

Abstract
The TH17 lineage is a novel CD4+ T cell effector subset that plays a key role in inflammatory and autoimmune responses, via its ability to produce IL-17 and IL-21. Given the potentially deleterious effects of TH17 cells, their generation needs to be strictly controlled. The regulatory pathways that prevent the inappropriate development of TH17 cells have not been fully elucidated. IRF-4 is a transcription factor that has recently emerged as a key regulator of TH17 differentiation. Our laboratory has isolated a protein, which interacts with IRF-4, that we have termed IBP (IRF-4 Binding Protein). Our studies previously demonstrated that IBP can act as an activator of Rho GTPases and that mice deficient in IBP develop a lupus-like syndrome upon aging. Here we show that TCR transgenic IBP deficient mice rapidly develop rheumatoid arthritis-like joint disease and large-vessel vasculitis. The pathology observed in the absence of IBP is associated with an enhanced responsiveness of T cells to low-levels of stimulation and with the inappropriate synthesis of IL-17 and IL-21. Furthermore, we demonstrate that the effect of IBP on cytokine production is due to its ability to sequester IRF-4 and prevent it from targeting the transcriptional regulatory regions of the IL-17 and IL-21 genes. Consistent with this finding, the enhanced ability of IBP deficient T cells to produce IL-17 and IL-21 is abolished by the concurrent lack of IRF-4. Taken together these studies suggest that IBP plays a key regulatory role in the prevention of T cell-mediated autoimmunity by ensuring that the production of IL-17 and IL-21 does not occur in response to self-antigens.
INTRODUCTION
Recent studies have uncovered the existence of a novel TH effector subset, the TH17 lineage, whose deregulation has been implicated in the pathogenesis of autoimmunity (Bettelli et al., 2007b; Weaver et al., 2006). In particular, TH17 cells are believed to play a key role in rheumatoid arthritis (RA) (McInnes and Schett, 2007; Toh and Miossec, 2007), a disease characterized by destructive inflammatory lesions affecting the synovial membranes of joints and by aberrant humoral responses that result in the production of autoantibodies like Rheumatoid Factor and anti-cyclic citrullinated peptide (CCP) antibodies. The ability of the TH17 subset to produce IL-17 is critical to their role in RA pathogenesis, since IL-17 can induce the production of proinflammatory cytokines such as TNF-α and IL-1 as well as stimulate MMP activity, matrix catabolism, and bone resorption (Koenders et al., 2006; Stamp et al., 2004). TH17 cells have also recently been shown to produce IL-21 (Korn et al., 2007; Nurieva et al., 2007; Zhou et al., 2007), a cytokine that can amplify the differentiation of TH17 cells in an autocrine manner as well as control T-dependent humoral responses (Leonard and Spolski, 2005; Mehta et al., 2004). TH17 cells develop via a pathway distinct from TH1 and TH2 cells. Induction of IL-17 production depends on the presence of Stat3 and RORγt (Ivanov et al., 2006; Laurence et al., 2007; Yang et al., 2007), while IL-21 expression requires the presence of Stat3 but not of RORγt (Nurieva et al., 2007). Given the potentially deleterious effects of the cytokines produced by TH17 cells, their production needs to be strictly controlled so that acquisition of these effector functions occurs only in response to the appropriate antigenic stimuli. The regulatory pathways that prevent the inappropriate production of IL-17 and IL-21 have, however, not been fully elucidated.
Interferon Regulatory Factor 4 (IRF-4) is a member of the IRF family of transcription factors whose absence leads to profound defects in the function and homeostasis of mature T and B cells (Mittrucker et al., 1997). Expression of IRF-4 is upregulated in response to T cell activation and we, as well as others, have shown that IRF-4 can regulate IL-4 production and TH2 differentiation (Hu et al., 2002; Rengarajan et al., 2002a). Interestingly, recent studies have demonstrated that IRF-4 is also a crucial regulator of TH17 differentiation (Brustle et al., 2007). During a yeast two-hybrid screen aimed at identifying proteins interacting with IRF-4, our laboratory isolated a human cDNA encoding a novel protein that we termed IBP (IRF-4 Binding Protein) (Gupta et al., 2003b). IBP shares significant homology with SWAP-70, a novel type of Rac activator. In contrast to SWAP-70 that is expressed mostly in B cells and mast cells, IBP is highly expressed in T cells. In unstimulated T cells, IBP is present in a “dormant“ conformation due to an inhibitory interaction between its N- and C-termini. TCR engagement leads to the tyrosine phosphorylation of the N-terminus of IBP, disrupting the autoinhibitory interaction and enabling IBP to be recruited to the immunological synapse (IS) where IBP activates Rac and Cdc42 (Gupta et al., 2003a). Our previous studies in mice deficient for IBP (IBPtrap/trap mice) have revealed that lack of IBP, with age, leads to the development of a lupus-like syndrome, which is characterized by the accumulation of CD44hiCD62Llo T cells and IgG+ B cells, profound hypergammaglobulinemia, autoantibody production, proteinuria, and glomerulonephritis (Fanzo et al., 2006). Consistent with the ability of IBP to act as an activator of Rho GTPases, IBP deficient T cells exhibited defects in ERK1/2 activation and in cytoskeletal reorganization including impaired assembly of the IS and actin polymerization.
We have recently generated TCR transgenic (DO11.10) IBP deficient mice. Surprisingly, these mice rapidly develop, with a high degree of penetrance, an autoimmune condition marked by the presence of rheumatoid arthritis-like joint disease and large-vessel vasculitis. In contrast to the SKG mouse, in which spontaneous development of arthritis is linked to the emergence of autoreactive T cell clones secondary to impairments in negative selection (Sakaguchi et al., 2003), thymocyte negative selection is not affected by the lack of IBP. The pathology observed in the absence of IBP, is instead, associated with an accumulation of TCR transgene positive T cells, which exhibit an altered pattern of responsiveness to pMHC complexes and aberrantly express both IL-17 and IL-21. Importantly, our studies demonstrate that the aberrant cytokine production observed in the absence of IBP is due to its ability to regulate IRF-4. These findings thus suggest that absence of IBP leads to autoimmunity due to the ability of IBP to control both the TCR signaling threshold as well as the acquisition of pathogenic TH effector functions.
RESULTS
Spontaneous development of rheumatoid arthritis-like joint disease and large-vessel vasculitis in IBPtrap/trap DO11.10 mice
To systematically analyze the effects of IBP deficiency on TCR-mediated signaling pathways, IBPtrap/trap mice were backcrossed onto a BALB/c background and then crossed to DO11.10 mice, which carry a I-Ad-restricted transgenic T cell receptor that recognizes a specific peptide (OVA323-339) derived from ovalbumin (Murphy et al., 1990; Robertson JM, 2000). Surprisingly, beginning at about 7 weeks of age, IBPtrap/trap DO11.10 mice but none of the IBP+/+ DO11.10 mice, started spontaneously developing joint erythema and swelling, which affected the wrist joints and fingers and, less frequently, the ankle joints and toes in a symmetrical manner (Fig. 1A). No obvious swelling of the knees, elbows, shoulders, or vertebral joints was observed. Approximately 90% of IBPtrap/trap DO11.10 mice developed a chronic progressive arthritis, which tended to be more severe in females and occasionally resulted in joint deformities and impaired mobility (Supplementary Fig. 1). Histopathologic analysis revealed synovitis with pannus formation, composed predominantly of fibroblasts, monocytes, lymphocytes, and a variable number of plasma cells and neutrophils (Fig. 1B and data not shown). The infiltrate invaded the adjacent joint structure with destruction of cartilage and increased osteoclastic activity leading to resorption of the subchondral bone and bone erosions. Serologic analysis demonstrated that IBPtrap/trap DO11.10 mice exhibited elevated titers of Rheumatoid Factor (RF), anti-Collagen II antibodies, and anti-cyclic citrullinated peptide antibodies (anti-CCP) in their serum but only a small increase in anti-dsDNA antibodies (Fig. 1C and Supplementary Fig. 1). IBPtrap/trap DO11.10 mice thus develop an inflammatory arthropathy that shares many clinical, histological, and serological features with human rheumatoid arthritis.
Figure 1.

Development of arthritis and large-vessel vasculitis in IBPtrap/trap DO11.10 mice. A. Shown are the wrist and ankle of a normal 16 wks old IBP+/+ DO11.10 (IBP+/+ DO) female mouse (left) and of an affected IBPtrap/trap DO11.10 (IBPtrap/trap DO) female mouse (right) that had developed swelling and erythema of the joints. B. Histopathologic analysis (hematoxylin/eosin staining) of wrist joints of a 12-week-old female IBP+/+ DO11.10 mouse (left) and an age- and sex- matched IBPtrap/trap DO11.10 mouse (right). Light microscopy images (magnification of 40 and 100×, as indicated) are shown. These findings are representative of 6 mice for each group. C. Serological analysis. Sera from IBP+/+ DO11.10 and IBPtrap/trap DO11.10 mice (6–25 weeks old, male and female, n=6–12) were collected and levels of rheumatoid factor (RF), anti-collagen II (CII) antibodies, and anti-cyclic citrullinated peptide (CCP) antibodies were analyzed by ELISA. *p<0.05. D. Histopathologic analysis (hematoxylin/eosin staining) of the root of the aorta of a 16 wk-old IBP+/+ DO11.10 female mouse (left panels) and an age-matched IBPtrap/trap DO11.10 female mouse (right panels). Light microscopy images (magnification of 40× and 100×, as indicated) are shown. These findings are representative of 6 mice for each group.
Beginning at ~3 months of age, IBPtrap/trap DO11.10 mice also started dying suddenly. Over 90% of IBPtrap/trap DO11.10 mice had died by 12 months of age with female IBPtrap/trap DO11.10 mice dying more rapidly than male IBPtrap/trap DO11.10 mice (Supplementary Fig. 1). Consistent with the idea that the sudden death of the IBPtrap/trap DO11.10 mice might be due to abnormalities of their cardiovascular system, histologic analysis revealed that all of the arthritic IBPtrap/trap DO11.10 mice also had developed severe inflammation of the root of the aorta with massive transmural infiltration of lymphocytes, monocytes, including occasional multinucleated giant cells, variable numbers of plasma cells and neutrophils, and reduplication or destruction of the internal elastic lamina (Fig. 1D and Supplementary Fig. 1). Occasionally, inflammation was also observed in the medium-sized arteries of the kidney and of the lung, but not veins or capillaries. No significant inflammation of other organs was noted, including the small and large intestine, pancreas, and liver.
IBP deficiency leads to abnormal TCR responsiveness and the spontaneous activation of TCR transgenic T cells
To explore the pathways underlying the pathology associated with IBP deficiency, we first investigated whether lack of IBP leads to abnormalities in central tolerance. Unlike what has been described for previous murine models, the emergence of spontaneous arthritis and vasculitis in the IBPtrap/trap DO11.10 mice was not due to a failure of eliminating autoreactive T cells within the thymus, as assessed by injecting IBP+/+ and IBPtrap/trap mice with high doses of anti-CD3 Ab (Supplementary Fig. 2) or by crossing IBPtrap/trap mice with HY transgenic mice, a well-characterized model of negative selection (Supplementary Fig. 2). Furthermore, no defects in the development or function of regulatory T cells could be observed in the absence of IBP (Supplementary Figs. 3 and 4).
These findings raised the possibility that the immunopathology observed in the IBPtrap/trap DO11.10 mice might be mediated by mature CD4+ T cells secondary to an enhanced recognition of self-peptides by the IBPtrap/trap DO11.10 TCR in the periphery. Consistent with this idea, expression of CD5, a gauge of the strength of TCR signaling upon interaction with self-ligands (Kieper et al., 2004), was higher on IBPtrap/trap DO11.10 CD4+ T cells than on IBP+/+ DO11.10 CD4+ T cells. Increased CD5 expression was observed in peripheral CD4+ T cells from adult mice as well as in thymic CD4SP cells from newborn IBPtrap/trap DO11.10 mice (Supplementary Fig. 5). To directly evaluate whether IBP deficiency leads to abnormalities in the recognition of pMHC complexes by the DO11.10 TCR, the responsiveness of purified naïve IBPtrap/trap DO11.10 CD4+ T cells to different doses of OVA323-339 peptide was assayed in vitro (Fig. 2A). As expected, the proliferative responses of the IBP+/+ DO11.10 CD4+ T cells increased upon exposure to increasing doses of OVA323-339 peptide. In contrast, IBPtrap/trap DO11.10 CD4+ T cells displayed a markedly abnormal pattern of proliferation, which was characterized by hyperresponsiveness to low-doses of OVA323-339 peptide but hyporesponsiveness to high-doses of the same peptide. Thus, in the absence of IBP, mature CD4+ T cells become hyperresponsive to low-levels of stimulation.
Figure 2.

Lack of IBP leads to abnormalities in the responsiveness of DO11.10 CD4+ T cells. A. Proliferative responses to OVA323-339 peptide. Naïve CD4+ T cells (CD44lowCD62Lhigh) were isolated from both IBP+/+ DO11.10 (blue) and IBPtrap/trap DO11.10 (red) mice and stimulated for 3 days with IBP+/+ APCs pulsed with increasing doses (0, 0.1, 1, 10 μM) of OVA323-339 peptide. Cell proliferation was assayed by BrdU ELISA. Experiment shown is representative of 3 independent experiments. B. Spontaneous acquisition of an effector phenotype in vivo in the absence of IBP. FACS analysis of CD44 and CD62L expression on CD4+ splenic T cells from a 6-week-old female IBP+/+ DO11.10 (left) and a sex- and age- matched IBPtrap/trap DO11.10 (right) mouse. C. Spontaneous activation of IBPtrap/trap DO11.10 T cells in vivo. The surface expression level of CD69, CD25, and ICOS on KJ1-26high IBP+/+ DO11.10 (blue) and KJ1-26high IBPtrap/trap DO11.10 (red) was analyzed by FACS and overlaid histograms are shown. Data are representative of three 6 week-old mice/group.
The abnormal responsiveness exhibited by the IBPtrap/trap DO11.10 T cells to pMHC complexes suggested that, in the absence of IBP, TCR transgenic T cells might become spontaneously activated in response to endogenous peptides. Indeed, by 6 weeks of age (before the onset of the autoimmune phenotype), ~30% of CD4+ T cells from IBPtrap/trap DO11.10 mice exhibited an effector phenotype (Fig. 2B). Most of the CD62LloCD44hi T cells were KJ1-26high and thus expressed the TCR transgene (Supplementary Fig. 6). Furthermore, already by 6 weeks of age, and even more markedly by 15 weeks of age, KJ1-26high IBPtrap/trap CD4+ T cells spontaneously upregulated the expression of CD69 and ICOS (Fig. 2C). Although an increase in KJ1-26low T cells was also observed in the IBPtrap/trap DO11.10 mice, these KJ1-26low T cells expressed a naïve phenotype (Supplementary Fig. 6).
To investigate whether transfer of the IBPtrap/trap DO11.10 T cells could mediate pathogenic effector functions, total splenocytes from the arthritic IBPtrap/trap DO11.10 mice were transferred into syngeneic SCID mice. These transfers were accompanied by the sudden demise of 6 out of 14 recipient mice by 3 months while no deaths were recorded in mice receiving IBP+/+ DO11.10 T cells (0/9 recipient mice). No premonitory signs of morbidity were evident in the recipients of IBPtrap/trap DO11.10 lymphocytes that suddenly died leading us to suspect that these mice succumbed to the profound cardiovascular abnormalities associated with the absence of IBP. Transfer of serum from arthritic IBPtrap/trap DO11.10 mice instead did not lead to any pathology (data not shown). Consistent with the finding that the abnormal acquisition of effector markers and costimulatory molecules mapped to the KJ1-26high rather than to the KJ1-26low T cell compartment, transfer of purified KJ1-26high IBPtrap/trap DO11.10 T cells but not of purified KJ1-26low IBPtrap/trap DO11.10 T cells into nude Balb/c mice also led to the sudden death of the recipient mice (Table I).
Table 1.
Survival of syngeneic nude mice transferred with CD4+ T cells derived from either IBP+/+DO11.10 or IBPtrap/trapDO11.10 mice.
| Donor cells | IBP+/+ Do CD4+T cells | IBPtrap/trap Do KJlow CD4+T cells | IBPtrap/trap Do KJhigh CD4+ T cells |
|---|---|---|---|
| Dead mice/Total mice | 0/6 | 0/6 | 3/7 |
IBP deficiency leads to the aberrant production of IL-17 and IL-21
The distinctive pathophysiology observed in the absence of IBP indicated that IBP, besides regulating TCR responsiveness, could also control T cell effector function. In particular, the presence of both inflammatory lesions and aberrant humoral responses in the IBPtrap/trap DO11.10 mice raised the possibility that IBP could regulate the production of IL-17 and IL-21. To investigate this hypothesis, cytokine production was assessed after obtaining naïve CD4+ T cells from 5-week old IBP+/+ DO11.10 or IBPtrap/trap DO11.10 mice by negative depletion (Lin et al., 2004) and culturing them with APCs pulsed with OVA323-339 peptide. When compared to wt T cells, IBPtrap/trap DO11.10 T cells exhibited an increased ability to produce IL-17 (Fig. 3A). Furthermore, in contrast to IBP+/+ DO11.10 T cells, IBPtrap/trap DO11.10 T cells also synthesized significant amounts of IL-21 (Fig. 3B). Deregulated production of IL-17 and IL-21 was not due to global upregulation in cytokine production since IBPtrap/trap DO11.10 CD4+ T cells produced less IL-2 than IBP+/+ DO11.10 CD4+ T cells (Fig. 3C) and variable levels of IL-4 and IFN-γ (Supplementary Fig. 7). A similar increase in IL-17 and IL-21 production was also detected with FACS sorted CD44loCD62LhiCD25−CD4+ IBPtrap/trap DO11.10 cells (Supplementary Fig. 8). The deregulated production of IL-17 and IL-21 observed upon stimulation of IBPtrap/trap DO11.10 CD4+ T cells under neutral conditions was accompanied by increased expression of RORγt but not by elevation of IL-22 (Fig. 3D).
Figure 3.


Lack of IBP leads to aberrant production of IL-17 and IL-21 in vitro and in vivo. A. Naïve CD4+ T cells derived from 6 wks. old IBP+/+ DO11.10 (white bars) or IBPtrap/trap DO11.10 (black bars) mice were cultured with IBP+/+ APCs pulsed with 1 μM OVA323-339 peptide for the times indicated. The production of IL-17 in the supernatants was measured by ELISA. Experiment shown is representative of 3 independent experiments. B. Naïve CD4+ T cells derived from 6 wks. old IBP+/+ DO11.10 (white bars) or IBPtrap/trap DO11.10 (black bars) mice were cultured as above. The production of IL-21 in the supernatants was measured by ELISA. Experiment shown is representative of 3 independent experiments. C. Naïve CD4+ T cells derived from 6 wks. old IBP+/+ DO11.10 (white bars) or IBPtrap/trap DO11.10 (black bars) mice were cultured as above. The production of IL-2 in the supernatants was measured by ELISA. Experiment shown is representative of 3 independent experiments. D. Naïve CD4+ T cells derived from 6 wks. old IBP+/+ DO11.10 (white bars) or IBPtrap/trap DO11.10 (black bars) mice were cultured for 3 days as described above and the mRNA expression of RORγt (left panel) and IL-22 (right panel) genes was measured by real-time RT-PCR. Experiment shown is representative of 3 independent experiments. E. Serum levels of IL-17 and IL-21 in IBP+/+ DO11.10 and IBPtrap/trap DO11.10 mice. Sera from IBP+/+ DO11.10 and IBPtrap/trap DO11.10 mice (12–25 weeks old, male and female, n=4) were collected and levels of IL-17 and IL-21 analyzed by ELISA. *p<0.05. F. Expression of IL-17 and IL-21 in the joints of arthritic IBPtrap/trap DO11.10 mice. Joints from 3 pairs of IBP+/+ DO11.10 and IBPtrap/trap DO11.10 mice (12–25 weeks old, male and female) were collected and IL-17 and IL-21 gene expression was analyzed by real-time RT-PCR. White bars represent IBP+/+ DO11.10 mice and black bars represent IBPtrap/trap DO11.10 mice. G. Blimp-1 and CD3 staining of spleens of IBPtrap/trap DO11.10 mice. Anti-Blimp-1 (blue) and anti-CD3 (red) staining was performed on spleens from IBP+/+ DO11.10 (left panel) and IBPtrap/trap DO11.10 (right panel) mice. Light microscopy images (magnification of 4×, upper panels, and 40×, lower panels) are shown.
To assess whether the aberrant synthesis of IL-17 and IL-21 by IBPtrap/trap DO11.10 cells detected in vitro led to deregulated production of IL-17 and IL-21 in vivo as well, we evaluated the levels of IL-17 and IL-21 in the sera of arthritic IBPtrap/trap DO11.10 mice (Fig. 3E). As compared to IBP+/+ DO11.10, IBPtrap/trap DO11.10 mice exhibited increased serum levels of both IL-17 and IL-21. Increased expression of IL-17 and IL-21 was also observed in the joints of the arthritic IBPtrap/trap DO11.10 mice (Fig. 3F). Furthermore, consistent with the fact that aberrant production of IL-21 by IBP deficient CD4+ T cells might enable these T cells to inappropriately drive the terminal differentiation of B cells, immunohistochemical staining of spleen sections revealed that, in the absence of IBP, numerous plasma cells could be observed within the T cell zones or perivascular lymphoid sheaths (Fig. 3G). Taken together, these data thus indicate that lack of IBP leads to the inappropriate synthesis of IL-17 and IL-21, whose production should be tightly controlled and occur only upon exposure to selected pathogens.
IRF-4 controls both IL-17 and IL-21 production
The deregulated production of IL-17 and IL-21 by IBPtrap/trap DO11.10 CD4+ T cells could be detected even under conditions where these cells displayed proliferative responses identical to those exhibited by the IBP+/+ DO11.10 CD4+ T cells (i.e. with 1μM OVA323-339 peptide) suggesting that the abnormal cytokine production was not simply the result of the aberrant TCR responsiveness displayed by these cells. Given that we had originally cloned IBP as a protein interacting with the transcription factor IRF-4 (Gupta et al., 2003b), we hypothesized that these effects might be linked to the ability of IBP to modulate IRF-4 function. To start exploring this possibility we first investigated whether IRF-4 could regulate not only the synthesis of IL-17 but also that of IL-21. For these studies we took advantage of previously generated IRF-4 deficient mice (Mittrucker et al., 1997). Naïve CD4+ T cells were purified from wt and IRF-4−/− mice and cultured in vitro under TH0 and TH17 conditions. Consistent with recent observations (Brustle et al., 2007), wt CD4+ T cells produced substantial amounts of IL-17 when cultured under TH17 conditions, but CD4+ T cells derived from IRF-4 deficient mice completely lost their ability to synthesize IL-17 (Fig. 4A). These results were further confirmed by performing intracellular FACS (Fig. 4B). In contrast to wt T cells, no IL-17 producing cells could be detected upon stimulation in the absence of IRF-4. The lack of IRF-4 instead resulted in an enhanced ability to produce IFN-γ under TH17 conditions. Importantly, when the production of IL-21 was investigated, no IL-21 synthesis could be observed when IRF-4 deficient T cells were cultured under TH17 conditions and then restimulated (Fig. 4C). Since the absence of IL-21 production in this setting could be secondary to the defective synthesis of IL-17, and since IL-21 can also be produced by TH2 cells (Leonard and Spolski, 2005; Mehta et al., 2004), we also investigated the effects of IRF-4 deficiency on IL-21 synthesis when CD4+ T cells were differentiated under TH2 conditions. As shown in Fig. 4D, production of IL-21 under these culture conditions was also abrogated by the absence of IRF-4.
Figure 4.

IRF-4 is a critical regulator of both IL-17 and IL-21 production. A. IL-17 production by wt and IRF-4−/− CD4+ T cells. Purified naïve CD4+ T cells were stimulated under either TH0 (white bars) or TH17 (black bars) conditions for 4 days. IL-17 levels in culture supernatants were determined by ELISA. The experiment is representative of four independent experiments. B. Intracellular IL-17 and IFN-γ production by wt and IRF-4−/− CD4+ T cells. Cells were cultured under TH17 conditions as above and then stimulated with PMA and ionomycin for 5 hours. Intracellular IFN-γ and IL-17 production was measured by FACS. The experiment is representative of three independent experiments. C. IL-21 production by wt and IRF-4−/− CD4+ T cells cultured under TH17 conditions. Purified naïve CD4+ T cells were stimulated under TH17 conditions for 4 days and then restimulated for either 24 hrs (white bars) or 48 hrs (black bars). IL-21 levels in culture supernatants were determined by ELISA. The experiment is representative of four independent experiments. D. IL-21 production by wt and IRF-4−/−CD4+ T cells cultured under TH0 and TH2 conditions. Purified naïve CD4+ T cells were stimulated under TH0 (white bars) or TH2 (black bars) conditions for 7 days and then restimulated for 48 hrs. IL-21 levels in culture supernatants were determined by ELISA. The experiment is representative of four independent experiments. E. Effects of IRF-4 on the transactivation of IL-21. Jurkat cells that express an empty vector or Jurkat cells expressing IRF-4 were transfected with a luciferase reporter construct driven either by the murine IL-21 promoter (IL-21 LUC), by a murine IL-21 promoter containing mutations within the putative IRF-4 binding site (IL-21 MUT LUC) or with a control luciferase reporter construct (pGL3) in the presence or absence of PMA and ionomycin as indicated. Cotransfection with a renilla-luciferase construct was performed to normalize the transfection efficiency of the different samples. Data are representative of 3 independent experiments.
A survey of the IL-21 promoter revealed that it contains potential IRF-4 binding elements, we thus proceeded to directly test whether IRF-4 could function as a transactivator of the IL-21 promoter. To this end, we performed transient transfection assays in Jurkat cells that either lacked or contained IRF-4, which we had utilized in previous studies (Hu et al., 2002). Consistent with studies demonstrating that NFAT proteins participate in the regulation of the IL-21 promoter (Kim et al., 2005; Mehta et al., 2005), inducibility of a luciferase reporter construct driven by the IL-21 promoter could be observed in Jurkat cells lacking IRF-4 upon stimulation with PMA and ionomycin (Fig. 4E). Importantly however, under these stimulatory conditions, Jurkat cells expressing IRF-4 demonstrated a greater level of luciferase activity when transfected with the IL-21 promoter construct (IL-21 LUC) but not with an IL-21 promoter construct containing mutations within the IRF-4 binding site (IL-21 MUT LUC). These findings thus indicate that IRF-4 can act as a transactivator of the IL-21 promoter and suggest that NFAT proteins and IRF-4 cooperate in the transcriptional regulation of this gene.
Absence of IBP leads to enhanced targeting of the IL-17 and IL-21 promoters by IRF-4
We had previously reported that IBP can be found in the cytoplasm and can be rapidly recruited to the immunological synapse (Gupta et al., 2003a). IBP, however, also contains putative NLS motifs (Gupta et al., 2003b) raising the possibility that it could also translocate to the nucleus. Subcellular fractionation experiments indeed demonstrated that IBP could be detected not only in the cytoplasm but also within the nuclear compartment of primary CD4+ T cells (Fig. 5A). Coimmunoprecipitation experiments confirmed that nuclear IBP and IRF-4 can physically interact (Fig. 5B).
Figure 5.

The absence of IBP leads to enhanced targeting of the IL-17 and IL-21 regulatory regions. A. IBP can be found within the nuclear compartment. Purified CD4+ T cells were either left unstimulated or stimulated with anti-CD3 and anti-CD28 Abs for 48 hrs. Nuclear and cytoplasmic extracts were then prepared, resolved by 8% SDS-PAGE, and then analyzed by Western blotting using an anti-IBP antiserum (upper panel). The blot was later stripped and reprobed with an anti-Lamin B (middle panel) or an anti-β-tubulin (lower panel) antibody to assess the purity of the different fractions. B. Nuclear IBP interacts with IRF-4. Purified CD4+ T cells were stimulated with anti-CD3 and anti-CD28 Abs for 48 hrs. Nuclear extracts were then prepared and immunoprecipitated with an anti-IRF-4 antibody (IRF-4 IP) or with a control antibody (Control Ab IP). The immunoprecipitates were resolved by 8% SDS-PAGE, and then analyzed by Western blotting using an anti-IBP antiserum (upper panel). The blot was later stripped and reprobed with an anti-IRF-4 antibody (lower panel). C. Lack of IBP leads to enhanced targeting of the IL-17 and IL-21 promoter by IRF-4 in vivo. CD4+ T cells were purified from either IBP+/+ (wt) or IBPtrap/trap mice, cultured under TH0 conditions for 7 days, and then either left unstimulated (unst) or restimulated for 24 hrs (st) with anti-CD3 and anti-CD28 Abs. Chromatin immunoprecipitation assays were then carried out on these cells with either an anti-IRF-4 antibody or a control Ab, and PCR for the IL-21 and IL-17 promoters was performed as indicated. The total input control is shown on the left. Actin was used as a negative control. D. Lack of IBP leads to enhanced binding of IRF-4 to the IRF-4 binding site within the IL-21 promoter. CD4+ T cells were purified from either IBP+/+ (wt) or IBPtrap/trap mice, cultured as described in C. and subjected to an oligonucleotide precipitation assay (ONP) with a biotin-labelled oligonucleotide corresponding to the IRF-4 binding site within the IL-21 promoter. Precipitates were separated by 8% SDS-PAGE and analyzed by Western blotting with an IRF-4 antibody (top panel). The levels of IRF-4 in the input samples are shown on the right (due to the high levels of IRF-4 in the input extracts a shorter exposure time of the IRF-4 input is shown). The blot was then stripped and reprobed with an anti-IBP antibody (lower panel). The levels of IBP in the input samples are shown on the right.
Since we had observed that, in addition to the IL-21 promoter, the IL-17 promoter also contains a potential binding site for IRF-4, ChIP assays with an anti-IRF-4 Ab were conducted next to examine whether the absence of IBP could lead to deregulated recruitment of IRF-4 containing complexes to the IL-17 and IL-21 promoters in vivo. As shown in Fig. 5C, when T cells were cultured under unskewed conditions, the lack of IBP resulted in markedly enhanced targeting of these regulatory regions by IRF-4 containing complexes. To further confirm these findings, we utilized an oligonucleotide corresponding to the IRF-4 binding element within the IL-21 promoter to perform oligonucleotide precipitation assays (ONPs) on extracts obtained from wt or IBPtrap/trap CD4+ T cells that had either been left unstimulated or stimulated under neutral conditions (Fig. 5D). Consistent with the ChIP findings, the levels of IRF-4 that were precipitated from stimulated IBPtrap/trap CD4+ T cells were much greater than those precipitated from stimulated wt CD4+ T cells despite similar input levels of IRF-4 (Figure 5D, upper panel). Interestingly, reblotting with an IBP antibody did not demonstrate any binding of IBP to this oligonucleotide in wt T cells (Figure 5D, lower panel). Taken all together these data thus support the notion that nuclear IBP can regulate the ability of IRF-4 to access the transcriptional regulatory regions of the IL-17 and IL-21 genes.
IBP prevents the ability of IRF-4 to bind and transactivate the IL-21 promoter
To explore the mechanisms by which IBP controls IRF-4 we first investigated whether IBP could regulate the translocation of IRF-4. Subcellular fractionation experiments (Fig. 6A) failed to demonstrate any effect of the absence of IBP on the cellular localization of IRF-4 suggesting that the differential ability of IRF-4 to target the IL-17 and IL-21 promoters was not simply the result of changes in its subcellular localization.
Figure 6.

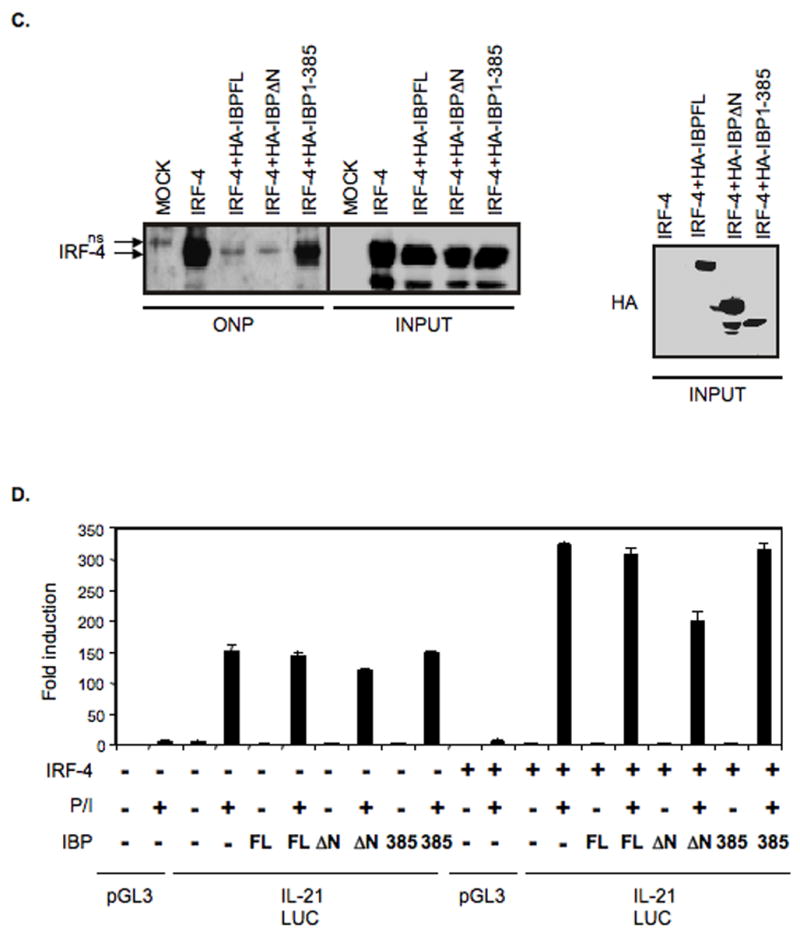
IBP inhibits the DNA binding and transactivating activity of IRF-4. A. The absence of IBP does not alter the expression or localization of IRF-4. CD4+ T cells were purified from either IBP+/+ (wt) or IBPtrap/trap mice, cultured under TH0 conditions for 7 days, and then either left unstimulated (unst) or restimulated for 24 hrs (st) with anti-CD3 and anti-CD28 Abs. Nuclear and cytoplasmic extracts were then prepared, resolved by 8% SDS-PAGE, and then analyzed by Western blotting using an anti-IRF-4 antiserum (upper panel). The blot was later stripped and reprobed with an anti-Lamin B (middle panel) or an anti-β-tubulin (lower panel) antibody to assess the purity of the different fractions. B. Mapping of the IBP domain responsible for the interaction with IRF-4. 293T cells were cotransfected with an IRF-4 expression construct together with either HA-tagged full length IBP or the panel of HA-tagged IBP mutants described in the diagram. Extracts were immunoprecipitated with an anti-HA antibody, precipitates were separated by 8% SDS-PAGE and analyzed by Western blotting with an IRF-4 antibody (top panel). The blot was later stripped and reprobed with an anti-HA antibody (bottom panel). C. IBP can block the binding of IRF-4 to an oligonucleotide containing the IRF-4 binding site within the IL-21 promoter. An oligonucleotide precipitation assay (ONP) was performed on 293T cells that were either mock transfected or transfected with IRF-4 in the presence/absence of HA-tagged FL IBP, HA-tagged IBPΔN, or HA-tagged IBP 1-385. Precipitates were separated by 8% SDS-PAGE and analyzed by Western blotting with an IRF-4 antibody (left panel). The levels of IRF-4 in the input samples are also shown (due to the high levels of IRF-4 in the extracts a shorter exposure time of the IRF-4 input is shown). A nonspecific band detected in the ONP from mock transfected 293 T cells is indicated as ns. The blot was later stripped and reprobed with an anti-HA antibody to assess the levels of the different IBP constructs in the input samples (right panel). D. IBP can inhibit the transactivating activity of IRF-4. Jurkat cells that express an empty vector or Jurkat cells expressing IRF-4 were transfected with a luciferase reporter construct driven by the murine IL-21 promoter (IL-21 LUC) together with an empty vector, HA-tagged FL IBP, HA-tagged IBPΔN, or HA-tagged IBP 1-385 as indicated. Cells were either left unstimulated or stimulated with PMA and ionomycin as indicated. Transfections with a control luciferase reporter construct (pGL3) in the presence/absence of PMA and ionomycin were also carried out. Cotransfection with a renilla-luciferase construct was performed to normalize the transfection efficiency of the different samples. Data are representative of 3 independent experiments.
We next examined the possibility that nuclear IBP may be able to sequester IRF-4 by determining whether IBP could prevent IRF-4 from targeting its DNA binding site within the IL-21 promoter. To facilitate these experiments we first conducted a mutational analysis to better define the region of IBP involved in the interaction with IRF-4 (Fig. 6B). This analysis revealed that a mutant of IBP (IBP 1-385) lacking a large portion of the carboxy-terminus of IBP (between aa 386 and 631) lost its ability to interact with IRF-4. Consistent with the finding that the N-terminal domain can exert an autoinhibitory effect, a mutant lacking this region (IBPΔN) instead exhibited an enhanced interaction with IRF-4. Oligonucleotide precipitation (ONP) experiments with an oligonucleotide containing the IRF-4 binding element within the IL-21 promoter were next performed on extracts of 293T cells transfected with IRF-4 in the presence or absence of FL IBP, IBPΔN, or IBP 1-385 (Figure 6C). IRF-4 was robustly precipitated from 293T cells transfected with IRF-4 alone but not from mock transfected 293T cells. The presence of either FL IBP or of IBPΔN markedly diminished the amount of precipitated IRF-4. In contrast, strong binding of IRF-4 to this site could still be detected in 293T cells cotransfected with IBP 1-385. Input levels of IRF-4 in the different samples were similar. IBP can thus directly inhibit the ability of IRF-4 to bind to the IL-21 promoter.
The effects of IBP on the transactivating activity of IRF-4 were also examined. Jurkat cells that either lack or contain IRF-4 were transiently transfected with the IL-21 luciferase reporter construct in the presence or absence of FL IBP, IBPΔN, or IBP 1-385 (Figure 6D). Cells were then either left unstimulated or stimulated with PMA and ionomycin. Similar to the results in Figure 4, presence of IRF-4 augmented the inducibility of this construct upon PMA and ionomycin stimulation. Consistent with our previous findings that, in T cells, FL IBP needs to be posttransationally modified in order to become activated, coexpression of FL IBP was unable to affect the transactivating ability of IRF-4, while cotranfection of a mutant lacking the autoinhibitory domain (IBPΔN) blocked the IRF-4-mediated transactivation. Importantly, the IBP 1-385 mutant that loses its ability to physically interact with IRF-4 was unable to significantly affect the transactivating ability of IRF-4 confirming the functional relevance of the IBP-IRF-4 interaction. To ensure that the inability of the IBP 1-385 mutant to interfere with the transactivating activity of IRF-4 was not due to the presence of the N-terminal autoinhibitory domain, an IBP mutant lacking both the autoinhibitory domain and the IRF-4 interacting domain was also generated (ΔN-385). Cotransfection of (ΔN-385) with IRF-4 also failed to block the IRF-4-mediated transactivation of the IL-21 luciferase reporter construct (Supplementary Fig. 9), confirming that the ability of IBP to interfere with the ability of IRF-4 to drive the expression of IL-21 is dependent on its physical interaction with IRF-4.
To finally ascertain whether the ability of IBP to regulate IRF-4 was indeed responsible for the aberrant production of IL-17 and IL-21 observed in IBPtrap/trap mice, we generated mice deficient in both IBP and IRF-4. As expected from previous studies demonstrating that neither a deficiency of IRF-4 nor a lack of IBP leads to obvious developmental defects (Fanzo et al., 2006; Mittrucker et al., 1997), IBPtrap/trapIRF-4−/− mice were viable, fertile and did not exhibit any significant defects in the maturation of the immune system (Supplementary Table I). To assess whether lack of IRF-4 could alter the deregulated production of IL-17 observed in the absence of IBP, naïve CD4+ T cells were cultured under unskewed conditions and IL-17 production in the supernatants was assessed by ELISA. The lack of IRF-4 completely abolished the increased ability of IBPtrap/trap T cells to synthesize IL-17 (Fig. 7A). In line with prior observations that IRF-4 controls the expression of RORγt, the aberrant RORγt expression detected in the absence of IBP was also dependent on the presence of IRF-4 (Fig. 7B). Consistent with a critical role for IRF-4 not only in the control of IL-17 but also in the regulation of IL-21, absence of IRF-4 also prevented the enhanced ability of IBPtrap/trap T cells to produce IL-21 when the T cells were cultured under either unskewed conditions (Supplementary Figure 10) or TH2 conditions (Fig. 7C). These defects were not due to abnormalities in proliferation, since CD4+ T cells from mice of all four genotypes exhibited similar CFSE profiles (Fig. 7D). Furthermore, differences in IL-17 production under these conditions did not simply correlate with differential ability of the four T cell populations to produce TGF-β or IL-6 (Supplementary Fig. 10). Interestingly, in contrast to the findings with IL-17 and IL-21, neither the absence of IBP nor the lack of IRF-4 affected the induction of ICOS upon T cell stimulation in vitro (Supplementary Fig. 10). These results thus indicate that the enhanced ability of IBP deficient T cells to aberrantly produce IL-17 and IL-21 is critically dependent on the presence of IRF-4.
Figure 7.

The deregulated IL-17 and IL-21 production observed in the absence of IBP is dependent on IRF-4. A. IL-17 production by wt, IBPtrap/trap, IRF-4−/−, and IRF-4−/−IBPtrap/trap CD4+ T cells. Purified naïve CD4+ T cells were stimulated under TH0 conditions for 4 days. IL-17 levels in culture supernatants were determined by ELISA. B. RORγt expression by wt, IBPtrap/trap, IRF-4−/−, and IRF-4−/−IBPtrap/trap CD4+ T cells. Purified naïve CD4+ T cells were stimulated as above and RORγt gene expression was determined by real-time RT-PCR. C. IL-21 production by wt, IBPtrap/trap, IRF-4−/−, and IRF-4−/−IBPtrap/trap CD4+ T cells. Purified naïve CD4+ T cells were stimulated under TH2 conditions for 7 days and then restimulated for 48 hrs. IL-21 levels in culture supernatants were determined by ELISA. D. CFSE profiles of wt, IBPtrap/trap, IRF-4−/−, and IRF-4−/−IBPtrap/trap CD4+ T cells that were either left unstimulated (red) or stimulated under TH0 conditions for 2 days (blue) or for 4 days (green). Experiments shown are representative of 3 independent experiments.
DISCUSSION
Although T cells play a key role in the pathogenesis of many autoimmune diseases such as RA and SLE, the molecular machinery preventing the emergence of autoreactive T cells has not been fully elucidated. Our studies suggest that deregulation of IBP-controlled pathways represents an important pathogenic mechanism leading to the spontaneous development of autoimmunity. The ability of IBP to play a critical role in this process is likely due to its capacity to control the responsiveness of T cells to pMHC complexes and to ensure that the production of IL-17 and IL-21 does not occur in the absence of the proinflammatory conditions known to drive TH17 differentiation.
One of the crucial aspects underlying the spontaneous development of autoimmunity in IBP deficient mice is the inability of IBPtrap/trap T cells to accurately sense the strength of TCR engagement. As shown in Fig. 2A, IBPtrap/trap DO11.10 T cells exhibit a highly abnormal pattern of responsiveness. Consistent with our previous findings that polyclonal IBP deficient T cells exposed to strong stimulatory conditions proliferate to a lesser extent than wt T cells (Fanzo et al., 2006), IBPtrap/trap DO11.10 T cells exhibit defective proliferative responses upon exposure to high doses of OVA323-339 peptide. IBPtrap/trap DO11.10 T cells, however, exhibit enhanced proliferative responses to low-levels of stimulation, an effect that we have also observed with polyclonal IBP deficient T cells (unpublished observations). The hyperresponsiveness of the IBPtrap/trap DO11.10 CD4+ T cells to low-levels of stimulation was further corroborated by their spontaneous activation in vivo in the absence of any exposure to ovalbumin and by their enhanced expression of CD5. Increased CD5 expression was also noted on CD4SP cells from newborn IBPtrap/trap DO11.10 mice suggesting that this phenotype was the consequence of intrinsic changes in the recognition of the DO11.10 TCR for its ligands rather than to exposure of these T cells to a pathogenic environment. Interestingly, preliminary studies from IBPtrap/trap RAG−/− DO11.10 mice that we have recently generated reveal that the TCR transgenic T cells from these mice still retain the observed abnormalities indicating that the arthritis and vasculitis occurring in IBPtrap/trap DO11.10 TCR transgenic mice are likely due to the inappropriate recognition of cross-reactive epitopes by the DO11.10 TCR rather than to the expression of a second TCR by these cells.
Importantly, our studies indicate that IBP, in addition to regulating the responsiveness of T cells to TCR engagement, controls the capacity of T cells to acquire a potentially pathogenic effector program. Indeed, one of the crucial abnormalities observed in the absence of IBP was increased production of IL-17, a cytokine with known proinflammatory effects relevant to the pathophysiology of rheumatoid arthritis in humans (Koenders et al., 2006; Stamp et al., 2004). We furthermore have demonstrated that the deregulated IL-17 production observed in the absence of IBP is critically dependent on IRF-4, a transcription factor recently shown to be necessary for TH17 differentiation (Brustle et al., 2007). Interestingly, IRF-4 is an important cellular target of the HTLV-I Tax oncoprotein (Mamane et al., 2002) and it is intriguing to speculate that deregulation of IRF-4 may also play a role in the autoimmune arthritis associated with the overexpression of Tax in mice (Habu et al., 1999). Consistent with previous studies showing that IRF-4 can regulate the expression of ROR-γt (Brustle et al., 2007), the enhanced production of IL-17 observed in the absence of IBP was accompanied by increased expression of ROR-γt, an effect that was dependent on the presence of IRF-4. It is, however, important to note that reconstitution of RORγt expression in IRF-4 deficient T cells only partially restores the deficient IL-17 production exhibited by these cells (Brustle et al., 2007). Thus our finding that IRF-4 containing complexes can also target the regulatory regions of the IL-17 gene indicates that the role of IRF-4 in TH17 differentiation is multifaceted and includes both direct effects on the transcription of IL-17 gene as well as indirect effects on the expression of other IL-17 transactivators.
The acquisition of a TH17 phenotype normally requires progression through a series of developmental stages during which TH17 cells are sequentially exposed to key cytokines (Bettelli et al., 2007a; Ivanov et al., 2007). Presence of IL-6 is crucial for the initiation of this process and leads to the induction of IL-21 production, which, in turn, acts in an autocrine manner and further amplifies commitment of TH cells toward the TH17 lineage. Our studies demonstrate that IBP deficient T cells can produce IL-21 even during the initial stimulation by antigen and in the absence of any TH17 skewing conditions. This abnormality may have profound pathophysiologic consequences since aberrant synthesis of IL-21 may not only reinforce the abnormal IL-17 production exhibited by these cells, but may also enable these CD4+ T cells to inappropriately promote the terminal differentiation of B cells at extrafollicular sites and, thus, provide help to autoreactive B cells (William et al., 2002). Consistent with this notion, the absence of IBP leads to the aberrant localization of Blimp1 positive plasma cells within peripheral lymphoid organs and the presence of autoantibodies. By controlling IL-21 production, IBP may thus play a critical role in ensuring that T cell help to B cells is provided only to appropriately selected B cell populations (Vinuesa et al., 2005). Unlike what we observed with IL-17 and IL-21, IBP deficiency did not affect the production of IL-22 in agreement with studies demonstrating that the regulation of IL-22 differs from that of IL-17 and that IL-22 may primarily function as a downstream target of IL-23 (Ouyang et al., 2008).
Similarly to IL-17, the ability of IBP to control IL-21 production is dependent on its interaction with IRF-4. At a mechanistic level, IBP does not affect either the expression or the subcellular localization of IRF-4. Consistent with the finding that SWAP-70 contains three nuclear localization signals and can be detected in the nucleus of activated B cells (Masat et al., 2000), IBP could be detected in the nuclear compartment of T cells where it “sequestered” IRF-4, preventing it from binding and transactivating the IL-21 promoter. The interaction of IBP and IRF-4 requires its carboxy-terminus, which is also important for the ability of IBP to activate Rho GTPases (Gupta et al., 2003a). Experiments are now in progress to determine whether the two functions of IBP map to distinct subdomains within its carboxy-terminal region. Unlike other master regulators of TH differentiation like GATA3 and RORγt, the expression of IRF-4 is upregulated by TCR stimulation and is not restricted to a specific TH effector lineage (Matsuyama et al., 1995; Rengarajan et al., 2002b). The presence of mechanisms that restrain the access of IRF-4 to selected regulatory regions may thus be particularly important to ensure that its activity can be controlled in a TH lineage-specific manner.
In contrast to the IBPtrap/trap DO11.10 mice, young IBPtrap/trap Balb/c mice do not develop obvious signs of arthritis, although evidence of a systemic autoimmune disease can be observed in these mice upon aging (unpublished observations). Interestingly, however, T cells derived from IBPtrap/trap Balb/c mice do exhibit abnormal production of both IL-21 and IL-17 upon in vitro stimulation (unpublished observations). These findings suggest that the pathogenic effector function observed in the absence of IBP becomes unmasked in the setting of a restricted TCR repertoire (i.e. in the DO11.10 TCR transgenic background) and/or of declining T cell numbers (i.e. upon aging). Given that the absence of IBP leads to an exaggerated ability to undergo homeostatic proliferation (unpublished observations), the pathogenicity of the IBP deficient T cells may become evident only after these T cells receive a signal to expand in order to replenish or maintain the mature T cell pool. It is also likely that interactions with other cellular constituents are required for IBP deficient T cells to fully acquire their pathogenic potential. Indeed, we have found that IBP deficient T cells exhibit markedly elevated ICOS expression in vivo but not in vitro suggesting that the deregulated ICOS expression observed in these mice is under complex regulation. We thus envision a scenario whereby the dysregulated production of IL-17 and IL-21 by IBP deficient T cells, via the effects of these cytokines on other cellular compartments, leads to additional aberrations within T cells eventually resulting in the development of autoimmunity.
Absence of IBP can thus result in the development of either a lupus-like syndrome or rheumatoid arthritis-like joint disease. The finding that IBP deficiency can lead to these distinct autoimmune manifestations is consistent with the fact that familial aggregation of RA and SLE has been reported (Alarcon-Segovia et al., 2005; Criswell et al., 2005). Deregulation of IL-17 and IL-21 production furthermore has been detected in murine models of RA and SLE as well as in patients affected by these two disorders (Afzali et al., 2007; Herber et al., 2007; McInnes and Schett, 2007; Sawalha et al., 2007). Blockade of the IL-21/IL-21R pathway has recently been reported to be efficacious in ameliorating disease in murine models of both lupus and RA (Herber et al., 2007; Young et al., 2007). The capacity of IBP to control both TCR responsiveness and the production of IL-17 and IL-21 thus suggest that deregulation of IBP-mediated pathways could function as a common pathogenic mechanism involved in the development of these two distinct autoimmune disorders.
MATERIALS AND METHODS
Mice
IBP deficient mice were generated by Lexicon Pharmaceuticals (Omnibank) utilizing a gene trapping strategy, and hence have been termed IBPtrap/trap mice (Fanzo et al., 2006). The original mice (on a mixed 129XC57BL/6 background) were backcrossed into either Balb/c mice or into C57BL/6 mice for >6 generations. IBPtrap/trap mice on a Balb/c background were then crossed to DO11.10 TCR transgenic mice (Jackson Laboratory) to generate IBPtrap/trapDO11.10 mice; C57BL/6 background were crossed to HY-TCR transgenic mice (Taconic) to generate IBPtrap/trapHY mice. IRF-4−/− mice on a C57BL6 background were obtained from Dr. T. Mak at Departments of Immunology and Medical Biophysics, University of Toronto and the Amgen Institute (Mittrucker et al., 1997). To generate IBPtrap/trapIRF-4−/− mice, IRF-4−/− mice were crossed with IBPtrap/trap that had been backcrossed onto the C57BL/6 background. Syngeneic SCID, Rag−/−, and nude mice were obtained from Jackson Laboratory. All mice used in the experiment were kept under specific pathogen-free conditions. The experimental protocols were approved by the Institutional Animal Care and Use Committee of Columbia University.
Flow Cytometry
Single cell suspensions from thymus, spleen, and lymph nodes were isolated, stained with fluorochrome-conjugated KJ1-26 (Caltag laboratories), CD3ε, CD4, CD5, CD8, B220, CD25, CD44, CD62L, CD69, (Pharmingen), T3.70, and ICOS antibodies (eBiosciences) and analyzed by FACS. Data were analyzed using FlowJo (Treestar) software.
Clinical assessment of arthritis
Joint swelling was monitored by inspection and scored as follows: 0, no evidence of erythema and swelling; 1, erythema and mild swelling confined to the wrist or ankle; 2, erythema and mild swelling extending from the wrist or ankle to the mid-paw; 3, erythema and moderate swelling extending from the wrist or ankle to the mid-paw; 4, erythema and severe swelling encompassing the wrist or ankle, paws, and digits. Score for all 4 paws were totaled for each mouse (Brand et al., 2004).
Histopathology and immunohistochemical staining
Tissue specimens were fixed in 10% neutral buffered formalin and embedded in paraffin. Tissue sections were stained with hematoxylin and eosin (H&E) and analyzed by light microscopy. Joints were fixed in 10% phosphate-buffered formalin, decalcified in 10% EDTA-4Na, and embedded in paraffin. Immunostaining of spleen sections with antibodies against CD3, Blimp-1, and Syndecan-1 was performed as previously described (Angelin-Duclos et al., 2000; Fanzo et al., 2006).
Serum autoantibodies
Serum levels of Rheumatoid Factor (RF) and of antibodies against Collagen II, cyclic citrullinated peptide (CCP), and double-stranded DNA (dsDNA) were determined by ELISA (Alpha Diagnostics for RF, CII, and dsDNA Abs and Axis-Shield Diagnostics for CCP Abs) as described (Kuhn et al., 2006; Sakaguchi et al., 2003). Data were analyzed using Student’s t-test.
In vivo thymocyte deletion
6-week-old mice were injected with 250 μg of anti-CD3 antibody i.p. as previously described (Sakaguchi et al., 2003). 72 hours later, mice were sacrificed and the thymus collected. Total thymocytes were counted, stained for CD4 and CD8, and analyzed by FACS.
Proliferation studies and cytokine production
Naïve CD4+ T cells were purified as previously described (Lin et al., 2004) or, in selected experiments, by sorting CD44loCD62LhiCD25−CD4+ T cells. For proliferation assays, cells were cultured at 1×105 per well in 96-well-plate together with 4×105 syngeneic IBP+/+ APCs loaded with increasing doses (0, 0.1, 1, 10 μM) of OVA323-339 peptide for 48 hours as described (Song et al., 2005) and then pulsed with BrdU for 18 hours. Incorporated BrdU was measured by BrdU ELISA (Roche-Applied-Science). For cytokine analysis 1×106 cells/ml were stimulated with 4×106 APC loaded with OVA323-339 peptide at 1μM concentration in 24-well-plates. TH0 and TH2 differentiation was conducted as previously described (Fanzo et al., 2006). For TH17 differentiation, naïve purified CD4 T cells were activated by plate-bound anti-CD3ε and soluble anti-CD28 in the presence of anti-IFNγ (10μg/ml, BD Biosciences), anti-IL4 (10μg/ml, BD Biosciences), IL6 (20ng/ml, Peprotech) and TGF-β (5ng/ml, Peprotech) as previously described (Ivanov et al., 2006). Supernatants were analyzed for IL-21 (R&D Systems), IL-17, IFN-γ (eBioscience), TGF-β (eBioscience) and IL-2 (eBioscience) production by ELISA.
Transient transfection assays
Transient transfection assays were performed as previously described (Hu et al., 2002). Briefly, 10 × 106 control or IRF4-expressing Jurkat cells were transfected with 5 μg of a murine IL-21 promoter luciferase reporter plasmid (Mehta et al., 2005) by electroporation at 260V and 960μF with a BTX electroporator. 100-200 ng of the pRL-TK reporter plasmid expressing renilla luciferase under the control of the thymidine kinase promoter was added to each transfection as a transfection efficiency control. After transfection, the cells were allowed to recover for 16hr at 37°C, 6% CO2. The cells were then collected and resuspended in 3 ml of media and equally split into two 1.5 ml aliquots. The aliquots were then cultured in the presence or absence of PMA (50 ng/ml) and ionomycin (1 μM) for 4 hrs. The transfected cells were then harvested, lysed, and assayed for luciferase activities with the Dual Luciferase Assay System (Promega) according to the manufacturer’s instructions. The firefly luciferase activity was normalized on the basis of renilla luciferase activity.
Cell extracts, protein assays
Nuclear and cytoplasmic extracts were prepared utilizing the NE-PER Nuclear and Cytoplasmic extraction reagent kit as previously described (So et al., 2006). The purity of the nuclear and cytoplasmic fractions was verified by probing with antibodies against Lamin B (sc-6217, Santa Cruz Biotechnology) and b-tubulin (clone D66, Sigma-Aldrich). Cell lysates were immunoprecipitated with an anti-IBP antibody or anti-IRF-4 Ab as previously described (Gupta et al., 2003a). The immunoprecipitates were resolved by 8% SDS-PAGE, transferred to a nitrocellulose membrane, and then immunoblotted with an anti-IRF-4 antibody or the anti-IBP antiserum. The protein bands were visualized by ECL (Amersham). Chromatin immunoprecipitation (ChIP) assays were performed as previously described (Sciammas et al., 2006). Briefly, CD4+ T cells were purified from wt mice and IBPtrap/trap mice and either left unstimulated or stimulated for 24 hrs under unskewed conditions. After harvesting, the cells were cross-linked with formaldehyde and chromatin extracts were prepared by standard methods. The DNA/protein complexes were immunoprecipitated with an IRF-4 specific antibody or a control antibody. After cross-linking was reversed and proteins digested, the DNA was purified from the immunoprecipitates as well as from input extracts, and then analyzed by PCR using primers specific for the murine IL-21 promoter (5′CCCTTGTGAATGCTGAAAACTG3′ and 5′GGCCTTGGTCTGGTTCTCACT3′), the IL-17 promoter (5′GCGGTACTCAGTTAAATAGAACGT3′ and 5′ATTAGTGCAGGACTCACCACAGA3′) or β-actin (5′GCTCCTCCTGAGCGCAAGT3′ and 5′TCGTCATACTCCTGCTTGCTGAT3′). Oligonucleotide precipitation (ONP) assays were conducted as previously described (Azam et al., 1997). Briefly, nuclear extracts were precleared with streptavidin-agarose beads and then incubated with biotinylated double-stranded oligonucleotide corresponding to the trimerized IRF-4 binding site within the IL-21 promoter (5′CCTTGGTGAATGCTGAAAACTGGAATTCACCCAT3′). Proteins bound to the biotin-labeled DNA were collected by streptavidin-agarose beads, separated by 8% SDS-PAGE and then analyzed by Western blotting.
Real-time RT-PCR
Total RNA was isolated from cells or organs using RNeasy Mini Kit (Qiagen GmbH). cDNAs were prepared and analyzed for the expression of the gene of interest by real-time PCR using a Sybr-Green PCR master mix kit (Applied Biosystems). The primers for IL-17, IL-21, IL-22, and RORγt genes have been previously described (Chung et al., 2006; Lubberts et al., 2005; Wurster et al., 2002; Xi et al., 2006). The expression of each gene was normalized to the expression of β-Actin.
Adoptive transfers
For splenocyte transfers, cells suspensions were obtained from either arthritic IBPtrap/trapDO11.10 mice or age- and sex- matched IBP+/+DO11.10 mice, and 2×107 cells were injected i.v. into syngeneic SCID mice. For T cell transfers, lymphocytes were pooled from peripheral lymph nodes and spleen, total CD4+ T cells were positively selected by MACS magnetic beads sorting (Miltenyl Biotec) and then stained with the KJ1-26 Ab (Caltag laboratories). KJ1-26high and KJ1-26low cells were sorted by FACSDiva (BD Biosciences). The sorted cells were then washed and 2×106 cells were injected i.v. into syngeneic nude mice.
Regulatory T cell suppression assays
CD4+ T cells (5 × 104) from DO11.10 IBP+/+ mice were cultured in 96-well plates with T cell depleted Mitomycin C (Sigma) treated APCs (2 × 105) pulsed with OVA323-339 (1 μM) in the presence/absence of increasing numbers of FACS sorted CD4+CD25+ regulatory T cells from IBP+/+ DO11.10 or IBPtrap/trap DO11.10 mice for 72 hrs as previously described (Thornton and Shevach, 2000). For proliferation assays, cultures were pulsed with [3H]-thymidine (PerkinElmer, MA) for the last 12 h of culture.
Supplementary Material
Acknowledgments
We would like to thank Dr. C. Schindler. Dr. Hua Gu and Dr. Steve Greenberg for their critical reading of the manuscript and discussions. The authors also thank Dr. G. Cattoretti for help with immunohistochemical staining. The research was supported by NIH grants R01 HL-62215, the Lupus Research Institute, the Alliance for Lupus Research, a Pilot Award from the National Multiple Sclerosis Society, and an American Heart Association Grant-in-Aid to A. P. Support to Q.C. has been provided by an SLE Lupus Foundation Fellowship.
Abbreviations
- IRF-4
Interferon Regulatory Factor 4
- IBP
IRF-4 Binding Protein
- SLE
Systemic Lupus Erythematosus
- RA
Rheumatoid Arthritis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
BIBLIOGRAPHY
- Afzali B, Lombardi G, Lechler RI, Lord GM. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol. 2007;148:32–46. doi: 10.1111/j.1365-2249.2007.03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon-Segovia D, Alarcon-Riquelme ME, Cardiel MH, Caeiro F, Massardo L, Villa AR, Pons-Estel BA. Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum. 2005;52:1138–1147. doi: 10.1002/art.20999. [DOI] [PubMed] [Google Scholar]
- Angelin-Duclos C, Catoretti G, Lin K-I, Calame K. Commitment of B lymphocytes to a plasma cell fate is associated with Blimp-1 expression in vivo. J Immunol. 2000;165:5462–5471. doi: 10.4049/jimmunol.165.10.5462. [DOI] [PubMed] [Google Scholar]
- Azam M, Lee C, Strehlow I, Schindler C. Functionally distinct isoforms of STAT5 are generated by protein processing. Immunity. 1997;6:691–701. doi: 10.1016/s1074-7613(00)80445-8. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Korn T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol. 2007a;19:652–657. doi: 10.1016/j.coi.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007b;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- Brand DD, Kang AH, Rosloniec EF. The mouse model of collagen-induced arthritis. Methods Mol Med. 2004;102:295–312. doi: 10.1385/1-59259-805-6:295. [DOI] [PubMed] [Google Scholar]
- Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, Arpaia E, Mak TW, Kamradt T, Lohoff M. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol. 2007;8:958–66. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- Chung Y, Yang X, Chang SH, Ma L, Tian Q, Dong C. Expression and regulation of IL-22 in the IL-17-producing CD4+ T lymphocytes. Cell Res. 2006;16:902–907. doi: 10.1038/sj.cr.7310106. [DOI] [PubMed] [Google Scholar]
- Criswell LA, Pfeiffer KA, Lum RF, Gonzales B, Novitzke J, Kern M, Moser KL, Begovich AB, Carlton VE, Li W, et al. Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. Am J Hum Genet. 2005;76:561–571. doi: 10.1086/429096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanzo JC, Yang W, Jang SY, Gupta S, Chen Q, Siddiq A, Greenberg S, Pernis AB. Loss of IRF-4-binding protein leads to the spontaneous development of systemic autoimmunity. J Clin Invest. 2006;116:703–714. doi: 10.1172/JCI24096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Fanzo J, Hu C, Cox D, Jang S, Lee A, Greenberg S, Pernis A. T cell receptor engagement leads to the recruitment of IBP, a novel guanine nucleotide exchange factor, to the immunological synapse. J Biol Chem. 2003a;278:43451–43459. doi: 10.1074/jbc.M308960200. [DOI] [PubMed] [Google Scholar]
- Gupta S, Lee A, Hu C, Fanzo J, Goldberg I, Cattoretti G, Pernis AB. Molecular cloning of IBP, a SWAP-70 homologous GEF, which is highly expressed in the immune system. Human Immunol. 2003b;64:389–401. doi: 10.1016/s0198-8859(03)00024-7. [DOI] [PubMed] [Google Scholar]
- Habu K, Nakayama-Yamada J, Asano M, Saijo S, Itagaki K, Horai R, Yamamoto H, Sekiguchi T, Nosaka T, Hatanaka M, Iwakura Y. The human T cell leukemia virus type I-tax gene is responsible for the development of both inflammatory polyarthropathy resembling rheumatoid arthritis and noninflammatory ankylotic arthropathy in transgenic mice. J Immunol. 1999;162:2956–2963. [PubMed] [Google Scholar]
- Herber D, Brown TP, Liang S, Young DA, Collins M, Dunussi-Joannopoulos K. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J Immunol. 2007;178:3822–3830. doi: 10.4049/jimmunol.178.6.3822. [DOI] [PubMed] [Google Scholar]
- Hu CM, Jang SY, Fanzo JC, Pernis AB. Modulation of T cell cytokine production by interferon regulatory factor-4. J Biol Chem. 2002;277:49238–49246. doi: 10.1074/jbc.M205895200. [DOI] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19:409–417. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieper WC, Burghardt JT, Surh CD. A role for TCR affinity in regulating naive T cell homeostasis. J Immunol. 2004;172:40–44. doi: 10.4049/jimmunol.172.1.40. [DOI] [PubMed] [Google Scholar]
- Kim HP, Korn LL, Gamero AM, Leonard WJ. Calcium-dependent activation of interleukin-21 gene expression in T cells. J Biol Chem. 2005;280:25291–25297. doi: 10.1074/jbc.M501459200. [DOI] [PubMed] [Google Scholar]
- Koenders MI, Joosten LA, van den Berg WB. Potential new targets in arthritis therapy: interleukin (IL)-17 and its relation to tumour necrosis factor and IL-1 in experimental arthritis. Ann Rheum Dis. 2006;65(Suppl 3):iii29–iii33. doi: 10.1136/ard.2006.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, Holers VM. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest. 2006;116:961–973. doi: 10.1172/JCI25422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Leonard WJ, Spolski R. Interleukin-21: a modulator of lymphoid proliferation, apoptosis and differentiation. Nat Rev Immunol. 2005;5:688–698. doi: 10.1038/nri1688. [DOI] [PubMed] [Google Scholar]
- Lin L, Spoor MS, Gerth AJ, Brody SL, Peng SL. Modulation of Th1 activation and inflammation by the NF-kappaB repressor Foxj1. Science. 2004;303:1017–1020. doi: 10.1126/science.1093889. [DOI] [PubMed] [Google Scholar]
- Lubberts E, Schwarzenberger P, Huang W, Schurr JR, Peschon JJ, van den Berg WB, Kolls JK. Requirement of IL-17 receptor signaling in radiation-resistant cells in the joint for full progression of destructive synovitis. J Immunol. 2005;175:3360–3368. doi: 10.4049/jimmunol.175.5.3360. [DOI] [PubMed] [Google Scholar]
- Mamane Y, Sharma S, Graandvaux N, Hernandez E, Hiscott J. IRF-4 activities in HTLV-I-induced T cell leukemogenesis. J Interf Cytok Res. 2002;22:135–143. doi: 10.1089/107999002753452746. [DOI] [PubMed] [Google Scholar]
- Masat L, Caldwell J, Armstrong R, Khoshnevisan H, Jessberger R, Herndier B, Wabl M, Ferrick D. Association of SWAP-70 with the B cell antigen receptor complex. Proc Natl Acad Sci USA. 2000;97:2180–2184. doi: 10.1073/pnas.040374497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama T, Grossman A, Mittrucker H, Siderovski D, Kiefer F, Kawakami T, Richardson C, Taniguchi T, Yoshinaga S, Mak T. Molecular cloning of LSIRF, a lymphoid-specific member of the ineterferon regulatory factor family that binds the interferon-stimulated response element (ISRE) Nuc Ac Res. 1995;23:2127–2136. doi: 10.1093/nar/23.12.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- Mehta DS, Wurster AL, Grusby MJ. Biology of IL-21 and the IL-21 receptor. Immunol Rev. 2004;202:84–95. doi: 10.1111/j.0105-2896.2004.00201.x. [DOI] [PubMed] [Google Scholar]
- Mehta DS, Wurster AL, Weinmann AS, Grusby MJ. NFATc2 and T-bet contribute to T-helper-cell-subset-specific regulation of IL-21 expression. Proc Natl Acad Sci U S A. 2005;102:2016–2021. doi: 10.1073/pnas.0409512102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittrucker H, Matsuyama T, Grossman A, Kundig T, Potter J, Shahinian A, Wakeham A, Patterson B, Ohashi P, Mak T. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science. 1997;275:540–543. doi: 10.1126/science.275.5299.540. [DOI] [PubMed] [Google Scholar]
- Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan J, Mowen KA, McBride KD, Smith ED, Singh H, Glimcher LH. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med. 2002a;195:1003–1012. doi: 10.1084/jem.20011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan J, Mowen KA, Mcbride KD, Smith ED, Singh H, Glimcher LH. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med. 2002b;195:1003–1012. doi: 10.1084/jem.20011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JMJP, Evavold BD. DO11.10 and OT-II T cells recognize a C-terminal ovalbumin 323-339 epitope. J Immunol. 2000;164:4706–4712. doi: 10.4049/jimmunol.164.9.4706. [DOI] [PubMed] [Google Scholar]
- Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, Sakihama T, Matsutani T, Negishi I, Nakatsuru S, Sakaguchi S. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 2003;426:454–460. doi: 10.1038/nature02119. [DOI] [PubMed] [Google Scholar]
- Sawalha AH, Kaufman KM, Kelly JA, Adler AJ, Aberle T, Kilpatrick J, Wakeland EK, Li QZ, Wandstrat AE, Karp DS, et al. Genetic association of IL-21 polymorphisms with systemic lupus erythematosus. Ann Rheum Dis. 2007;67:458–61. doi: 10.1136/ard.2007.075424. [DOI] [PubMed] [Google Scholar]
- Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. 2006;25:225–236. doi: 10.1016/j.immuni.2006.07.009. [DOI] [PubMed] [Google Scholar]
- So T, Song J, Sugie K, Altman A, Croft M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc Natl Acad Sci U S A. 2006;103:3740–3745. doi: 10.1073/pnas.0600205103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, So T, Cheng M, Tang X, Croft M. Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity. 2005;22:621–631. doi: 10.1016/j.immuni.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Stamp LK, James MJ, Cleland LG. Interleukin-17: the missing link between T-cell accumulation and effector cell actions in rheumatoid arthritis? Immunol Cell Biol. 2004;82:1–9. doi: 10.1111/j.1440-1711.2004.01212.x. [DOI] [PubMed] [Google Scholar]
- Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- Toh ML, Miossec P. The role of T cells in rheumatoid arthritis: new subsets and new targets. Curr Opin Rheumatol. 2007;19:284–288. doi: 10.1097/BOR.0b013e32805e87e0. [DOI] [PubMed] [Google Scholar]
- Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol. 2005;5:853–865. doi: 10.1038/nri1714. [DOI] [PubMed] [Google Scholar]
- Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- Wurster AL, Rodgers VL, Satoskar AR, Whitters MJ, Young DA, Collins M, Grusby MJ. Interleukin 21 is a T helper (Th) cell 2 cytokine that specifically inhibits the differentiation of naive Th cells into interferon gamma-producing Th1 cells. J Exp Med. 2002;196:969–977. doi: 10.1084/jem.20020620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H, Schwartz R, Engel I, Murre C, Kersh GJ. Interplay between RORgammat, Egr3, and E proteins controls proliferation in response to pre-TCR signals. Immunity. 2006;24:813–826. doi: 10.1016/j.immuni.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- Young DA, Hegen M, Ma HL, Whitters MJ, Albert LM, Lowe L, Senices M, Wu PW, Sibley B, Leathurby Y, et al. Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum. 2007;56:1152–1163. doi: 10.1002/art.22452. [DOI] [PubMed] [Google Scholar]
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–74. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.