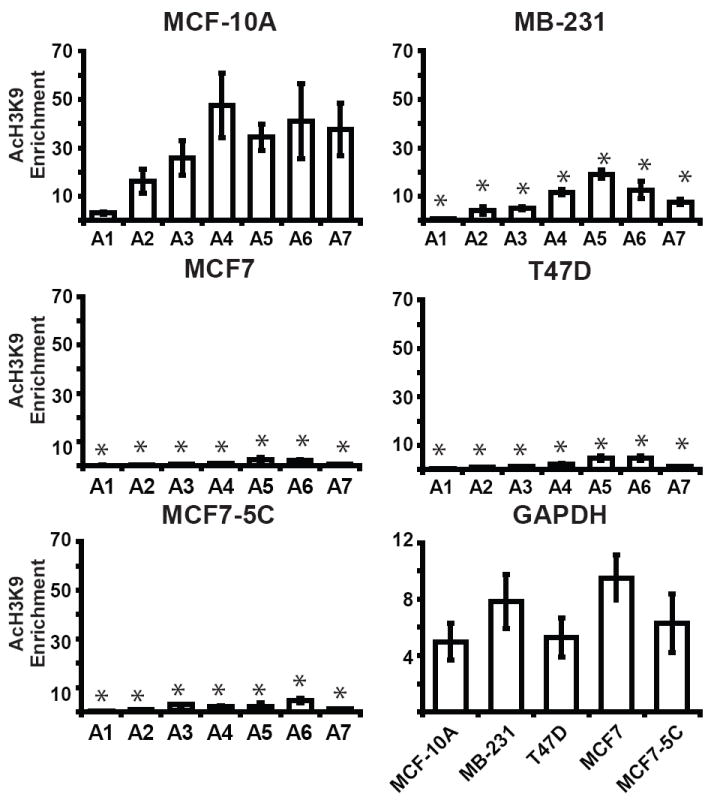
The SOD2 promoter of breast cancer cells is associated with hypoacetylated histones. The association of acetylated H3K9 with SOD2 and GAPDH in breast cell lines was determined using chromatin immunoprecipitation (ChIP). The level of acetylated H3K9 at seven regions (A1-A7) of the SOD2 and GAPDH promoters in breast cell lines were calculated as described in methods and materials. Values given are the averages of three independent experiments ± the standard error. (* p < 0.05 between MCF-10A and breast cancer cell lines at respective amplicons. For individual statistical analysis between MCF-10A and breast cancer cell lines see Supplemental Table S2).
Breast cancer cell lines have condensed chromatin structure at SOD2
To establish whether the decrease in H3K4 dimethylation and H3K9 acetylation might form a closed chromatin structure at SOD2, we queried its chromatin accessibility using DNase I hypersensitivity. Chromatin accessibility at the six transcriptional elements of SOD2 were in good agreement with their level of histone acetylation in MCF-10A and statically different from amplicon A1 (Fig. 5) (p < 0.05 ANOVA, for individual statistical analysis see Supplemental Table S1). Comparing the average accessibility of these six regions in MCF-10A with MB-231, T47D, MCF7 and MCF7-5C revealed that breast cancer cell had considerably condensed chromatin at SOD2 relative to breast epithelial cells. Overall our findings revealed that an inaccessible chromatin structure at SOD2 could be decreasing the level of MnSOD in these cell lines.
Fig. 5.
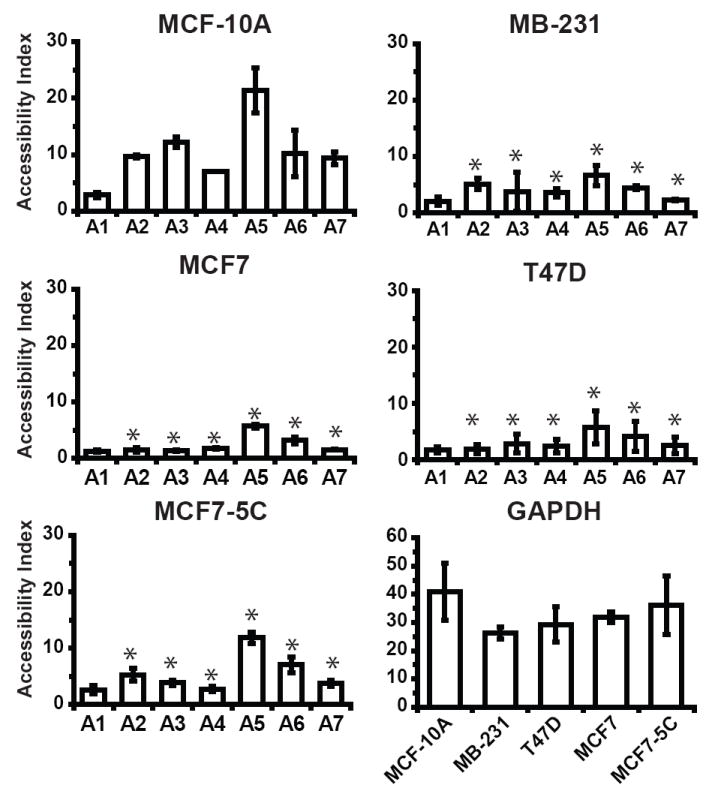
The SOD2 gene of breast cancer cell lines is less accessible than immortalized breast epithelial cells. Chromatin accessibility of the seven regions (A1-A7) of the SOD2 promoter and GAPDH was determined by DNAse I hypersensitivity. Accessibility index of each region was determined using SYBR green real-time PCR. Accessibility index for each amplicon was then as described in methods and materials. Values given are the averages of three independent experiments ± the standard error. (* p < 0.05 between MCF-10A and breast cancer cell lines at respective amplicons. For individual statistical analysis between MCF-10A and breast cancer cell lines see Supplemental Table S3).
The binding of transcription factors at SOD2 is altered in breast cancer cell lines in vivo
The existence of repressive histone modifications and chromatin structure can impede transcription by limiting transcription factor binding. Overall, the pattern of SP-1 AP-1 and NFκB binding at SOD2 is in good agreement with previous reports (Fig 6A-B). SP-1 binding was limited to the basal promoter region (amplicons 4 and 5) and was decreased in breast cancer cell lines compared to MCF-10A (Fig. 6A top panel). Enrichment of amplicons 2, 4 and 5 was observed in AP-1 immunoprecipitations; however only amplicon 5 enrichment was decreased in breast cancer cell lines (Fig. 6A, middle panel). Breast cancer cell lines also exhibited decreased levels of NFκB binding at amplicons 2, 3 and 7 (Fig. 6A, bottom panel). Taken together these results support the overall hypothesis that an altered chromatin environment can inhibit the transcription of SOD2.
Fig. 6.
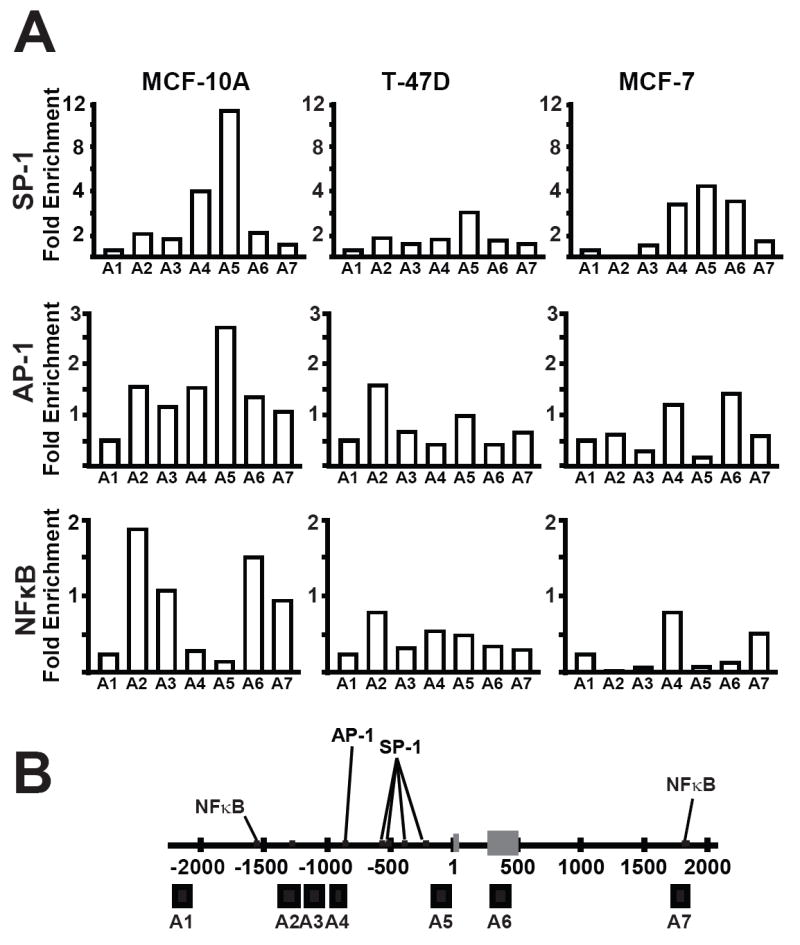
Binding of AP-1, SP-1 and NFκB are decreased at SOD2 in breast cancer cell lines. The placement and level of SP-1, NFκB, and AP-1 binding at SOD2 was determined using (ChIP). Fold enrichments of these transcription factors SOD2 was calculated as described under methods and materials.
Histone deacetylase inhibitors increase SOD2 transcription
Epigenetic repression by histone hypoacetylation can be reversed through the pharmacological inhibition of histone deacetylases (HDACs). To test if histone hypoacetylation was causally responsible for repressing SOD2 in breast cancer we treated our panel of cell lines with trichostatin A (TSA) and sodium butyrate. Treatment of MB-231, MCF7 and MCF7-5C with 200 ng/ml TSA for 12 hours moderately increased their steady state levels of SOD2 mRNA, no response was elicited from T47D (Fig. 7). However, treatment with 5 mM sodium butyrate did increase SOD2 expression in MB-231, MCF7, and T47D. Moreover, such treatment increased the chromatin accessibility index of amplicon 4 of SOD2 in MB-231 and MCF7-5C from 4-6 in untreated controls (Fig. 5) to 23-25 in TSA treated cells, consistent with chromatin remodeling to a more transcriptionally accessible structure. Together, these data suggest that histone hypoacetylation is, at least in part, causally responsible for repressing SOD2 expression in breast cancer.
Fig. 7.
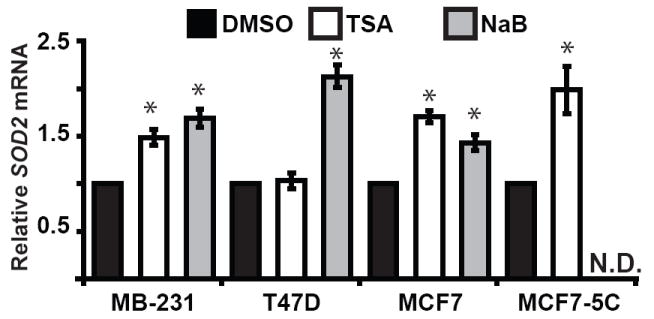
The histone deacetylase inhibitors trichostatin A (TSA) and sodium butyrate increased SOD2 expression in breast cancer cell lines. MB-231, T47D, MCF7 and MCF7-5C breast cancer cell lines were treated 200 ng/ml trichostatin A or 5 mM sodium butyrate for 12 hours. Total RNA was then harvest and subjected to quantitative real time RT-PCR analysis to determine changes in SOD2 expression. The levels of SOD2 mRNA were normalized using 18S rRNA. The relative level of SOD2 mRNA in each sample were determined by comparing TSA and sodium butyrate treated cells with their matched DMSO controls using the ΔΔCt method. Values shown are the average of three independent experiments ± stand are error of mean (* p < 0.5 versus untreated control). N.D.; not determined.
Discussion
SOD2 expression is often attenuated in breast cancer, and can usually be attributed to a defect in its transcription [8]. The SOD2 gene is not commonly deleted or mutated in breast cancer, thus the mechanism(s) by which MnSOD levels become decreased in breast cancer remains elusive [15]. Previous studies have described the repression of SOD2 by DNA methylation [16-20]. We sought to investigate if changes in histone modifications and chromatin structure could constitute another means by which epigenetic processes could decrease SOD2 expression in human breast carcinoma cells.
Epigenetic alterations are just as capable as mutations at changing the expression of tumor suppressor genes and metastasis suppressors. Mutations in the basal promoter of SOD2 have been shown to alter transcription factor function and decrease MnSOD levels in colon cancer, glioblastoma, and fibrosarcoma [27]. At this time no such mutations have been identified at SOD2 in breast cancer. Transcriptional repression by epigenetic processes can occur without mutation. SOD2 contains a large CpG island, which makes it a likely candidate for repression by DNA methylation. DNA methylation has been shown to decrease MnSOD levels in immortalized fibroblasts, multiple myeloma, and pancreatic cancer [18, 28, 29]. We have recently reported that SOD2 can become aberrantly hypermethylated at CpGs in breast cancer cells with decreased levels of MnSOD [20]. DNA methylation and histone acetylation often collaborate to repress gene expression [30, 31]. However, we did not detect DNA methylation at SOD2 in the breast cancer cell lines used in this study (Supplemental Figure 1 and data not shown).
The pattern of histone modifications changes during carcinogenesis [32]. Hypoacetylation of H3K9 and hypomethylation of H3K4 can both silence gene expression independent of DNA methylation [23]. When MnSOD expression is induced, histones associated with SOD2 become hyperacetylated [24, 33]. Here we have investigated the decreased expression of SOD2 in breast cancer. We compared and contrasted the level and placement of acetylated H3K9 and methylated H3K4 at SOD2 between breast cancer cell lines and immortalized epithelial cells. These results demonstrate that breast cancer cells have significantly lower levels of both acetylated H3K9 and methylated H3K4 at SOD2 which is consistent with their decreased level of MnSOD.
Histone acetylation often coincides with an accessible chromatin structure and active transcription. When chromatin is accessible, transcription factors have admittance to their recognition sites. By measuring chromatin accessibility, we found that chromatin at SOD2 in breast cancer cells is in a closed state that correlates with their level of acetylated H3K9 histones. We also determined that the level of accessibility differs between discrete transcriptional regulatory elements within SOD2. For example, accessibility and histone acetylation are lowest at amplicons A2 and A5 in breast cancer cells. Our transcription factor ChIP experiments concluded that AP-1 and NFκB binding was also significantly reduced at these regions. Others have demonstrated that transcriptional activation of SOD2 by NFκB can affect histone acetylation, chromatin structure and SP-1 binding at the basal promoter [33]. SP-1 binding at the basal promoter is not a prerequisite for SOD2 expression in mouse embryonic fibroblasts. However, SP-1 is required to mediate the transcriptional activation of SOD2 by the intronic enhancer and other 5′ regulatory elements. Based on this knowledge, it seems likely that epigenetic inactivation of these other regulatory elements might lead to the decreased histone acetylation, methylation chromatin accessibility and decreased SP-1 binding we observed at the basal promoter (amplicon A5) of breast cancer cells.
Epigenetic silencing is perpetuated by the activity of enzymes that add and remove epigenetic marks. The activity of these enzymes can be interrupted by treatment with pharmacological inhibitors. Many of these inhibitors have been aimed at influencing the activity of HDACs. Trichostatin A and sodium butyrate are two such pharmacological agents that have been widely used to inhibit the activity of class I and class II HDACs in cancer [34]. Treatment with TSA was able to slightly increase the level of SOD2 mRNA in MB-231, MCF7, and MCF7-5c in association with acquisition of a more accessible chromatin structure, but not in T47D. However, sodium butyrate was able to elicit a SOD2 transcription response in T47D. Reports from others have established that TSA increases MnSOD and H3K9 acetylation at SOD2 in mouse myoblasts [10]. Together these findings suggest that SOD2 could be repressed in breast cancer through the activity of class I and II HDACs in the breast cancer cell lines that exhibited a response. Previous reports suggest that SP-1 can recruit HDACs to SOD2 [10]. Our ChIP results reveal that SP-1 binding is decreased at SOD2 in breast cancer cell lines. This implies that some as yet unknown mechanism could be recruiting HDAC activity to SOD2. Taken together, these results demonstrate that the SOD2 is a potential target for pharmacological reactivation by TSA and sodium butyrate in some breast cancers.
Epigenetic regulation of SOD2 may also be responsible for increased levels MnSOD in metastatic disease [35, 36]. It is counter intuitive that mutations at SOD2 would result in increased MnSOD. Specific histone modifications at genes are inheritable after cell division; however they can exhibit a certain plasticity. Increased H3K9 acetylation and chromatin accessibility could change at SOD2 to increase MnSOD expression to suit the needs of tumor cells at metastatic sites. Our study has focused on examining the influence of epigenetic processes on SOD2 in cell cells derived from human tumors. Nevertheless, when our findings are considered together with previous studies describing chromatin changes during SOD2 transcriptional activation it seems likely that epigenetic control of SOD2 expression is consistent with all the existing data.
In conclusion, our results show changes in chromatin function at SOD2 is associated with its decreased expression in human breast cancer. The relationship between SOD2 expression, histone acetylation, histone methylation and chromatin accessibility are represented in the radar graphs in Fig. 8. Depicting the data in this manner illustrates the sharp contrasts of SOD2 function in immortalized breast epithelial and breast cancer cell lines. From these observations we suggest the following model for how SOD2 is regulated in breast cells (Fig. 9). In our model normal breast epithelial have high levels of acetylated H3K9 and H3K4 dimethylation associated with SOD2. In kind, the chromatin structure at SOD2 is accessible, making cis-regulatory elements open for transcription factor binding (Figure 8a). Human breast cancer cells have decreased levels of both histone modifications at SOD2, which forms a closed chromatin structure which ultimately inhibits the function of AP-1, NFκB, and SP-1 (Figure 8b). The contrasting differences in the chromatin structure between immortalized breast epithelial and breast cancer cell lines suggest that epigenetic processes may, at least in part, be responsible for the altered level of SOD2 transcription in breast cancer.
Fig. 8.

Changes in MnSOD expression correlate with chromatin function at SOD2. The relationship between SOD2 expression and the chromatin parameters in breast cells represented as radar graphs. The average values of relative SOD2 mRNA (north axis) were plotted against enrichment of H3K4 methylation at amplicon 4 (east axis), enrichment of acetylated H3K9 at amplicon 4 (south axis) and chromatin accessibility (west axis). These graphs contrast the differences in chromatin biology at SOD2 to clearly relate of the changes between MCF-10A and MB-231, T47D, MCF7, and MC7-5C breast cancer cell lines.
Fig. 9.
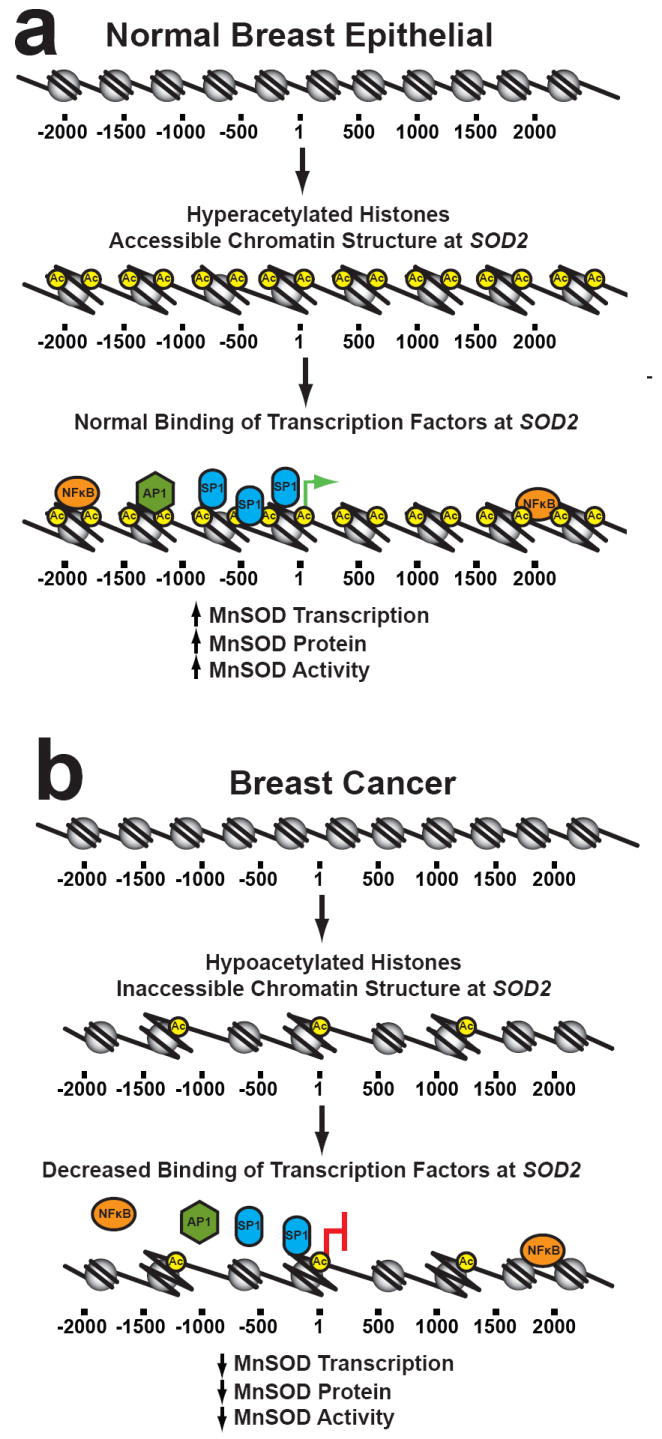
Proposed model for how changes in histone modifications alter SOD2 expression. The epigenetic state of SOD2 when MnSOD levels are high. Hyperacetylated histones create an accessible chromatin structure at SOD2 that allows transcription factors to bind. (A) When MnSOD is epigenetically repressed by histone hypoacetylation, an inaccessible chromatin structure forms at SOD2. This change in chromatin structure inhibits SP-1, NFκB, and AP-1 binding at SOD2 which decreases MnSOD transcription, protein and activity. This process is reversible and increased SOD2 expression in metastatic disease may be effected by further epigenetic alterations during progression leading to the epigenetic reactivation depicted in part (B).
Supplementary Material
Acknowledgments
This work was supported by NIH grants CA073612 to F.E.D. and CA115438 to L.W.O. M.J.H. received salary support from T32 CA078586.
Abbreviations
- MnSOD
manganese superoxide dismutase
- mRNA
messenger RNA
- ChIP
chromatin immunoprecipitation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Oberley LW, Oberley TD, Buettner GR. Cell division in normal and transformed cells: the possible role of superoxide and hydrogen peroxide. Med Hypotheses. 1981;7:21–42. doi: 10.1016/0306-9877(81)90018-9. [DOI] [PubMed] [Google Scholar]
- 2.Li JJ, Oberley LW, St Clair DK, Ridnour LA, Oberley TD. Phenotypic changes induced in human breast cancer cells by overexpression of manganese-containing superoxide dismutase. Oncogene. 1995;10:1989–2000. [PubMed] [Google Scholar]
- 3.Zhong W, Oberley LW, Oberley TD, St Clair DK. Suppression of the malignant phenotype of human glioma cells by overexpression of manganese superoxide dismutase. Oncogene. 1997;14:481–490. doi: 10.1038/sj.onc.1200852. [DOI] [PubMed] [Google Scholar]
- 4.Ough M, Lewis A, Zhang Y, Hinkhouse MM, Ritchie JM, Oberley LW, Cullen JJ. Inhibition of cell growth by overexpression of manganese superoxide dismutase (MnSOD) in human pancreatic carcinoma. Free Radic Res. 2004;38:1223–1233. doi: 10.1080/10715760400017376. [DOI] [PubMed] [Google Scholar]
- 5.Weydert C, Roling B, Liu J, Hinkhouse MM, Ritchie JM, Oberley LW, Cullen JJ. Suppression of the malignant phenotype in human pancreatic cancer cells by the overexpression of manganese superoxide dismutase. Mol Cancer Ther. 2003;2:361–369. [PubMed] [Google Scholar]
- 6.Weydert CJ, Waugh TA, Ritchie JM, Iyer KS, Smith JL, Li L, Spitz DR, Oberley LW. Overexpression of manganese or copper-zinc superoxide dismutase inhibits breast cancer growth. Free Radic Biol Med. 2006;41:226–237. doi: 10.1016/j.freeradbiomed.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 7.Bravard A, Sabatier L, Hoffschir F, Ricoul M, Luccioni C, Dutrillaux B. SOD2: a new type of tumor-suppressor gene? Int J Cancer. 1992;51:476–480. doi: 10.1002/ijc.2910510323. [DOI] [PubMed] [Google Scholar]
- 8.Borrello S, De Leo ME, Galeotti T. Defective gene expression of MnSOD in cancer cells. Mol Aspects Med. 1993;14:253–258. doi: 10.1016/0098-2997(93)90012-3. [DOI] [PubMed] [Google Scholar]
- 9.Lomberk G, Urrutia R. The family feud: turning off Sp1 by Sp1-like KLF proteins. Biochem J. 2005;392:1–11. doi: 10.1042/BJ20051234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maehara K, Uekawa N, Isobe K. Effects of histone acetylation on transcriptional regulation of manganese superoxide dismutase gene. Biochem Biophys Res Commun. 2002;295:187–192. doi: 10.1016/S0006-291X(02)00646-0. [DOI] [PubMed] [Google Scholar]
- 11.Zhu CH, Huang Y, Oberley LW, Domann FE. A family of AP-2 proteins down-regulate manganese superoxide dismutase expression. J Biol Chem. 2001;276:14407–14413. doi: 10.1074/jbc.M009708200. [DOI] [PubMed] [Google Scholar]
- 12.Zhu C, Huang Y, Weydert CJ, Oberley LW, Domann FE. Constitutive activation of transcription factor AP-2 is associated with decreased MnSOD expression in transformed human lung fibroblasts. Antioxid Redox Signal. 2001;3:387–395. doi: 10.1089/15230860152409031. [DOI] [PubMed] [Google Scholar]
- 13.St Clair DK, Porntadavity S, Xu Y, Kiningham K. Transcription regulation of human manganese superoxide dismutase gene. Methods Enzymol. 2002;349:306–312. doi: 10.1016/s0076-6879(02)49345-7. [DOI] [PubMed] [Google Scholar]
- 14.Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 15.St Clair DK, Holland JC. Complementary DNA encoding human colon cancer manganese superoxide dismutase and the expression of its gene in human cells. Cancer Res. 1991;51:939–943. [PubMed] [Google Scholar]
- 16.Hodge DR, Xiao W, Peng B, Cherry JC, Munroe DJ, Farrar WL. Enforced expression of superoxide dismutase 2/manganese superoxide dismutase disrupts autocrine interleukin-6 stimulation in human multiple myeloma cells and enhances dexamethasone-induced apoptosis. Cancer Res. 2005;65:6255–6263. doi: 10.1158/0008-5472.CAN-04-4482. [DOI] [PubMed] [Google Scholar]
- 17.Hurt EM, Thomas SB, Peng B, Farrar WL. Integrated molecular profiling of SOD2 expression in multiple myeloma. Blood. 2007;109:3953–3962. doi: 10.1182/blood-2006-07-035162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hurt EM, Thomas SB, Peng B, Farrar WL. Molecular consequences of SOD2 expression in epigenetically silenced pancreatic carcinoma cell lines. Br J Cancer. 2007;97:1116–1123. doi: 10.1038/sj.bjc.6604000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Y, Peng J, Oberley LW, Domann FE. Transcriptional inhibition of manganese superoxide dismutase (SOD2) gene expression by DNA methylation of the 5’ CpG island. Free Radic Biol Med. 1997;23:314–320. doi: 10.1016/s0891-5849(97)00095-6. [DOI] [PubMed] [Google Scholar]
- 20.Hitchler MJ, Wikainapakul K, Yu L, Powers K, Attatippaholkun W, Domann FE. Epigenetic Regulation of Manganese Superoxide Dismutase Expression in Human Breast Cancer Cells. Epigenetics. 2006;1:163–171. doi: 10.4161/epi.1.4.3401. [DOI] [PubMed] [Google Scholar]
- 21.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 22.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 23.Musri MM, Corominola H, Casamitjana R, Gomis R, Parrizas M. Histone H3 lysine 4 dimethylation signals the transcriptional competence of the adiponectin promoter in preadipocytes. J Biol Chem. 2006;281:17180–17188. doi: 10.1074/jbc.M601295200. [DOI] [PubMed] [Google Scholar]
- 24.Ranjan P, Boss JM. C/EBPbeta regulates TNF induced MnSOD expression and protection against apoptosis. Apoptosis. 2006;11:1837–1849. doi: 10.1007/s10495-006-9530-0. [DOI] [PubMed] [Google Scholar]
- 25.Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 26.Rose SL, Fitzgerald MP, White NO, Hitchler MJ, Futscher BW, De Geest K, Domann FE. Epigenetic regulation of maspin expression in human ovarian carcinoma cells. Gynecol Oncol. 2006;102:319–324. doi: 10.1016/j.ygyno.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 27.Xu Y, Krishnan A, Wan XS, Majima H, Yeh CC, Ludewig G, Kasarskis EJ, St Clair DK. Mutations in the promoter reveal a cause for the reduced expression of the human manganese superoxide dismutase gene in cancer cells. Oncogene. 1999;18:93–102. doi: 10.1038/sj.onc.1202265. [DOI] [PubMed] [Google Scholar]
- 28.Huang Y, He T, Domann FE. Decreased expression of manganese superoxide dismutase in transformed cells is associated with increased cytosine methylation of the SOD2 gene. DNA Cell Biol. 1999;18:643–652. doi: 10.1089/104454999315051. [DOI] [PubMed] [Google Scholar]
- 29.Hodge DR, Peng B, Pompeia C, Thomas S, Cho E, Clausen PA, Marquez VE, Farrar WL. Epigenetic Silencing of Manganese Superoxide Dismutase (SOD-2) in KAS 6/1 Human Multiple Myeloma Cells Increases Cell Proliferation. Cancer Biol Ther. 2005;4:585–592. doi: 10.4161/cbt.4.5.1704. [DOI] [PubMed] [Google Scholar]
- 30.Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 32.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 33.Guo Z, Boekhoudt GH, Boss JM. Role of the intronic enhancer in tumor necrosis factor-mediated induction of manganous superoxide dismutase. J Biol Chem. 2003;278:23570–23578. doi: 10.1074/jbc.M303431200. [DOI] [PubMed] [Google Scholar]
- 34.Taddei A, Roche D, Bickmore WA, Almouzni G. The effects of histone deacetylase inhibitors on heterochromatin: implications for anticancer therapy? EMBO Rep. 2005;6:520–524. doi: 10.1038/sj.embor.7400441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malafa M, Margenthaler J, Webb B, Neitzel L, Christophersen M. MnSOD expression is increased in metastatic gastric cancer. J Surg Res. 2000;88:130–134. doi: 10.1006/jsre.1999.5773. [DOI] [PubMed] [Google Scholar]
- 36.Lewis A, Du J, Liu J, Ritchie JM, Oberley LW, Cullen JJ. Metastatic progression of pancreatic cancer: changes in antioxidant enzymes and cell growth. Clin Exp Metastasis. 2005;22:523–532. doi: 10.1007/s10585-005-4919-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.