

Abstract
Type I diabetes (T1D) is a T cell-mediated autoimmune disease characterized by loss of tolerance to islet autoantigens, leading to the destruction of insulin-producing beta cells. Peripheral tolerance to self is maintained in health through several regulatory mechanisms, including a population of CD4+CD25hi naturally occurring regulatory T cells (Tregs), defects in which could contribute to loss of self-tolerance in patients with T1D. We have reported previously that near to T1D onset, patients demonstrate a reduced level of suppression by CD4+CD25hi Tregs of autologous CD4+CD25− responder cells. Here we demonstrate that this defective regulation is also present in subjects with long-standing T1D (> 3 years duration; P = 0·009). No difference was observed in forkhead box P3 or CD127 expression on CD4+CD25hi T cells in patients with T1D that could account for this loss of suppression. Cross-over co-culture assays demonstrate a relative resistance to CD4+CD25hi Treg-mediated suppression within the CD4+CD25− T cells in all patients tested (P = 0·002), while there appears to be heterogeneity in the functional ability of CD4+CD25hi Tregs from patients. In conclusion, this work demonstrates that defective regulation is a feature of T1D regardless of disease duration and that an impaired ability of responder T cells to be suppressed contributes to this defect.
Keywords: autoimmunity, diabetes, immune regulation, regulatory T cells
Introduction
Type 1 diabetes (T1D) is a T cell-mediated disease in which insulin-producing pancreatic islet beta cells are destroyed selectively, resulting in a loss of insulin production and a subsequent inability to control glucose metabolism [1]. Autoreactive T cells specific for the islet autoantigens insulin, glutamic acid decarboxylase (GAD65) and the islet tyrosine phosphatase IA-2 have been isolated from patients with T1D [2–5]. Several lines of evidence suggest that these cells are involved in disease pathogenesis, including their presence in islet mononuclear infiltrates, delay of disease progression using T cell-inhibiting agents [6,7] and their ability to transfer disease in the murine model of T1D – the non-obese diabetic (NOD) mouse [1]. Islet autoreactive T cells are also present in normal non-diabetogenic individuals [3] suggesting that, in the normal immune system, mechanisms must operate in the periphery to control the potentially autoreactive cells.
The CD4+CD25hi regulatory T cell population is essential for maintenance of peripheral tolerance in vivo, as highlighted by animals and humans in which a natural or experimentally induced deficit in this population exists. Deficiency of this T regulatory (Treg) population leads to the development of a range of autoimmune and lymphoproliferative disorders, including autoimmune diabetes [8–10]. Tregs can also control the responses of autoreactive T cells stimulated in vitro[11–13]. Much evidence has been collected to suggest an important role for regulatory T cells in the control of islet autoreactivity in both mouse and man [14–17].
Initial studies in human patients with T1D suggested that there may be a reduction in the frequency of CD4+CD25hi regulatory T cells in these individuals [15]. This finding was not confirmed by subsequent studies, of which some used more reliable markers to identify Tregs and appropriately age-matched control subjects [14,16,18,19]. However, careful analysis of Treg function indicated that the suppression of autologous responder T cells by CD4+CD25hi Tregs from individuals with newly diagnosed T1D is reduced significantly compared with that observed in age-matched control subjects [16], findings confirmed subsequently by Brusko and colleagues [14]. In the present study, we examined whether this defective regulation was due to changes in the immune system close to diagnosis of T1D [20], or represented a stable phenotype also present in individuals with long-standing T1D (L/S T1D). In addition, we sought to determine whether the reduced regulation that we observe in vitro could be the result of a reduced frequency of ‘true’ regulatory T cells within the CD25hi population by using additional markers of the regulatory T cell lineage, such as the transcription factor forkhead box P3 (FoxP3) [8,10,21–23] and the interleukin (IL)-7Rα chain CD127 [24,25]. Finally, we analysed the contribution that both responder and regulatory T cells make to defective in vitro regulation, using cross-over co-culture assays.
Materials and methods
Subjects
Peripheral blood samples were obtained from 44 patients with T1D and 44 control subjects. Long-standing disease was defined as T1D duration of > 3 years and control subjects had no family history of T1D. Eleven patients with L/S T1D [mean ± standard deviation (s.d.), age 43·7 years ± 14·4] and 12 age- and human leucocyte antigen (HLA)-matched healthy control subjects (age 37·2 years ± 13·1) were recruited for analysis of Treg frequency and function. Thirteen patients with L/S T1D (age 40·0 years ± 8·6) and 13 age- and HLA-matched control subjects (age 34 years ± 11·4) were recruited for analysis of FoxP3 expression. Fifteen patients with L/S T1D (age 41·5 years ± 14·4) were recruited for analysis of CD127 expression along with 15 age- and HLA-matched healthy control subjects (age 34·8 years ± 10·6). Finally, five patients with L/S T1D (age 39·2 years ± 8) were recruited for cross-over functional analysis along with four control subjects (age 39·2 years ± 13·4), forming five pairs of age- and HLA-matched subjects (one healthy control subject was paired with two patients with T1D). Peripheral blood mononuclear cells (PBMC) were obtained by density gradient centrifugation (Lymphoprep; Axis-Shield PoC AS, Oslo, Norway) as described previously [26]. Ethical approval for this study was granted by the local ethics committee and informed consent obtained.
Monoclonal antibodies
Fluorescein isothiocyanate (FITC)-conjugated anti-CD3 (clone SK7), phycoerythrin (PE)-conjugated anti-CD127, peridin-chlorophyll protein (PerCP)-conjugated anti-CD4 (clone L200) and allophycocyanin (APC)-labelled anti-CD4 (clone RPA-T4; BD Pharmingen, San Diego, CA, USA), PE-labelled anti-CD25 (clone MEM-181), Alexa Fluor 647-conjugated anti-CD25 (clone MEM-181) antibodies (Serotec, Oxford, UK), as well as APC-labelled anti-FoxP3 (clone PCH101; eBioscience, San Diego, CA, USA) and relevant isotype- and fluorochrome-matched control antibodies, were used in this study. Antibody concentrations used were based on the manufacturers' recommendations and initial optimization studies.
Cell separation
CD4+ T cells were isolated from PBMCs by negative selection using magnetic cell sorting technology (MACS; Miltenyi Biotec, Bisley, UK). CD25hi T cells were isolated from the CD4+ populations obtained using 50% of the manufacturer's recommended concentration of anti-CD25 microbeads (Miltenyi Biotec). The CD25− T cell population was isolated from the resulting CD25−/lo T cells using 150% of the manufacturer's recommended concentration of anti-CD25 microbeads. T cell depleted accessory cells were isolated from PBMC by negative selection using anti-CD3 microbeads (Miltenyi Biotec) and then irradiated at 3000 rad. The purity of all cell populations isolated was determined by flow cytometry using anti-CD3, anti-CD4 and anti-CD25 antibodies as described below and was routinely > 90%.
Cell stimulation and suppression assays
Cell stimulation and suppression assays were performed by culturing CD4+CD25− (5 × 103/well) with CD4+CD25hi T cells at various ratios (0:1, 1:0 and 1:1) in the presence of 5 × 104 irradiated CD3 depleted accessory cells. Cells were stimulated using plate-bound anti-CD3 (clone UCHT1) and soluble anti-CD28 (clone CD28·2) antibodies (BD Pharmingen). Briefly, plates were incubated with 50 µl/well phosphate-buffered saline (PBS) that contained 1 µg/ml anti-CD3 antibody for 4 h at 37°C and then washed twice in PBS. Cells were cultured in RPMI-1640 Glutamax 25 mM HEPES media supplemented with 100 µg/ml penicillin/streptomycin (all from Invitrogen, Paisley, UK) and 5% AB serum (PAA Laboratories, Yeovil, UK).
All cell culture conditions were carried out in triplicate. On day 5 of culture, 100 µl of supernatant was removed from each well and 100 µl of fresh medium that contained 0·5 µCi of [3H]-thymidine was added for the final 16 h of culture before harvesting. Percentage suppression was calculated as 100-{[counts per minute (cpm) in co-cultures/cpm in CD4+CD25− cultures] × 100}.
Flow cytometry
Immunofluorescence staining was performed on 0·5–1 × 106 PBMC or 1 × 105 MACS isolated populations after washing twice in cold wash buffer [PBS, 2 mM ethylenediamine tetraacetic acid, 1% fetal calf serum; PAA Laboratories]. Staining with anti-CD3, anti-CD4, anti-CD25 and anti-CD127 was carried out in a final volume of 100 µl on ice for 15 min. For investigation of intracellular FoxP3 expression, PBMC were stained with FITC-labelled anti-CD3, PerCP-labelled anti-CD4 and PE-conjugated anti-CD25 as described above, fixed and permeabilized according to the manufacturer's instructions (anti-human FoxP3 staining set; eBioscience,) stained with APC-labelled anti-FoxP3 for 30 min at 4°C, washed twice in wash buffer and analysed. PBMC were gated on their forward- and side-scatter properties, CD3+CD4+ cells were gated using the relevant antibodies. The appropriate isotype- and fluorochrome-matched isotype control antibodies were used to determine the percentage of positive cells. A minimum of 100 000 events were acquired and analysed on a fluorescence activated cell sorter (FACSCalibur) (Becton Dickinson, Mountain View, CA, USA) using CellQuest (Becton Dickinson) and FlowJo (Tree Star Inc., Ashland, OR, USA) software.
Statistical analysis
Statistical significance was determined using Student's two-tailed t-test after data sets were determined not to be significantly different from a Gaussian distribution. P-values of less than 0·05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Frequency of CD4+CD25hi T cells
A clear CD4loCD25hi population was visible in the majority of donors and this was used to define CD4+CD25hi Tregs (Fig. 1a). We found no significant difference in the number of CD4+ T cells expressing high levels of CD25 between patients with L/S T1D (mean ± s.d.; 5·4% ± 1·3) and control subjects (5·0% ± 1·3; Fig. 1b). Furthermore, no difference was observed between the patient and control group in the frequency of CD25hi cells within the CD4+ T cell population, regardless of the CD25hi definition utilized [CD25 fluorescence intensity (MFI) of over 100, the top 2% of CD25+ cells, CD4+ T cells expressing CD25 at a level higher than that of CD4− T cells; data not shown].
Fig. 1.
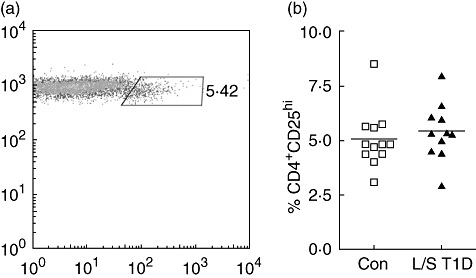
Isolated populations of CD4-positive cells were stained for expression of CD3, CD4 and CD25 and analysed by flow cytometry. CD25hi T cells were defined as those with a slightly lowered expression of CD4 (a). The percentage of CD4+ T cells in control subjects (squares) and patients with long-standing T1D (triangles) which were CD25hi. Each point represents an individual and mean values are indicated with a horizontal line (b).
Suppressive function of CD4+CD25+ T cells
CD4+CD25− and CD4+CD25hi T cell populations were isolated using magnetic bead technology and a previously described optimized protocol [16]. There was no difference in the purity or levels of CD25 expression of either population between patients with L/S T1D and control subjects (data not shown). As expected, CD4+CD25− T cells isolated from both groups proliferated well in response to stimulation with anti-CD3 and anti-CD28 antibodies and to a similar extent in patients with T1D (mean ± s.d.; 85 385 c.p.m. ± 23 859) and controls (67 567 cpm ± 28 141; Fig. 2a). In contrast, CD4+CD25hi T cells isolated from both subject groups were relatively anergic to this stimulation [mean ± s.d. in the patient group, 13 473 cpm ± 9294; control subjects 8350 cpm ± 8447; Fig. 2b; P = not significant (n.s.)]. However, when CD4+CD25− and autologous CD4+CD25hi T cells were co-cultured at a 1 : 1 ratio a significant reduction in the level of suppression of proliferation was observed in the co-cultures of patients with L/S T1D (37·8% ± 14·5) in comparison with control subjects (56·3% ± 15·9; P = 0·0086; Fig. 2c). When co-cultures were stimulated with lower levels of T cell receptor (TCR) stimulation (0·3 µg/ml anti-CD3 antibody) suppression was observed in the patient group at similar levels to that in controls (L/S T1D, 51·1% ± 35; control, 49·4% ± 32·3; data not shown).
Fig. 2.
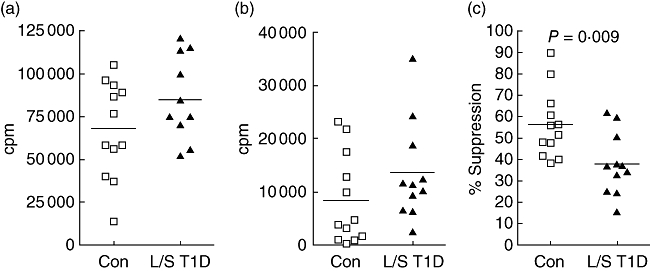
Proliferation of single cultures of CD4+CD25− (a) and CD4+CD25hi (b) T cells from control subjects (squares) and patients with long-standing T1D (triangles) was assessed by tritiated thymidine incorporation. The percentage suppression of proliferation observed in co-cultures of CD4+CD25− and CD4+CD25hi T cells was calculated (c), P-values were calculated using Student's t-test. Cells were stimulated with 1 µg/ml anti-CD3 and 5 µg/ml anti-CD28 antibodies for 5 days. Each point represents mean counts per minute (cpm) in triplicate wells from one individual. Mean values are indicated with a horizontal line.
Expression of FoxP3 and CD127 in CD4+CD25hi T cells
We observed no difference in the percentage of CD4+CD25hi cells co-expressing FoxP3 between patients with L/S T1D (mean ± s.d.; 91·7% ± 5·3) and control subjects (93·3% ± 3·5; Fig. 3a). Furthermore, no difference in the level of FoxP3 (as judged by MFI) was observed (data not shown). The lack of a standard definition of the CD4+CD25hi T cell population led us to examine the levels of FoxP3 expression throughout the top 10% (90th–99th centile) of CD25+ cells within the CD4+ population, within which the regulatory population is expected to be contained. This showed that there was no difference in the percentage of cells co-expressing FoxP3 between patients and control subjects, regardless of the level of CD25 expression (Fig. 3b).
Fig. 3.
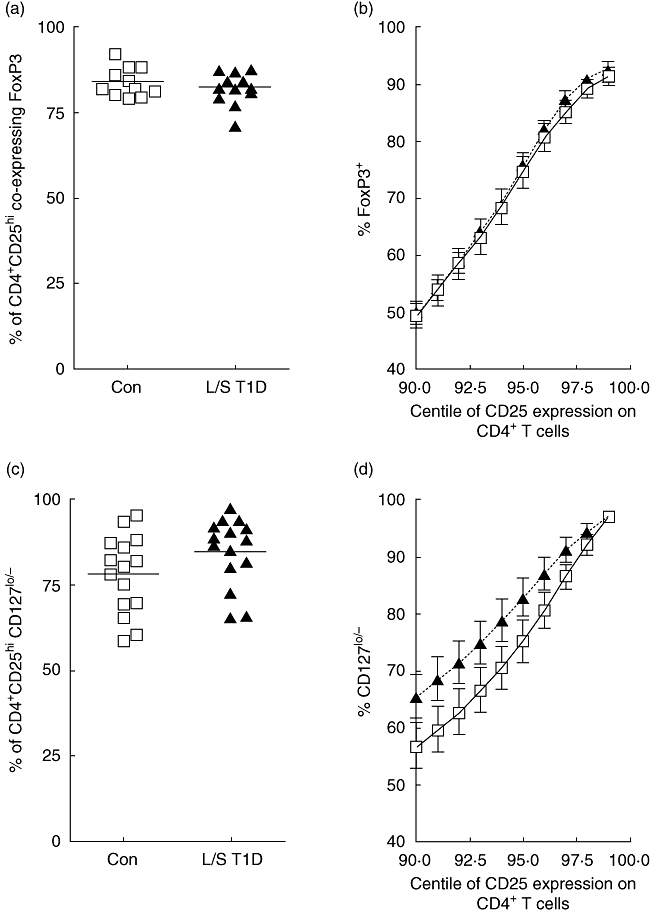
Peripheral blood mononuclear cells (PBMC) were stained using anti-CD3, anti-CD4, anti-CD25 and anti-forkhead box P3 (FoxP3) or anti-CD127 antibodies and analysed by fluorescence activated cell sorter (FACS). The CD25hi population was defined as those with a lowered expression of CD4 and this population was analysed for expression of FoxP3 or CD127. The percentage of FoxP3+ (a) or CD127lo/− (c) cells in the CD4+CD25hi population was determined in control subjects (squares) and patients with long-standing T1D (triangles). Each point represents one individual and mean values are indicated by a horizontal line. The percent of FoxP3-expressing cells (b) or CD127lo/− cells (d) in the control (squares, solid line) and patient (triangles, dotted line) groups within the top 10% of CD25 expression within the CD4+ T cell compartment is shown. Each point with error bars represents the mean and standard error of the mean of each group.
Similarly, no difference in the frequency of CD127lo/− cells within the CD4+CD25hi population was observed between patients (mean ± s.d.; 84·8% ± 9·9) and control subjects (78·1% ± 11·3; Fig. 3c). When the top 10% (90th–99th centile) of CD25-expressing cells within the CD4+ population was analysed, no significant difference in CD127lo/− expression was observed (Fig. 3d).
Suppression in cross-over co-cultures
In order to dissect further the loss of suppression in patients with T1D, cross-over co-culture suppression assays were carried out. Control subjects and patients with L/S T1D were analysed as pairs. There was no significant difference in the proliferation levels observed in single cultures of CD4+CD25− or CD4+CD25hi T cells from either group, nor was there any difference in the purity of the isolated T cell populations (data not shown). In accordance with previous results, suppression levels in autologous co-cultures of CD4+CD25− and CD4+CD25hi T cells were reduced in the patients with T1D (mean ± s.d.; 39·8% ± 20·4) when compared with control subjects (52·8% ± 23; P = 0·017; Fig. 4a). When CD4+CD25− T cells isolated from patients with T1D were co-cultured with CD4+CD25hi T cells from control subjects, significantly reduced levels of suppression (30% ± 26·5) were observed in all cases when compared with the control co-cultures (P = 0·0022; Fig. 4b). When CD4+CD25− T cells from control subjects and CD4+CD25hi from patients with T1D were co-cultured, levels of suppression (36·3% ± 28) were not significantly different to levels observed in control autologous co-cultures (52·8% ± 23; Fig. 4c). However, some individuals did show reduced suppressive function in this population suggesting, that there may be heterogeneity within the human T1D population.
Fig. 4.
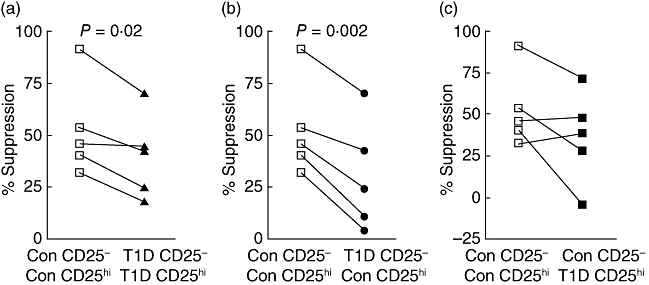
CD4+CD25− and CD4+CD25hi T cells were isolated using magnetic bead technology from paired sets of control subjects and patients with long-standing T1D. Control autologous (open squares; a, b, c) and patients with long-standing T1D autologous (closed triangles; a) co-cultures and cross-over co-cultures of control CD4+CD25hi and patient CD4+CD25− T cells (closed circles; b) and patient CD4+CD25hi and control CD4+CD25− T cells (closed squares; c) were set up. Each set of joined points represents one pair of subjects. P-values were calculated using Student's t test.
Discussion
In agreement with the majority of published studies [14,16,18,19], we found no significant difference in the frequency of CD4+CD25hi T cells in patients with T1D. We chose to define the CD25hi regulatory population as those cells expressing the highest levels of CD25 with a slightly lowered expression of CD4, as this population was clearly visible in the majority of individuals studied. However, even when other commonly used definitions were employed no significant difference was noted. These results suggest strongly that the frequency of regulatory T cells in patients with T1D is not abnormal. However, this will remain an unresolved question until the discovery of definitive phenotypic marker(s) of regulatory T cells.
Our previous report [16] demonstrated a reduced level of suppression in co-cultures of cells isolated from patients with recent onset T1D when compared with healthy control subjects and was confirmed by Brusko et al. using a mixed cohort of newly diagnosed and long-standing patients [14]. However, no difference was found in a subsequent study by Putnam and colleagues using only patients with long-standing T1D [18]. The current study suggests that defective regulation is a feature of T1D regardless of disease duration. There are several possible reasons for the seeming discrepancies between these studies, including differences in isolation technique and the T cell stimulation conditions employed during the suppression assay. This study and our previous report isolated T cell populations using magnetic bead technology, whereas the analyses by Putnam and Brusko isolated populations by FACS sorting, suggesting that it is unlikely that this could be the sole cause of the inconsistency. A more attractive possibility is that the in vitro stimulation conditions determine whether a difference in suppression is observed between the patients with T1D and controls. The level of TCR stimulation is known to affect both the regulatory T cell's ability to suppress and the effector T cell's susceptibility to regulation [27]. The study by Putnam et al., in which no reduction in suppression was observed, used relatively low levels of anti-CD3 and anti-CD28 antibody stimulation [18]. Similarly, in the present study we observed similar suppression in both T1D and control individuals when a lower level of stimulation was used. Our conclusion is that the stimulation conditions are critical for exposing the regulatory failure in patients with T1D.
The CD4+CD25hi population is known to be a heterogeneous mixture of cells containing recently activated effector and regulatory T cells. Functional Tregs have been demonstrated as expressing high levels of the transcription factor FoxP3 and low levels of the IL-7Rα chain, CD127 [10,23–25]. We therefore examined the CD4+CD25hi population using these more precise markers to identify ‘true’ Tregs to determine whether the defective regulation in T1D may be due to increased levels of non-Tregs in the CD4+CD25hi population. Moreover, we examined the expression of CD127 and FoxP3 across the full range of CD25 expression levels. However, no differences in CD127 or FoxP3 expression in the CD4+CD25hi population were observed. This suggests that an alteration in the balance of Tregs and activated T cells does not contribute to the defective suppression observed in T1D.
In the absence of any obvious alterations in the percentage of regulatory T cells within the CD4+CD25hi T cell population which could account for the loss of suppression in patients with T1D, cross-over co-culture experiments were performed to determine which T cell population contributes to this phenomenon in these individuals. Accessory cells were always isolated from control donors to minimize any affect these cells may have on the outcome of in vitro co-culture suppression assays. These studies revealed a significant resistance to suppression in CD4+CD25− responder T cells isolated from patients with T1D when CD4+CD25hi regulatory T cells isolated from control subjects were used. In addition it appears that, in the majority of cases, regulatory T cells isolated from T1D patients were capable of suppressing the proliferation of responder T cells isolated from control donors. This suggests that the reduced levels of regulation observed in autologous co-cultures of cells isolated from patients is due to reduced susceptibility of the responder T cell population to regulation.
To our knowledge, these are the first studies to demonstrate reduced susceptibility of responder T cells to regulation in human T1D, but similar findings have been reported in the NOD mouse [28,29] and a TCR transgenic model of autoimmune diabetes [30]. Overt diabetes in the NOD mouse model is preceded by a prolonged period of insulitis, and it has been suggested that during this time a ‘battle’ ensues between diabetogenic effector T cells and regulatory T cells with the eventual defeat of the regulatory population leading to diabetes [17]. You et al. performed in vitro and in vivo cross-over experiments with cells isolated from diabetic and prediabetic NOD mice and found that responder T cells isolated from diabetic mice were less susceptible to regulatory T cell control. CD4+CD25+ regulatory T cells isolated from diabetic donors were still capable of suppression of responder T cells from prediabetic donors [28]. Therefore, a progressive, qualitative difference in the responder T cell populations emerges as diabetes-prone mice progress towards disease onset, implying that defective regulation is a progressive disorder rather than an intrinsic defect. It remains to be established, using prediabetic human subjects, whether the same is true in man. This differs from studies in patients with multiple sclerosis and autoimmune polyglandular syndrome type II [31–33], which indicate a reduced ability of the regulatory population to suppress. Therefore, while the observed phenotype (loss of suppression in vitro) is similar, it may have different causes in different diseases. It is noteworthy that a subset of the patients analysed on cross-over assays in the present study have reduced regulatory T cell function while others did not. This apparent heterogeneity in the T1D patient population merits further study, including an analysis of potential contributory gene polymorphisms, such as IL-2RA and CTLA-4[34,35].
In conclusion, the results presented in this study suggest that defective regulation is a feature of T1D regardless of disease duration and that it is the result of a lowered susceptibility to regulation in the CD4+CD25− responder T cell population. It will be important to discern whether this reduced sensitivity to regulation is present at a cell intrinsic or population level in order to tailor regulatory T cell therapy for the human T1D population [36].
Acknowledgments
This work was supported by grants from Diabetes UK BDA:RD03/0002727 and BDA:RD04/0002877. The authors acknowledge financial support from the Department of Health via the National Institute for Health Research comprehensive Biomedical Research Centre award to Guy's and St Thomas' National Health Service Foundation Trust in partnership with King's College London. We thank Amanda Bishop and Samantha Archibald for phlebotomy services and also gratefully acknowledge patients and control subjects for blood donation.
References
- 1.Castano L, Eisenbarth GS. Type-I diabetes: a chronic autoimmune disease of human, mouse and rat. Annu Rev Immunol. 1990;8:647–79. doi: 10.1146/annurev.iy.08.040190.003243. [DOI] [PubMed] [Google Scholar]
- 2.Di Lorenzo TP, Peakman M, Roep BO. Translational mini-review series on type 1 diabetes: systematic analysis of T cell epitopes in autoimmune diabetes. Clin Exp Immunol. 2007;148:1–16. doi: 10.1111/j.1365-2249.2006.03244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arif S, Tree TI, Astill TP, et al. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest. 2004;113:451–63. doi: 10.1172/JCI19585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peakman M, Stevens EJ, Lohmann T, et al. Naturally processed and presented epitopes of the islet cell autoantigen IA-2 eluted from HLA-DR4. J Clin Invest. 1999;104:1449–57. doi: 10.1172/JCI7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tree TI, Peakman M. Autoreactive T cells in human type 1 diabetes. Endocrinol Metab Clin North Am. 2004;33:113–33. doi: 10.1016/S0889-8529(03)00081-1. [DOI] [PubMed] [Google Scholar]
- 6.Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–8. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 7.Feutren G, Papoz L, Assan R, et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Results of a multicentre double-blind trial. Lancet. 1986;2:119–24. doi: 10.1016/s0140-6736(86)91943-4. [DOI] [PubMed] [Google Scholar]
- 8.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 9.Gavin MA, Torgerson TR, Houston E, et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci USA. 2006;103:6659–64. doi: 10.1073/pnas.0509484103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 11.Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW. Autoreactive T cells in healthy individuals. J Immunol. 2004;172:5967–72. doi: 10.4049/jimmunol.172.10.5967. [DOI] [PubMed] [Google Scholar]
- 12.Taams LS, Vukmanovic-Stejic M, Smith J, et al. Antigen-specific T cell suppression by human CD4+CD25+ regulatory T cells. Eur J Immunol. 2002;32:1621–30. doi: 10.1002/1521-4141(200206)32:6<1621::AID-IMMU1621>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 13.Afzali B, Lombardi G, Lechler RI, Lord GM. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol. 2007;148:32–46. doi: 10.1111/j.1365-2249.2007.03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional defects and the influence of age on the frequency of CD4+CD25+ T-cells in type 1 diabetes. Diabetes. 2005;54:1407–14. doi: 10.2337/diabetes.54.5.1407. [DOI] [PubMed] [Google Scholar]
- 15.Kukreja A, Cost G, Marker J, et al. Multiple immuno-regulatory defects in type-1 diabetes. J Clin Invest. 2002;109:131–40. doi: 10.1172/JCI13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes. 2005;54:92–9. doi: 10.2337/diabetes.54.1.92. [DOI] [PubMed] [Google Scholar]
- 17.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–85. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 18.Putnam AL, Vendrame F, Dotta F, Gottlieb PA. CD4+CD25high regulatory T cells in human autoimmune diabetes. J Autoimmun. 2005;24:55–62. doi: 10.1016/j.jaut.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 19.Brusko T, Wasserfall C, McGrail K, et al. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes. 2007;56:604–12. doi: 10.2337/db06-1248. [DOI] [PubMed] [Google Scholar]
- 20.Glisic-Milosavljevic S, Wang T, Koppen M, et al. Dynamic changes in CD4+ CD25+(high) T cell apoptosis after the diagnosis of type 1 diabetes. Clin Exp Immunol. 2007;150:75–82. doi: 10.1111/j.1365-2249.2007.03475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bacchetta R, Passerini L, Gambineri E, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest. 2006;116:1713–22. doi: 10.1172/JCI25112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 23.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–42. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 24.Liu W, Putnam AL, Xu-yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ Treg cells. J Exp Med. 2006;203:1701–11. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seddiki N, Santner-Nanan B, Martinson J, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203:1693–700. doi: 10.1084/jem.20060468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schloot NC, Meierhoff G, Karlsson Faresjö M, et al. Comparison of cytokine ELISpot assay formats for the detection of islet antigen autoreactive T cells: report of the third immunology of diabetes society T-cell workshop. J Autoimmun. 2003;21:365. doi: 10.1016/s0896-8411(03)00111-2. [DOI] [PubMed] [Google Scholar]
- 27.Baecher-Allan C, Viglietta V, Hafler DA. Inhibition of human CD4(+)CD25(+high) regulatory T cell function. J Immunol. 2002;169:6210–7. doi: 10.4049/jimmunol.169.11.6210. [DOI] [PubMed] [Google Scholar]
- 28.You S, Belghith M, Cobbold S, et al. Autoimmune diabetes onset results from qualitative rather than quantitative age-dependent changes in pathogenic T-cells. Diabetes. 2005;54:1415–22. doi: 10.2337/diabetes.54.5.1415. [DOI] [PubMed] [Google Scholar]
- 29.Gregori S, Giarratana N, Smiroldo S, Adorini L. Dynamics of pathogenic and suppressor T cells in autoimmune diabetes development. J Immunol. 2003;171:4040–7. doi: 10.4049/jimmunol.171.8.4040. [DOI] [PubMed] [Google Scholar]
- 30.Clough LE, Wang CJ, Schmidt EM, et al. Release from regulatory T cell-mediated suppression during the onset of tissue-specific autoimmunity is associated with elevated IL-21. J Immunol. 2008;180:5393–401. doi: 10.4049/jimmunol.180.8.5393. [DOI] [PubMed] [Google Scholar]
- 31.Kriegel MA, Lohmann T, Gabler C, Blank N, Kalden JR, Lorenz HM. Defective suppressor function of human CD4+ CD25+ regulatory T cells in autoimmune polyglandular syndrome type II. J Exp Med. 2004;199:1285–91. doi: 10.1084/jem.20032158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–9. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu GZ, Gomes AC, Fang LB, Gao XG, Hjelmstrom P. Decreased 4-1BB expression on CD4+CD25 high regulatory T cells in peripheral blood of patients with multiple sclerosis. Clin Exp Immunol. 2008;154:22–9. doi: 10.1111/j.1365-2249.2008.03730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowe CE, Cooper JD, Brusko T, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet. 2007;39:1074–82. doi: 10.1038/ng2102. [DOI] [PubMed] [Google Scholar]
- 35.Ueda H, Howson JMM, Esposito L, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 36.Staeva-Vieira T, Peakman M, von Herrath M. Translational mini-review series on type 1 diabetes: immune-based therapeutic approaches for type 1 diabetes. Clin Exp Immunol. 2007;148:17–31. doi: 10.1111/j.1365-2249.2007.03328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]