

Abstract
The concept of cell-based therapy has been advocated as a novel approach for treating diseases or conditions where regeneration of cells, tissue and/or potentially organs is required. A promising source for cell-replacement therapies is provided by stem cells, but the success of this approach will ultimately rely on the ability to isolate primary stem or progenitor cells. Cell-surface protein markers will play a critical role in this step. Current methodologies for the identification of cell-surface protein markers rely primarily on antibody availability and flow cytometry, but many cell-surface proteins remain undetectable. Proteomic technologies now offer the possibility to specifically identify and investigate the cell-surface subproteome in a quantitative and discovery-driven manner. Once a cell surface protein marker panel has been identified by MS and the antibodies become available, the panel should permit the identification, tracking, and/or isolation of stem or progenitor cells that may be appropriate for therapeutics. This review provides a context for the use of proteomics in discovering new cell-surface markers for stem cells.
Keywords: Cell surface protein marker, Stem cells
1 Introduction
1.1 Stem cells and their potential role in therapeutics
One of the most critical problems in transplantation medicine is the lack of suitable donor organs or tissues. In the case of intractable diseases like Parkinsonism, diabetes and spinal injury, the problems are even greater, since there are no readily available therapies capable of curing these syndromes. For these reasons, the concept of cell-replacement or cell-supplement therapy has been advocated as a novel therapeutic approach. Importantly, treatments to replace, repair or enhance the biological function of damaged tissue or organs through cell-transplantation/replacement therapy have proven successful in the case of a few syndromes, the best examples of which are bone marrow transplantations for the treatment of leukemia after myeloablative therapies, breast cancer, or congenital immunodeficiencies, and cultured epidermal autographs in the case of severe burns (reviewed in [1-5]). The ability to isolate, cultivate, multiply and manipulate these cells for therapeutics has, however, either limited or encouraged their use [1, 2, 6]. Currently, only allogeneic or matched donor-derived stem cells have been routinely used in human cell-grafting therapies, thus confirming that one of the best potential sources for cell-replacement therapies is stem cells.
A stem cell is unique in that it has the ability to self-renew indefinitely and to differentiate into specialized cells under appropriate physiological or experimental conditions. In fact, a cell’s ability to self-renew and differentiate into as few as one or even many specialized cell types is the functional basis for defining a stem cell. Two types of stem cells, embryonic and adult (tissue specific or cord blood) have been classified. Embryonic stem (ES) cells derived from the pre-implantation blastocyst can give rise to all cells of an embryo proper, including the germ line. Because of this robust developmental potential, the introduction of undifferentiated ES cells into an inappropriate environment can lead to the formation of teratomas, which therapeutically is unacceptable. ES cells may, however, have therapeutic potential if more committed cells (stem or otherwise) without tumorogeneic potential can be identified. The key will be to isolate these cells from the heterogeneous cultures that usually are generated with ES cell differentiation. Adult stem cells, in contrast, are generally believed to be much more restricted in their potential and generally have a lower tumorogeneic potential. For example, several types of stem cells are present in the bone marrow, including hematopoietic stem cells (HSC) and mesenchymal stem cells (MSC). Importantly, only primary isolates of long-term repopulating HSC can fully reconstitute the hematopoietic system of a myeloablated host. MSC in contrast [7, 8] readily form chondrocytes, adipocytes and osteocytes and are expandable in vitro. Despite their potential heterogeneity, MSC have been employed in early clinical studies, including some designed to treat human myocardial infarctions (MI) [9]. Similarly, the hematopoietic stem cell marker for c-Kit1 has been used to isolate putative stem cells from heart, which appear to have reparative capacities. Although some evidence has been presented to suggest that both MSC and c-Kit1 heart cells may form functional cardiac myocytes in vivo, many of the reparative effects seem to be linked more with either anti-inflammatory effects or their potential to produce local paracrine factors. Numerous other types of stem cells are also found in tissues throughout the body in brain, skin, muscle, gut, liver, and all of these stem cells have different properties and unique differentiation capabilities that are affected by their microenvironment, commitment and origin [10, 11].
Moreover, recent studies have challenged the dogma that adult stem cells are restricted to the generation of only the type of cells present in the tissue where they reside. In fact, a number of groups have reported that adult stem cells have a greater differentiation potential than previously thought (i.e. these cells could be coaxed to differentiate into cells not normally associated with their ‘committed’ state) [12]. Examples include HSC that reputedly develop into neural, myogenic and hepatic cell types; neural or skeletal muscle stem cells that develop into the hematopoietic lineage [12-18]; and stromal stem cells that differentiated into cardiac muscle cells (myocytes) [19]. The reputed trans-differentiation potential and increased developmental potential of most of these adult stem cells has, however, been challenged [20-23]. Their altered differentiation potential has been attributed to be at least partly a consequence of culture conditions [24, 25], which may have led to epigenetic changes, cell contaminations, cell fusion events [26, 27], and even transformation events. Cells put in culture for several passages before transplant, therefore, may have changed their original properties [28], and as such may not be apt for any therapeutic applications. What is lacking, is a clear set of markers that can be employed to identify and isolate to homogeneity primary isolates of adult stem cells (i.e. cells that have not been cultured).The need for surface markers is essential for the study of stem cell research and their potential therapeutic use. The focus of this review is to discuss proteomic approaches that are particularly well suited to identifying new protein markers on the surface of stem cells. This specialized topic cannot be adequately addressed by more-global reviews that focus on whole-proteome analyses. Instead, we will outline those methods that are most appropriate for the discovery of new cell-surface protein markers and the need for further technology development. We will conclude by outlining a workflow that should prove valuable for the development of validated protein marker panels.
1.2 Stem cells and the need for cell-surface protein markers
Practically, it has proven difficult to definitively discern stem and progenitor cells with therapeutic potential from other cells with in vitro differentiation potential, including some that may have undergone transformation or epigenetic modifications. In the most conservative case, stem cells need to be defined as single cells that are clonal precursors of more stem cells of the same type as well as differentiated progeny [3, 4]. Accordingly, only when stem or progenitor cells have been purified to homogeneity as a primary isolate can one know with certainty that the generation of expected (or unexpected) progeny is a property of a known cell type, barring culturing issues, of course. Based on these stringent criteria, only rarely have stem cells been identified as clonogenic precursors (e.g. long-term repopulating HSC) that include in their progeny both self-renewed stem cells and differentiated progeny.
One question is how to isolate a single primary cell that can be defined as an authentic stem cell or progenitor cell and reintroduced into a tissue/organ without passaging in tissue culture. One approach, and the topic of this review, involves the identification of specific protein markers found on the cell surface (Fig. 1). Although the list of known stem cell markers (compiled at http://stemcells.nih.gov/info/scireport/appendixe, for example) contains both cell-surface and intracellular markers, antibody availability and accessibility determines whether they are suitable for isolating and sorting defined populations. While cell-surface markers can include proteins, lipids, and carbohydrates, for example, proteomics focuses on identifying the proteins and their potential modifications. Cells generally express distinct assortments of proteins and lipids on their plasma membrane (PM), including those commonly known as CD molecules (for cluster of differentiation) [29]. The presence of the antigens or other proteins may reflect either unique stages of lineage-specific differentiation or different states of activation or inactivation. These cell-surface protein markers can be used to distinguish different types of cells (see example of HSC discussed below). Because the populations of cells, or their derivatives, sorted by surface markers are most often heterogeneous [30], they could include cells that are true, long-term self-renewing stem cells, shorter-term progenitors, and/or some non-stem cells. Therefore, a functional assay is necessary to confirm their authenticity. To date, no single marker has thus far proved sufficient to unambiguously define an authentic stem cell or a unique stem-cell function. The goal, therefore, is to ultimately correlate the function of a pure population of stem cells with a panel of defined cell-surface protein markers. Thus, when the functional assays have been adequately tested, the use of a well-defined panel of markers for positive and negative selection can be employed to identify authentic stem cell populations that have therapeutic potential. This concept proved highly effective in the case of primary bone marrow-derived HSC, which are composed of long-term and shortterm repopulating cells and myeloid and lymphoid progenitors. Importantly, only the long-term repopulating HSC, which display a unique set of markers (Murine: cKit+, Sca1+, Lin-; Human: CD34+Thy-1+Lin-), correspond to the authentic HSC that are suitable for long-term replacement therapy [31, 32]. Regrettably, very few other stem cells have been adequately defined by markers for therapeutic interventions, in part due to the challenges in identifying new potential protein markers.
Figure 1.
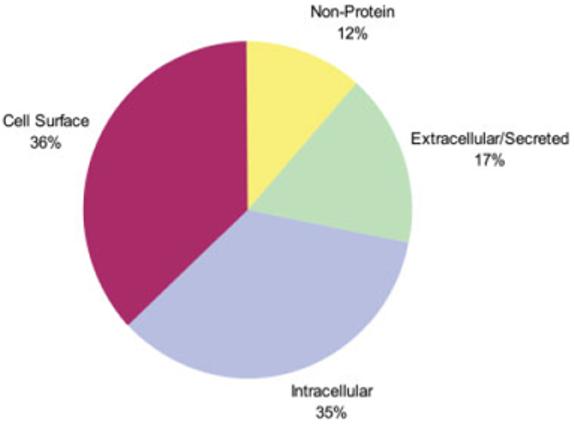
Known stem cell markers. All known stem cell markers listed at http://stemcells.nih.gov/info/scireport/appendixe were classified as either intracellular, extracellular, or cell surface based upon UniProt/Swiss-Prot annotation.
Thus, there is an urgent need for the identification of unique sets of cell-surface protein markers that can be employed to identify and purify to homogeneity stem and progenitor cells from the person/model in question. Although it is possible to use techniques such as flow cytometry, antibody arrays, and microscopy to probe for known proteins on the cell surface in discrete populations (Fig. 2), these methodologies require the use of specific antibodies and as such are knowledge-driven. The quantitative proteomic approach that we describe here is, however, discovery-driven. One advantage of this approach is that it does not require a priori knowledge of the proteins on the cell surface or antibodies in order to discover new protein markers that are present. MS-based proteomics enables the identification of cell-surface proteins within a specific sub-proteome, and the identification of regulatory PTM, such as protein phosphorylation sites, which cannot be detected in gene microarrays [33]. However, gene microarrays are an invaluable tool for the definition of a range of active genes, which must be considered in order to understand stem cells and their differentiation potential [34, 35]. Ultimately, knowledge about the cell-surface proteome in combination with gene-expression signatures should allow for the discovery of cell-surface protein markers, and aid in understanding the biology, regulation, and development of stem cells. In other words, understanding the cell-surface subproteome will enhance our understanding of which signals can be processed by stem cells (e.g. signaling controls of self-renewal [36-38]).
Figure 2.
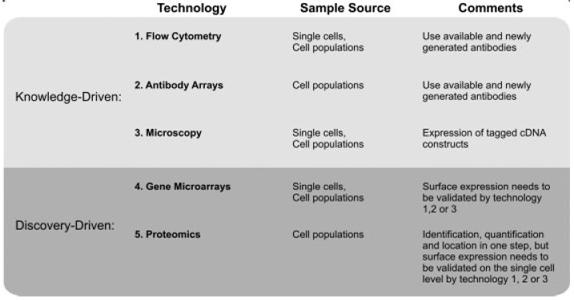
Technologies for the identification of cell-surface markers. Knowledge-driven methods seek to determine whether known proteins can be used as markers, while discovery-driven approaches seek to identify new proteins.
1.3 Challenges to identifying new cell-surface markers
Cell-surface markers are a class of PM proteins that are able to respond to or sense the environment around the cell and contain an extracellular domain, which has the benefit of being available for detection by an antibody. The relatively small number of available CD cell-surface markers (∼350) [29], compared to the number of predicted human transmembrane (∼13 000) [39, 40] and PM proteins (∼2700) [40](see Fig. 3) illustrates the gap between the currently available cell-surface markers vs. potential markers.
Figure 3.
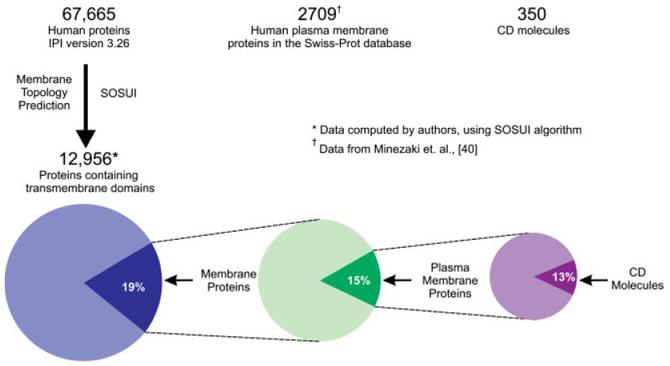
Current coverage of potential cell-surface markers.
Currently, using classical biochemical cell fractionation techniques, it is difficult to obtain purified PM proteins without contamination from membranes from other intracellular organelles, such as the nucleus, mitochondria, ER, Golgi, and lysosome, for example. Furthermore, the difficulty in obtaining sufficient quantities of low-abundance membrane proteins for subsequent proteomic protein identification purposes is magnified when membrane purity is not optimal. Finally, since solubilizing the entire PM protein is difficult due to the hydrophobic nature of the transmembrane domain, common proteomic approaches to identifying PM proteins are peptide-centric approaches and rely on identification of the soluble portions of the PM proteins. In addition to challenges related to the properties of the cell-surface proteins themselves, there are other limitations for cell-surface proteomics.
The major advantage of using quantitative proteomics in a search for new stem cell-surface markers is due to its ability to establish the presence of a particular protein in a specific location without the use of an antibody. Quantitative proteomics, thus, will be useful in helping to bridge the gap and identify novel cell-surface protein markers, eventually as panels of new cell differentiation markers. In this way, the lack of a specific antibody does not limit the potential to identify a particular protein of interest. However, the cell surface is in constant flux as the cell senses and responds to its environment, yet currently available quantitative proteomic methods can reveal only ‘snapshots’ of the cell surface in a population of cells. Single-cell surface proteomics is not on the horizon yet. Therefore, complementary methods must also be included for the dynamic state analysis of the cell surface on single cells. Current proteomic technologies require relatively high cell numbers and will yield “averaged” information about the proteins expressed in a population of cells. Thus, rare events such as those on only a few cells within a larger population are not detected by today’s quantitative proteomic workflows and cannot be followed in short time intervals. Consequently, for the functional validation and tracking of cell-surface proteins in single cells of heterogeneous primary cell populations, antibodies are needed that can recognize these newly MS-identified cell-surface proteins, and eventually their PTM.
2 Proteomic discovery methods
2.1 Enrichment methods - shrinking the haystack
Proteomics has evolved tremendously during the past 10 years. Much of the progress is due to technological advances in MS and chromatographic separations of peptides and proteins. However, in recent years, the field is also advancing due to inventive methods for carrying out proteomics studies that target a specific subset or class of proteins. Proteomic approaches that employ enrichment methods have developed out of necessity, due to the realization that the dynamic range of protein concentrations and proteome complexity eliminate the possibility of finding all proteins within a proteome by using global approaches with current MS technology. By enriching for the protein class of interest based on a particular chemical/physical characteristic(s), these focused proteomics approaches offer the advantage of reducing sample complexity and access to lower abundance proteins in a discovery-driven experimental approach. In particular, PM proteins are inherently difficult to solubilize and, therefore, they may be under-represented when analyzed in conjunction with more soluble proteins in proteomic workflows. Thus, by enriching for PM proteins early on in the proteomic workflow, one can eliminate the bias towards more soluble proteins. Additionally, the enrichment will reduce the amount of protein or peptide separation required, and therefore, reduce the potential protein loss associated with such strategies. Recently published proteomic approaches for the discovery of new stem cell-surface proteins that are reviewed here are summarized in Table 1.
Table 1.
Summary of proteomics methods focused on identifying plasma membrane proteins in stem cells
| Citation | Cell type/source | Chemical tag | Method of physical PM protein enrichment | Method of protein separation | Method of peptide separation/MS | No. of proteins identified | % of proteins that are PMa) | Notes |
|---|---|---|---|---|---|---|---|---|
| Jeong et al. [41] | hMSC from umbilical cord blood | None | Ultracentrifugation of cell lysate to obtain water-insoluble fraction | 1-D SDS-PAGE, 2-D SDS-PAGE | MALDI-TOF MS | 35 in water-insoluble fraction | 5% | |
| Sidibe et al. [58] | ESC-derived smooth muscle cell and adult smooth muscle cells | CyDye and biotin - labeled proteins on intact cells | Avidin capture of labeled proteins | 1-D SDS-PAGE | 1-D LC-MS/MS | 228 | 6% IM + TM; 23% associated, based on Swiss-Prot annotation | |
| Foster et al. [42] | hMSC-TERT | None | Multiple centrifugation steps for subcellular fractionation of cell lysate | None | 1-D LC-MS/MS | 463 | 32% IM+TM; 66% IM + TM + associated, based on Swiss-Prot annotation | Peptide ion volumes for quantitative MS |
| Lund et al. [43] | Umbilical cord blood stem cell (CD34+38-Lin-) derived NK cells and adult NK cells | None | Multiple centrifugation steps for subcellular fractionation of cell lysate | None | 2-D LC-MS/MS | 137 | 33%, based on PANTHER gene ontology function | iTRAQ for quantitative MS |
| Nunomura et al. [56] | mES (D3) | Biotin - labeled proteins on intact cells | Multiple centrifugation steps for subcellular fractionation of cell lysate followed by avidin capture of labeled peptides | None | 2-D LC-MS/MS | 324 (biotin labeled only) | 62%, based on SOSUI analysis, includes TM domain | Orientation of protein in PM determined by where label observed |
When provided by the authors, PM proteins are designated as integral membrane (IM), transmembrane (TM), or membrane associated (associated).
2.1.1 Physical approaches for PM protein enrichment
Subcellular fractionation techniques that employ a combination of centrifugation steps are a common choice for preparing PM-enriched fractions including detergent-resistant membrane fractions (DRM), commonly known as lipid rafts (also known as cell-membrane rafts). These methods can offer a significant improvement in specificity for PM proteins over approaches that do not perform any subcellular fractionation, but rather use whole-cell or tissue preparations. An approach that used a single-step centrifugation to separate the cell lysate of human MSC from umbilical cord blood into water-soluble and water-insoluble fractions identified 35 proteins as shared between two different MSC populations, including two that were classified as cell-surface proteins [41]. Methods that more specifically enrich for the PM typically achieve a higher percentage of PM proteins. For example, Foster et al. [42] compared hMSC-TERT cells before and after induction of osteoblast lineage commitment by using a traditional differential centrifugation strategy. The resulting membrane pellet was enzymatically digested followed by LC-MS/MS. Changes in relative protein abundance were defined by peptide ion volumes and gene quantitation was performed by RT-PCR. Of the 463 proteins identified by at least two unique peptides in two out of three analyses, 26% were integral membrane proteins, 5% were membrane-anchored, 34% membrane-interacting, and 5% were mitochondrial proteins, as classified in UniProt and Gene Ontology references. Importantly, all known protein markers for MSC were found, most of which are CD molecules. Another quantitative proteomics study used a traditional differential centrifugation strategy to compare proteins in natural killer (NK) cells derived from umbilical cord blood stem cells vs. adult NK cells [43]. The membrane pellet was enzymatically digested and labeled with iTRAQ reagent and subsequently analyzed by 2-D LC-MS/MS. The experiment was performed in triplicate and proteins were identified by at least two unique peptides with quantitation requiring three peptides. The number of proteins identified among the three experiments spanned 129 to 325, with 33% involved in cell adhesion, trafficking, or signaling, as defined by the PANTHER ontology classification. The iTRAQ quantitation data were confirmed using Western blot analysis for some proteins, and FACS for several CD molecules.
In addition to examining the whole PM, approaches that examine the lipid raft portion of the PM may offer complementary information and provide understanding of cell localization and/or trafficking. A study by Osterhues et al. [44] compared multipotent CD34- cells to a transfected cell line characterized by malignant transformation and tumor growth, in order to differentiate stem cells from experimental leukemia cells. Lipid rafts of both cell types were isolated and subsequently separated by 1-D and 2-DE SDS-PAGE followed by MALDI-TOF/TOF. Five proteins (caveolin-1, flotillin-1, vimentin, GAPDH, and galectin-3) were identified as being differentially expressed. Unfortunately, for cell-surface protein marker studies, while these proteins are associated with the lipid raft, none of these proteins contains extracellular domains. Two other methods that employ physical sub-fractionation are worth mentioning here, though to date there are no publications that employ them to study stem cells. The method of using high pH to induce beta sheet formation, followed by proteinase K digestion might be of interest for identifying integral membrane proteins [45], though may have limited use for identifying cell-surface markers. Additionally, the use of colloidal silica [46-50] to physically enrich for the PM is appealing because of its ability to use a lower number of cells and primary tissue. This may be of particular interest whether looking for native stem cells in adult tissue or in vitro studies using a small number of cultured cells.
There are several reasons why most of the current literature using proteomics does not discuss the identification of unknown cell-surface proteins. The term ‘unknown’ can refer to the fact that there is no evidence for its existence at the protein level, or that that the protein is known protein, but has not been previously shown to be on the cell surface. First, a majority of the MS methods described above do not allow for the unambiguous determination of whether the membrane proteins identified are truly on the cell surface. Typically, the information regarding subcellular localization included in proteomics datasets are annotated by cross-referencing the protein sequences to available protein and gene ontology databases. In this case, the evidence for a protein being localized to the cell surface is thus based on anecdotal annotations (which may be cross-referenced to primary literature sources), not based on first-hand experimental evidence obtained via the MS. Consequently, by relying only on what is known, this approach limits the possibility of finding new information. It is for these reasons that chemical-tagging approaches are becoming more desirable, as information regarding the true localization to the cell surface can be gained experimentally, independently of information in the databases.
2.1.2 Chemical-tagging approaches for PM protein enrichment
Chemical-tagging methods (for review see [50]) have been a more recently applied technique used to enrich for PM proteins and are often used in conjunction with physical separation strategies like those discussed above. Chemical tagging, in general, allows for a specific class of protein or modification of interest to be physically separated from other, non-tagged proteins. Importantly, when chemical tags are attached to the extracellular domain of PM proteins on intact cells, they offer an unrivaled specificity for PM proteins, because they offer a manner to distinguish true PM proteins from intracellular contaminants that are typically present due to the inability to obtain an absolutely pure PM isolation by subcellular fractionation methods. Cell-surface biotinylation, the covalent attachment of a biotin tag to the extracellular domain of PM proteins, is a popular choice [51-55]. Biotin can be coupled either via a cleavable or non-cleavable sulfo-NHS ester to primary amine groups, on proteins for example. The specificity of the labeling procedure for PM proteins depends on the concentration of the labeling reagent used, the cell type, the temperature of the reaction and the duration of the labeling. It is essential that a viable population of cells with intact membranes be used as any membrane permeability of the reagent in combination with necrotic and/or apoptotic cells can lead to the tagging of unwanted intracellular proteins. The choice of biotinylation reagent (size, charge and hydrophobicity) used is also important, as the cleavable di-sulfide bridge containing sufo-NHS biotin has been reported to be more specific for cell-surface proteins compared to the non-cleavable reagent [55].
Such cell-surface biotinylation has been used to study mouse ES cell (cell line D3) PM proteins [56]. The labeled membrane proteins were enriched using centrifugation followed by acetone precipitation. Following enzymatic digestion, the biotinylated peptides were captured using avidin chromatography and subsequently analyzed by 2-D LC-MS/MS. Combining data from two experiments yielded 965 biotinylated peptides resulting in the identification of 324 proteins, 47% of which were identified by multiple peptides. Of these 324 proteins, 200 (62%) were classified as integral membrane proteins based on the SOSUI algorithm (for description of algorithm see [57]) and the number of transmembrane domains per protein ranged from 1-13. Included in the identified proteins were 59 CD molecules. Cell-surface biotinylation has also been combined with fluorescent dye labeling (CyDye) to facilitate the discrimination of true PM proteins from intracellular contaminants in a study to compare adult vascular smooth muscle cells to ESC-derived smooth muscle cells [58]. Following lysis of the labeled cells, cellular debris was removed by low-speed centrifugation. Labeled proteins were captured from the clarified cell lysate using avidin chromatography and subsequently eluted and separated by 1-D SDS-PAGE prior to digestion and LC-MS/ MS. In total, 228 proteins were identified by multiple peptides, with 6% classified as membrane proteins, 23% membrane-associated, as defined in the Swiss-Prot database.
2.2 Glycoprotein enrichment methods - a sweet source for stem cell-surface markers?
Approximately half of all proteins [59] and a majority of animal cell PM proteins are predicted glycoproteins [60]. Cell-surface glycoproteins have important roles in cell adhesion, signaling, and immune recognition, among others (for examples see reviews [61, 62]), and glycoconjugates have already been shown to be markers for stem cells (for examples see reviews [63, 64]). It is important for the study of cell-surface glycoproteins to extend beyond identifying the proteins themselves, further to establish the glycosylation site and eventually the structure of the glycan (glycomics). The occupancy of potential glycosylation sites on the extracellular domain will be important for antigen design and antibody development if new protein markers are found for which there are no available antibodies, as it may influence antibody binding and thus epitope selection. Secondly, the glycan structure itself and/or the occupancy of the glycosylation site can be biologically important [65-67]. Thirdly, knowledge of occupied glycosylation sites may help to identify the orientation of the protein in the membrane, if unknown, or otherwise help to confirm topology predictions. Several studies on stem cell glycoproteins have been published, and because the topic is especially relevant for a discussion of cell-surface proteins, it is being included here. In two reports by Wearne et al. [68, 69], cell-surface glycans present on BG01 hESC that had been differentiated for 12 days as embryoid bodies (EB) and the changes in the carbohydrates that occur between 12-28 days of differentiation as EB were analyzed using fluorescence microscopy with fluorescein-labeled lectins and antibodies specific for carbohydrate epitopes. In another study, MALDI-TOF MS profiling, NMR structural analysis, and lectin-based flow cytometry were used to compare the N-glycan structures of cord blood-derived CD133+ and CD133- cells [70]. The results were correlated to gene expression analysis of glycosyltransferases and glycosidases. Bi-antennary complex-type and high-mannose type glycans were found to be increased in CD133+ cells.
2.2.1 Cell-surface capturing technology
To overcome problems in identifying low-abundance proteins and peptides in complex mixtures via MS, several groups, including ours, have recently focused on a method for the selective chemical isolation of glycosylated proteins. A method developed by Zhang et al. [71] is based on the observation that many interesting plasma proteins are glycosylated [72-74]. The method is designed to provide a selective analysis of N-glycopeptides and has proven to be highly specific, and has led to an increased sensitivity for lower abundance serum proteins due to the reduction in sample complexity [71]. Since most cell-surface proteins, and potentially secreted proteins, can also be glycosylated, we further developed this technology for the selective detection of cell-surface glycoproteins. Initial results from the cell-surface capturing technology (CSC-technology) have yielded an unparalleled degree of specificity for the detection of low-abundance lipid raft-associated and PM proteins (Dr. Bernd Wollscheid, unpublished observations). Cell-surface proteins isolated from model cell lines via the CSC-technology were consistently identified by MS with less than 15% contamination from intracellular and non-glycosylated peptides/proteins. The CSC-technology overcomes inherent problems in analyzing PM proteins by the selective chemical tagging of glycoproteins, high-affinity enrichment and gel-free LC-MS/MS analysis of peptides derived from PM proteins.
2.3 Quantitative membrane proteomics
Classification of cell types, including stem cells, will be dependent not only on the set of cell-surface expressed proteins, but also on their individual cell-surface copy number. Therefore, it will be important to identify novel cell-surface proteins via MS as well as to quantify and compare their abundance on the cell surface throughout different developmental stages. MS technology is capable of relative and absolute quantification of proteins [75]. Relative and absolute quantitative proteomic technologies (MRM [76], label-free [77], SILAC [78], iTRAQ [79], ICAT [80]) are key technologies, which should be included into iterative proteomic workflows that are designed to identify markers useful for the classification of stem cells. While a thorough discussion of quantitative proteomics is outside the scope of this review, readers are encouraged to refer to an excellent review on quantitative MS in proteomics that was recently published by Bantscheff et al. [81].
3 Validation of stem cell-surface markers
Quantitative proteomics can be used in a discovery-based manner to identify new potential stem cell-surface protein markers. In order to validate the usefulness of any potential cell-surface markers (whether protein, lipid, carbohydrate, etc.), the candidate markers will then need to enter into an iterative cycle of validation and discovery (Fig. 4), which includes techniques other than proteomics. An essential question is how specific is the cell-surface protein marker, and separately, is the marker restricted to stem/progenitor cells or does it show lineage restrictions. To address these questions, an antibody that reacts with the antigen will have to be generated or obtained. Thus, it is essential that the proteomic efforts for discovering the candidate protein markers provide as much characterization as possible about the protein, so that epitope selection is efficient. This will aid in the development of antibodies with sufficient specificity, which is typically labor and time intensive. Secondarily and if not already established, it is essential to determine if the marker is quantitatively more or less abundant on original stem/progenitor cells relative to other cells. A variety of techniques is available for this, but the use of genomic and proteomic databases are a good place to begin. Once this has been established, it is critical to determine if the marker(s) is present on a subpopulation of cells present in vivo, particularly if the starting population of stem/progenitor cells involved in vitro cultivation. In vitro cultivation can lead to changes (epigenetic) or even abnormalities (transformations) in the cell’s properties. An in vivo correlate must therefore be found using immunostaining and/or flow cytometry. If the latter is successful, then fluorescence-activated cell sorting (FACS) or analogous techniques can be used to isolate antigen-presenting cells. Once the cells are isolated and characterized by a variety of potential in vitro assays (colony forming, proliferation, differentiation), it will then be possible to determine if the isolated cells can self-renew and differentiate (i.e. are really stem cells). Finally, the cells should be tested for therapeutic functionality in animal models. Primary isolates (but ideally with a marker gene) need to be introduced into an appropriate animal models (injury models and testing for repair, transgenic models with gene deletions) to ensure that they function like stem cells (i.e. participate in chimera formation, integrate into appropriate niche, or differentiate into the appropriate progeny). The ability of the isolated cells to self-renew and produced known (and unknown) progeny in vivo can then be verified, thus setting the stage for eventual therapeutic trials.
Figure 4.
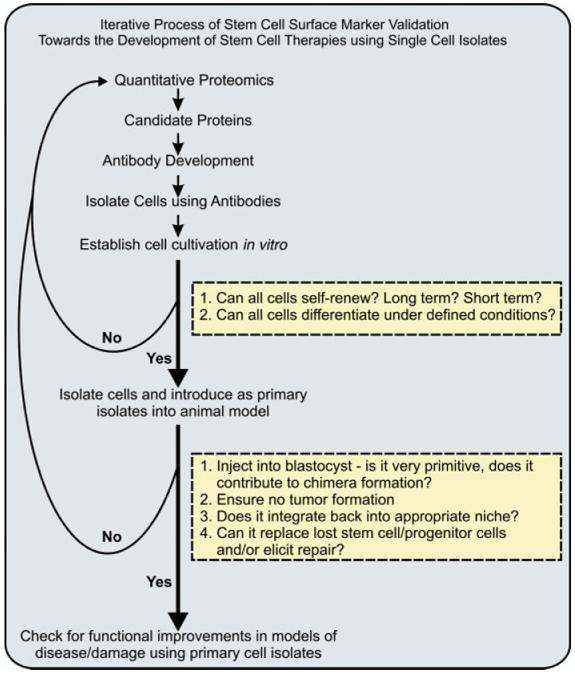
Roadmap to identifying and validating stem cell surface markers. Iterative process of discovery and validation required to define a panel of positive and negative selection markers useful in defining stem cells and purifying homogeneous cell populations.
It is through this rigorous process that stem cell-surface protein markers identified in the discovery phase become bona fide markers for future research or clinical applications. Ultimately, a panel of antibodies will be necessary to isolate cells to homogeneity that have a specific differentiation/developmental potential. It should then be possible to isolate resident stem or progenitor cells that are neither tumorogenic nor immunologically incompatible with a host. Once achieved, then specific types of stem cells my be available to improve motion or appendage movement in spinal cord injuries, improve function and ejection fraction in heart failure or after injury (myocardial infarction), or improve insulin responsiveness and normalization of glucose levels in diabetes. It is likely that the process from initial discovery to final validation will involve numerous iterations to arrive at a suitable panel of negative and positive selection markers and their respective antibodies. While this process is perhaps rather time consuming, it currently holds the best hope for identifying and isolating primary stem/progenitor cells capable of treating intractable diseases without inadvertent complications.
4 Conclusions
In conclusion, we have discussed proteomics as a tool for discovering new cell-surface protein candidates for antibody development within the context of a larger, iterative process that includes alternating between discovery and validation stages in order to define stem cell markers. The role of proteomics is threefold: to identify, quantify, and localize cell-surface proteins of interest. There are presently a number of excellent reviews that discuss various proteomic methods used for analyzing PM proteins [82-84] and lipid rafts [85]. Additionally, there are several recent comprehensive reviews on proteomics methods used for characterizing stem cells in general [86-88], but they do not specifically focus on discovering new cell-surface protein markers. Readers are encouraged to consider these reviews as a complement to the current review, which focuses specifically on proteomics methods that are especially suited for characterizing the cell-surface proteome with emphasis on the discovery of proteins that can serve as cell-surface markers for stem cell research. This review does not include a large number of publications that present proteomics studies of few/single known cell-surface proteins, but rather is limited to methods amenable for large-scale screening to identify new protein targets of interest. The limited number of publications that use proteomics for the discovery of new stem cell-surface protein markers highlights the need for novel quantitative proteomics methods that are designed to use fewer cells and are more specific for discovering bona fide novel cell-surface protein markers, to be established. MS technology is evolving fast and it can be expected that the increased sensitivity of the next generation of mass spectrometers, coupled to intelligent sample preparation workflows, will allow for experimental strategies with fewer cells required as starting material. It is clear that the amount of cells needed for current proteomic workflows is critical and sometimes limiting. Nevertheless, selected reaction monitoring (SRM) and multiple reaction monitoring (MRM) style experiments already allow for the identification of known peptides in the range of below 100 copies per Escherichia coli cell (Dr. Paola Picotti, unpublished observations). Finally, we have outlined an iterative approach towards the classification and isolation of stem cells required for clinical stem cell therapy using cell-surface protein markers. When integrated into existing stem cell research strategies, proteomics offers a new avenue of investigation towards the molecular understanding of stem cell renewal and differentiation by scratching the surface of the cell.
Acknowledgments
The authors would like to sincerely thank Damaris BauschFluck for a careful review of this manuscript. This research was supported in part by funding from the Intramural Research Program of the National Institute on Aging/NIH (KRB), NCCR Neural Plasticity and Repair (BW), and NHLBI (RLG, JEV).
Abbreviations
- CD
cluster of differentiation
- ES cell
embryonic stem cell
- HSC
hematopoietic stem cell
- MSC
mesenchymal stem cell
- PM
plasma membrane
Footnotes
The authors have declared no conflict of interest.
5 References
- [1].Gage FH. Cell therapy. Nature. 1998;392:18–24. [PubMed] [Google Scholar]
- [2].Boheler KR, Fiszman MY. Can exogenous stem cells be used in transplantation? Cells Tissues Organs. 1999;165:237–245. doi: 10.1159/000016684. [DOI] [PubMed] [Google Scholar]
- [3].Weissman IL, Anderson DJ, Gage F. Stem and progenitor cells: origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol. 2001;17:387–403. doi: 10.1146/annurev.cellbio.17.1.387. [DOI] [PubMed] [Google Scholar]
- [4].Kondo M, Wagers AJ, Manz MG, Prohaska SS, et al. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu. Rev. Immunol. 2003;21:759–806. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- [5].Blanpain C, Fuchs E. Epidermal stem cells of the skin. Annu. Rev. Cell Dev. Biol. 2006;22:339–373. doi: 10.1146/annurev.cellbio.22.010305.104357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fodor WL. Tissue engineering and cell based therapies, from the bench to the clinic: the potential to replace, repair and regenerate. Reprod. Biol. Endocrinol. 2003;1:102. doi: 10.1186/1477-7827-1-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- [8].Herzog EL, Chai L, Krause DS. Plasticity of marrow-derived stem cells. Blood. 2003;102:3483–3493. doi: 10.1182/blood-2003-05-1664. [DOI] [PubMed] [Google Scholar]
- [9].Zimmet JM, Hare JM. Emerging role for bone marrow derived mesenchymal stem cells in myocardial regenerative therapy. Basic Res. Cardiol. 2005;100:471–481. doi: 10.1007/s00395-005-0553-4. [DOI] [PubMed] [Google Scholar]
- [10].Fuchs S, Sommer L. The neural crest: understanding stem cell function in development and disease. Neurodegener. Dis. 2007;4:6–12. doi: 10.1159/000100354. [DOI] [PubMed] [Google Scholar]
- [11].Matthews VB, Yeoh GC. Liver stem cells. IUBMB Life. 2005;57:549–553. doi: 10.1080/15216540500215606. [DOI] [PubMed] [Google Scholar]
- [12].Fuchs E, Segre JA. Stem cells: a new lease on life. Cell. 2000;100:143–155. doi: 10.1016/s0092-8674(00)81691-8. [DOI] [PubMed] [Google Scholar]
- [13].Clarke D, Vegiopoulos A, Crawford A, Mucenski M, et al. In vitro differentiation of c-myb(-/-) ES cells reveals that the colony forming capacity of unilineage macrophage precursors and myeloid progenitor commitment are c-Myb independent. Oncogene. 2000;19:3343–3351. doi: 10.1038/sj.onc.1203661. [DOI] [PubMed] [Google Scholar]
- [14].Bjornson CR, Rietze RL, Reynolds BA, Magli MC, Vescovi AL. Turning brain into blood: a hematopoietic fate adopted by adult neural stem cells in vivo. Science. 1999;283:534–537. doi: 10.1126/science.283.5401.534. [DOI] [PubMed] [Google Scholar]
- [15].Gussoni E, Soneoka Y, Strickland CD, Buzney EA, et al. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature. 1999;401:390–394. doi: 10.1038/43919. [DOI] [PubMed] [Google Scholar]
- [16].Jackson KA, Mi T, Goodell MA. Hematopoietic potential of stem cells isolated from murine skeletal muscle. Proc. Natl. Acad. Sci. USA. 1999;96:14482–14486. doi: 10.1073/pnas.96.25.14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Galli R, Borello U, Gritti A, Minasi MG, et al. Skeletal myogenic potential of human and mouse neural stem cells. Nat. Neurosci. 2000;3:986–991. doi: 10.1038/79924. [DOI] [PubMed] [Google Scholar]
- [18].Petersen BE, Bowen WC, Patrene KD, Mars WM, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284:1168–1170. doi: 10.1126/science.284.5417.1168. [DOI] [PubMed] [Google Scholar]
- [19].Makino S, Fukuda K, Miyoshi S, Konishi F, et al. Cardiomyocytes can be generated from marrow stromal cells in vitro. J. Clin. Invest. 1999;103:697–705. doi: 10.1172/JCI5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Morshead CM, Benveniste P, Iscove NN, van der Kooy D. Hematopoietic competence is a rare property of neural stem cells that may depend on genetic and epigenetic alterations. Nat. Med. 2002;8:268–273. doi: 10.1038/nm0302-268. [DOI] [PubMed] [Google Scholar]
- [21].Wagers AJ, Sherwood RI, Christensen JL, Weissman IL. Little evidence for developmental plasticity of adult hematopoietic stem cells. Science. 2002;297:2256–2259. doi: 10.1126/science.1074807. [DOI] [PubMed] [Google Scholar]
- [22].Wagers AJ, Weissman IL. Plasticity of adult stem cells. Cell. 2004;116:639–648. doi: 10.1016/s0092-8674(04)00208-9. [DOI] [PubMed] [Google Scholar]
- [23].Anversa P, Leri A, Rota M, Hosoda T, et al. Concise review: stem cells, myocardial regeneration, and methodological artifacts. Stem Cells. 2007;25:589–601. doi: 10.1634/stemcells.2006-0623. [DOI] [PubMed] [Google Scholar]
- [24].Verfaillie CM. Multipotent adult progenitor cells: an update. Novartis Found. Symp. 2005;265:55–61. discussion 61-55, 92-57. [PubMed] [Google Scholar]
- [25].Jiang Y, Vaessen B, Lenvik T, Blackstad M, et al. Multipotent progenitor cells can be isolated from postnatal murine bone marrow, muscle, and brain. Exp. Hematol. 2002;30:896–904. doi: 10.1016/s0301-472x(02)00869-x. [DOI] [PubMed] [Google Scholar]
- [26].Terada N, Hamazaki T, Oka M, Hoki M, et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 2002;416:542–545. doi: 10.1038/nature730. [DOI] [PubMed] [Google Scholar]
- [27].Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, Fike JR, et al. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003;425:968–973. doi: 10.1038/nature02069. [DOI] [PubMed] [Google Scholar]
- [28].Frisen J. Stem cell plasticity? Neuron. 2002;35:415–418. doi: 10.1016/s0896-6273(02)00798-5. [DOI] [PubMed] [Google Scholar]
- [29].Zola H, Swart B, Banham A, Barry S, et al. CD molecules 2006 - human cell differentiation molecules. J. Immunol. Methods. 2007;319:1–5. doi: 10.1016/j.jim.2006.11.001. [DOI] [PubMed] [Google Scholar]
- [30].Barile L, Messina E, Giacomello A, Marban E. Endogenous cardiac stem cells. Prog. Cardiovasc. Dis. 2007;50:31–48. doi: 10.1016/j.pcad.2007.03.005. [DOI] [PubMed] [Google Scholar]
- [31].Shizuru JA, Negrin RS, Weissman IL. Hematopoietic stem and progenitor cells: clinical and preclinical regeneration of the hematolymphoid system. Annu. Rev. Med. 2005;56:509–538. doi: 10.1146/annurev.med.54.101601.152334. [DOI] [PubMed] [Google Scholar]
- [32].Bryder D, Rossi DJ, Weissman IL. Hematopoietic stem cells: the paradigmatic tissue-specific stem cell. Am. J. Pathol. 2006;169:338–346. doi: 10.2353/ajpath.2006.060312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cox J, Mann M. Is proteomics the new genomics? Cell. 2007;130:395–398. doi: 10.1016/j.cell.2007.07.032. [DOI] [PubMed] [Google Scholar]
- [34].Gallardo TD, Hammer RE, Garry DJ. RNA amplification and transcriptional profiling for analysis of stem cell populations. Genesis. 2003;37:57–63. doi: 10.1002/gene.10223. [DOI] [PubMed] [Google Scholar]
- [35].Ip JE, Wu Y, Huang J, Zhang L, et al. Mesenchymal stem cells use integrin beta1 not CXC chemokine receptor 4 for myocardial migration and engraftment. Mol. Biol. Cell. 2007;18:2873–2882. doi: 10.1091/mbc.E07-02-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fok EY, Zandstra PW. Shear-controlled single-step mouse embryonic stem cell expansion and embryoid body-based differentiation. Stem Cells. 2005;23:1333–1342. doi: 10.1634/stemcells.2005-0112. [DOI] [PubMed] [Google Scholar]
- [37].Davey RE, Zandstra PW. Spatial organization of embryonic stem cell responsiveness to autocrine gp130 ligands reveals an autoregulatory stem cell niche. Stem Cells. 2006;24:2538–2548. doi: 10.1634/stemcells.2006-0216. [DOI] [PubMed] [Google Scholar]
- [38].Kirouac DC, Zandstra PW. Understanding cellular networks to improve hematopoietic stem cell expansion cultures. Curr. Opin. Biotechnol. 2006;17:538–547. doi: 10.1016/j.copbio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- [39].Ahram M, Litou ZI, Fang R, Al-Tawallbeh G. Estimation of membrane proteins in the human proteome. In Silico Biol. 2006;6:379–386. [PubMed] [Google Scholar]
- [40].Minezaki Y, Homma K, Nishikawa K. Intrinsically disordered regions of human plasma membrane proteins preferentially occur in the cytoplasmic segment. J. Mol. Biol. 2007;368:902–913. doi: 10.1016/j.jmb.2007.02.033. [DOI] [PubMed] [Google Scholar]
- [41].Jeong JA, Lee Y, Lee W, Jung S, et al. Proteomic analysis of the hydrophobic fraction of mesenchymal stem cells derived from human umbilical cord blood. Mol. Cells. 2006;22:36–43. [PubMed] [Google Scholar]
- [42].Foster LJ, Zeemann PA, Li C, Mann M, et al. Differential expression profiling of membrane proteins by quantitative proteomics in a human mesenchymal stem cell line undergoing osteoblast differentiation. Stem Cells. 2005;23:1367–1377. doi: 10.1634/stemcells.2004-0372. [DOI] [PubMed] [Google Scholar]
- [43].Lund TC, Anderson LB, McCullar V, Higgins L, et al. iTRAQ Is a useful method to screen for membrane-bound proteins differentially expressed in human natural killer cell types. J. Proteome Res. 2007;6:644–653. doi: 10.1021/pr0603912. [DOI] [PubMed] [Google Scholar]
- [44].Osterhues A, Liebmann S, Schmid M, Buk D, et al. Stem cells and experimental leukemia can be distinguished by lipid raft protein composition. Stem Cells Dev. 2006;15:677–686. doi: 10.1089/scd.2006.15.677. [DOI] [PubMed] [Google Scholar]
- [45].Wu CC, MacCoss MJ, Howell KE, Yates JR., 3rd A method for the comprehensive proteomic analysis of membrane proteins. Nat. Biotechnol. 2003;21:532–538. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- [46].Chaney LK, Jacobson BS. Coating cells with colloidal silica for high yield isolation of plasma membrane sheets and identification of transmembrane proteins. J. Biol. Chem. 1983;258:10062–10072. [PubMed] [Google Scholar]
- [47].Oh P, Li Y, Yu J, Durr E, et al. Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature. 2004;429:629–635. doi: 10.1038/nature02580. [DOI] [PubMed] [Google Scholar]
- [48].Rahbar AM, Fenselau C. Integration of Jacobson’s pellicle method into proteomic strategies for plasma membrane proteins. J. Proteome Res. 2004;3:1267–1277. doi: 10.1021/pr040004t. [DOI] [PubMed] [Google Scholar]
- [49].Rahbar AM, Fenselau C. Unbiased examination of changes in plasma membrane proteins in drug resistant cancer cells. J. Proteome Res. 2005;4:2148–2153. doi: 10.1021/pr0502370. [DOI] [PubMed] [Google Scholar]
- [50].Zhang H, Yan W, Aebersold R. Chemical probes and tandem mass spectrometry: a strategy for the quantitative analysis of proteomes and subproteomes. Curr. Opin. Chem. Biol. 2004;8:66–75. doi: 10.1016/j.cbpa.2003.12.001. [DOI] [PubMed] [Google Scholar]
- [51].Perosa F, Luccarelli G, Neri M, De Pinto V, et al. Evaluation of biotinylated cells as a source of antigens for characterization of their molecular profile. Int. J. Clin. Lab. Res. 1998;28:246–251. doi: 10.1007/s005990050053. [DOI] [PubMed] [Google Scholar]
- [52].Castronovo V, Waltregny D, Kischel P, Roesli C, et al. A chemical proteomics approach for the identification of accessible antigens expressed in human kidney cancer. Mol. Cell. Proteomics. 2006;5:2083–2091. doi: 10.1074/mcp.M600164-MCP200. [DOI] [PubMed] [Google Scholar]
- [53].Scheurer SB, Rybak JN, Roesli C, Brunisholz RA, et al. Identification and relative quantification of membrane proteins by surface biotinylation and two-dimensional peptide mapping. Proteomics. 2005;5:2718–2728. doi: 10.1002/pmic.200401163. [DOI] [PubMed] [Google Scholar]
- [54].Zhao Y, Zhang W, Kho Y. Proteomic analysis of integral plasma membrane proteins. Anal. Chem. 2004;76:1817–1823. doi: 10.1021/ac0354037. [DOI] [PubMed] [Google Scholar]
- [55].Peirce MJ, Wait R, Begum S, Saklatvala J, Cope AP. Expression profiling of lymphocyte plasma membrane proteins. Mol. Cell. Proteomics. 2004;3:56–65. doi: 10.1074/mcp.M300064-MCP200. [DOI] [PubMed] [Google Scholar]
- [56].Nunomura K, Nagano K, Itagaki C, Taoka M, et al. Cell surface labeling and mass spectrometry reveal diversity of cell surface markers and signaling molecules expressed in undifferentiated mouse embryonic stem cells. Mol. Cell. Proteomics. 2005;4:1968–1976. doi: 10.1074/mcp.M500216-MCP200. [DOI] [PubMed] [Google Scholar]
- [57].Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- [58].Sidibe A, Yin X, Tarelli E, Xiao Q, et al. Integrated membrane protein analysisof mature and embryonic stem cell-derived smooth muscle cells using a novel combination of Cy-dye/biotin labelling. Mol. Cell. Proteomics. 2007:M600433–MCP600200. doi: 10.1074/mcp.M600433-MCP200. [DOI] [PubMed] [Google Scholar]
- [59].Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- [60].Gahmberg CG, Tolvanen M. Why mammalian cell surface proteins are glycoproteins. Trends Biochem. Sci. 1996;21:308–311. [PubMed] [Google Scholar]
- [61].Docheva D, Popov C, Mutschler W, Schieker M. Human mesenchymal stem cells in contact with their environment: surface characteristics and the integrin system. J. Cell Mol. Med. 2007;11:21–38. doi: 10.1111/j.1582-4934.2007.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Liu G, Zhao Y. Toll-like receptors and immune regulation: their direct and indirect modulation on regulatory CD4+ CD25+ T cells. Immunology. 2007;122:149–156. doi: 10.1111/j.1365-2567.2007.02651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Adewumi O, Aflatoonian B, Ahrlund-Richter L, Amit M, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 2007;25:803–816. doi: 10.1038/nbt1318. [DOI] [PubMed] [Google Scholar]
- [64].Yanagisawa M, Yu RK. The expression and functions of glycoconjugates in neural stem cells. Glycobiology. 2007;17:57R–74R. doi: 10.1093/glycob/cwm018. [DOI] [PubMed] [Google Scholar]
- [65].Nurcombe V, Cool SM. Heparan sulfate control of proliferation and differentiation in the stem cell niche. Crit. Rev. Eukaryot. Gene Expr. 2007;17:159–171. doi: 10.1615/critreveukargeneexpr.v17.i2.50. [DOI] [PubMed] [Google Scholar]
- [66].Haylock DN, Nilsson SK. The role of hyaluronic acid in hemopoietic stem cell biology. Regen. Med. 2006;1:437–445. doi: 10.2217/17460751.1.4.437. [DOI] [PubMed] [Google Scholar]
- [67].Stanley P. Regulation of Notch signaling by glycosylation. Curr. Opin. Struct. Biol. 2007;17:530–535. doi: 10.1016/j.sbi.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wearne KA, Winter HC, Goldstein IJ. Temporal changes in the carbohydrates expressed on BG01 human embryonic stem cells during differentiation as embryoid bodies. Glycoconj. J. 2007;25:121–136. doi: 10.1007/s10719-007-9064-x. [DOI] [PubMed] [Google Scholar]
- [69].Wearne KA, Winter HC, O’Shea K, Goldstein IJ. Use of lectins for probing differentiated human embryonic stem cells for carbohydrates. Glycobiology. 2006;16:981–990. doi: 10.1093/glycob/cwl019. [DOI] [PubMed] [Google Scholar]
- [70].Hemmoranta H, Satomaa T, Blomqvist M, Heiskanen A, et al. N-glycan structures and associated gene expression reflect the characteristic N-glycosylation pattern of human hematopoietic stem and progenitor cells. Exp. Hematol. 2007;35:1279–1292. doi: 10.1016/j.exphem.2007.05.006. [DOI] [PubMed] [Google Scholar]
- [71].Zhang H, Li X.-j., Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- [72].Zhou Y, Aebersold R, Zhang H. Isolation of N-linked glycopeptides from plasma. Anal. Chem. 2007;79:5826–5837. doi: 10.1021/ac0623181. [DOI] [PubMed] [Google Scholar]
- [73].Zhang H, Liu AY, Loriaux P, Wollscheid B, et al. Masss pectrometric detection of tissue proteins in plasma. Mol. Cell. Proteomics. 2007;6:64–71. doi: 10.1074/mcp.M600160-MCP200. [DOI] [PubMed] [Google Scholar]
- [74].Zhang H, Loriaux P, Eng J, Campbell D, et al. UniPep - a database for human N-linked glycosites: a resource for biomarker discovery. Genome Biol. 2006;7:R73. doi: 10.1186/gb-2006-7-8-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ong SE, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 2005;1:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- [76].Stahl-Zeng J, Lange V, Ossola R, Eckhardt K, et al. High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol. Cell. Proteomics. 2007;6:1809–1817. doi: 10.1074/mcp.M700132-MCP200. [DOI] [PubMed] [Google Scholar]
- [77].Mueller LN, Rinner O, Schmidt A, Letarte S, et al. SuperHirn - a novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics. 2007;7:3470–3480. doi: 10.1002/pmic.200700057. [DOI] [PubMed] [Google Scholar]
- [78].Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- [79].Ross PL, Huang YN, Marchese JN, Williamson B, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- [80].Gygi SP, Rist B, Gerber SA, Turecek F, et al. Quantitative analysis of complex protein mixtures using isotopecoded affinity tags. Nat. Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- [81].Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 2007;389:1017–1031. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- [82].Speers AE, Wu CC. Proteomics of integral membrane proteins-theory and application. Chem. Rev. 2007;107:3687–3714. doi: 10.1021/cr068286z. [DOI] [PubMed] [Google Scholar]
- [83].Macher BA, Yen TY. Proteins at membrane surfaces-a review of approaches. Mol. Biosyst. 2007;3:705–713. doi: 10.1039/b708581h. [DOI] [PubMed] [Google Scholar]
- [84].Josic D, Clifton JG. Mammalian plasma membrane proteomics. Proteomics. 2007;7:3010–3029. doi: 10.1002/pmic.200700139. [DOI] [PubMed] [Google Scholar]
- [85].Sprenger RR, Horrevoets AJ. The ins and outs of lipid domain proteomics. Proteomics. 2007;7:2895–2903. doi: 10.1002/pmic.200700189. [DOI] [PubMed] [Google Scholar]
- [86].Baharvand H, Fathi A, van Hoof D, Salekdeh GH. Concise review: trends in stem cell proteomics. Stem Cells. 2007;25:1888–1903. doi: 10.1634/stemcells.2007-0107. [DOI] [PubMed] [Google Scholar]
- [87].Vodicka P, Skalnikova H, Kovarova H. The characterization of stem cell proteomes. Curr. Opin. Mol. Ther. 2006;8:232–239. [PubMed] [Google Scholar]
- [88].Van Hoof D, Mummery CL, Heck AJ, Krijgsveld J. Embryonic stem cell proteomics. Expert Rev. Proteomics. 2006;3:427–437. doi: 10.1586/14789450.3.4.427. [DOI] [PubMed] [Google Scholar]