

Abstract
Alterations in the tissue microenvironment collaborate with cell autonomous genetic changes to contribute to neoplastic progression. The importance of the microenvironment in neoplastic progression is underscored by studies demonstrating that fibroblasts isolated from a tumor stimulate the growth of preneoplastic and neoplastic cells in xenograft models. Similarly, senescent fibroblasts promote preneoplastic cell growth in vitro and in vivo. Because senescent cells accumulate with age, their presence is hypothesized to facilitate preneoplastic cell growth and tumor formation in older individuals. To identify senescent stromal factors directly responsible for stimulating preneoplastic cell growth, we carried out whole genome transcriptional profiling and compared senescent fibroblasts to their younger counterparts. We identified osteopontin (OPN) as one of the most highly elevated transcripts in senescent fibroblasts. Importantly, reduction of OPN protein levels by RNAi did not impact senescence induction in fibroblasts; however, it dramatically reduced the growth-promoting activities of senescent fibroblasts in vitro and in vivo, demonstrating that OPN is necessary for paracrine stimulation of preneoplastic cell growth. In addition, we found that recombinant OPN was sufficient to stimulate preneoplastic cell growth. Finally, we demonstrate that OPN is expressed in senescent stroma within preneoplastic lesions that arise following DMBA/TPA treatment of mice, suggesting that stromal-derived OPN-mediated signaling events impact neoplastic progression.
Keywords: osteopontin, microenvironment, senescence, tumorigenesis, fibroblast
Introduction
Age is the greatest risk factor for the development of neoplasia (1, 2). The molecular and physiologic factors that underlie age-related increases in cancer incidence are diverse and include cell autonomous genetic and epigenetic alterations and coincident alterations in the tissue microenvironment. Genetic analysis of human cancers has revealed a myriad of mutations that disrupt diverse cellular signaling pathways. Cell culture systems and murine models have demonstrated that disruption of pathways that regulate tissue homeostasis, cell growth, cell survival/death and invasiveness cooperate to produce a transformed cell (3-5). While these cell autonomous alterations are critical to tumorigenesis, preneoplastic and neoplastic lesions arise within the context of a complex stromal compartment that consists of numerous cell types. Among these are epithelial cells, fibroblasts, immune cells, and cells that contribute to angiogenesis that together work to enable transformation.
The importance of the host microenvironment in the tumorigenic process is supported by numerous studies. For example, it has been shown that chickens infected with the Rous sarcoma virus develop tumors at sites of injury (6) and that activation of mast cells enhances transformation in a skin model of carcinogenesis (7). These studies suggest that the inflammatory response plays a key role in tumorigenesis. In further support of this hypothesis, other studies have demonstrated that the chronic inflammation caused by H. pylori infection or Crohn’s disease predisposes affected individuals to cancer (8, 9). In addition to inflammatory cells, fibroblasts can also promote tumorigenesis. Under normal conditions, fibroblasts are responsible for matrix deposition and reorganization and facilitate recruitment of cells that participate in tissue homeostasis. However, fibroblasts present within neoplastic lesions display altered expression profiles compared to fibroblasts isolated from normal tissues. Indeed, the gene expression profiles found in cancer associated fibroblasts (CAFs) are reminiscent of those found within a wound, and include expression of numerous growth factors, chemokines, and angiogenic factors (reviewed in (10, 11)). A role for CAFs in promoting transformation was demonstrated in one study in which CAFs isolated from prostate carcinomas were found to stimulate the in vitro and in vivo growth of immortalized prostate epithelial cells whereas prostate fibroblasts distal to tumors had no such capacity (12).
In addition to the increased mutational load and presence of preneoplastic cells found to arise in aging epithelia, (13, 14), the stromal compartment also undergoes age-related changes. Indeed, senescent cells accumulate in tissues with age (15-17). Analysis of senescent fibroblasts reveals a significantly altered gene expression profile relative to their younger counterparts (18, 19). Interestingly this altered gene expression profile mirrors that observed during wound repair (reviewed in (10, 11)). Prominent among genes whose expression is dramatically altered during wound repair are those that remodel the extracellular matrix as well as growth factors and inflammatory cytokines. Moreover, senescent fibroblasts function analogously to CAFs in that they promote the in vitro and in vivo growth of preneoplastic cells (19, 20). This latter observation suggests that the age-related accumulation of senescent stromal cells and the alterations that they produce within the tissue, collaborate with increasing numbers of preneoplastic cells to influence cancer incidence in older individuals. However, how senescent stromal cells function to promote tumorigenesis is still poorly understood because the panoply of factors secreted by these cells has yet to be fully identified.
Our goal was to identify senescent stromal factors that impact preneoplastic cell growth in order to begin to delineate the molecular mechanisms by which senescent stroma functions in tumorigenesis. To this end, we performed expression profiling of replicative senescent (RS) and stress-induced premature senescent (SIPS) fibroblasts and found that numerous factors were coordinately modulated. This study identifies one of these factors, osteopontin (OPN) as a critical mediator of stromal-epithelial interactions both in vitro and in vivo. We demonstrate that OPN is sufficient to stimulate the growth of preneoplastic cells. Finally, we show that senescent stroma expressing OPN can be found within premalignant lesions in vivo, supporting the physiologic relevance of these results and the potential role of OPN in promoting tumorigenesis.
Materials and Methods
Cell Lines
Human foreskin BJ fibroblasts and 293T cells were grown as previously described (REF). HaCaT cells are spontaneously immortalized human keratinocytes; are aneuploid, nontumorigenic and retain a near normal pattern of differentiation (21). They were grown in DME plus 10% heat-inactivated FCS. N.p.c.T human keratinocytes were a kind gift from James Rheinwald (Harvard, MA) and were grown as previously described (22). N.p.c.T. are human epidermal keratinocytes that were derived from newborn foreskin and transduced with human telomerase, mutant CDK4 (R24C), and a p53 mutant (p53DD). These cells are immortal but retain the ability to undergo an epidermis-type suprabasal differentiation program as evidenced by the ability to form a granular cell layer and an enucleated stratum corneum (22).
Virus production and retroviral infections
Microarray analysis
BJ fibroblasts were mock or Bleomycin sulfate-treated (100ug/ml, Sigma, St. Louis, MO) for 24 hrs. Replicatively senescent fibroblasts were obtained by continuous passage. After 72 hr serum-starvation, RNA was collected using TRIzol (Invitrogen, Carlsbad, CA). Biotinylated cRNA was hybridized to Affymetrix Human Genome U133 Plus 2.0 GeneChips (Affymetrix, Santa Clara, CA) by the Washington University Microarray Facility. There were 4, 6, and 6 samples for SIPS, RS, and young respectively. Microarray quality control, analysis, and clustering (UPGMA by Centroid) were performed using dChip (May, 2008 release) (23). All GeneChip comparisons had a cut-off basis of a lower bound of 90% confidence of fold-change ≥1.5 and expression intensity difference ≥75. Gene Ontological categorization of probe sets was performed using the Gene Ontology database, current to publication date. GO terms used: Immune & Inflammation (IDs: 6955, 9615, 42742, 6952, 6954), Mitogens and Regulation of Proliferation (IDs: 7067, 85, 45840, 86, 82, 45930, 7049, 45786, 187, 186, 1558, 8283), Extracellular Matrix and Secreted Factors (IDs: 7596, 7160, 7155, 5604, 8243, 42730, 7596, 5201, 5581, 4252, 8237, 5125, 8008).
Co-Culture conditions
1.2×104 fibroblasts were plated in 96 well plates (Fisher Scientific, Pittsburgh, PA) and treated as described above. HaCaT- (1.0 ×103) or N.p.c.T- (2.0×103) CBR cells were plated on fibroblasts and incubated for 96 hr. Luciferase (Chroma-Glo Luciferase System, Promega) readings were performed in a Bio Tek microplate reader (Bio Tek Instruments, Winooski, VT).
OPN shRNA and Real-Time PCR
Three shRNA plasmids targeting the human OPN gene sequence were provided in the pLKO.1 plasmid (sequences found on Mission shRNA website; Sigma, St. Louis, MO). Standard protocol was followed for cDNA and RT-PCR using the following primers: OPN, forward TTGCAGCCTTCTCAGCCAA and reverse CAAAAGCAAATCACTGCA ATTCTC, GAPDH, forward GCATGGCCTTCCGTGTCC, reverse AATGCCAGCCCCAGCGTCAAA.
OPN Immunoprecipitation
BJ fibroblasts plated in serum-free for 72 hrs. NETN lysis buffer was added to media (20 mM Tris-HCl (pH 7.9), 1 mM EDTA, 100 mM NaCl, and 0.5% NP40) and incubated overnight at 4°C with OPN antibody (AF1433; R&D, Minneapolis, MN).
Recombinant human OPN (rhOPN)
HaCaT cells were plated in a 96 well plate (1.0×103) and incubated in DME/F12 supplemented with 0.1% FCS for 24 hrs. 100 ng/ml rhOPN (R&D, Minneapolis, MN) was added for 72hrs. N.p.c.T cells were plated overnight (1.0×103) and 100ng/ml rhOPN was added in 5% K-sfm/DME/F12 (Invitrogen, Carlsbad, CA) for 72 hrs.
Xenograft model and chemical carcinogenesis protocol
Female NOD/SCID mice (NCI-Fredrick) were housed according to the guidelines of DCM, Washington University School of Medicine. Live in vivo imaging as described (24). Two-step chemical carcinogenesis protocol as previously described (35) on 129S6/SvEv female mice (Taconic, Germantown, NY). To obtain papillomas outbred transgenic mice (RBP-jflox/+ and K14CreERT; N1flox/+; RBP-jflox/+) were treated topically with DMBA (25ug) followed by twice weekly TPA treatments (4ug) for 15 weeks starting one week after DMBA treatment.
Senescent-associated β-galactosidase
Staining was performed on cells and 10um frozen sections as previously described (16).
Immunohistochemistry
Dissected tissue was fixed for 1hr in 4% paraformaldehyde and embedded in paraffin or OCT freezing medium (Sakura Finetek U.S.A, Torrance, CA) and stained with the following antibodies: OPN 1:400 (AF808, R&D, Minneapolis, MN); p16 1:50 (antigen retrieval in pH 9 Tris-buffer; sc-1661, Santa Cruz Biotechnology, Santa Cruz, CA); CD45 1:60 (550539, BD PharMingen, San Diego, CA); F4/80 (MCA497G, Serotec, Raleigh, NC). Detection was visualized with Dako and Vector kits (Dako, Carpinteria, CA and Vector, Burlingame, CA)
Statistical Analysis
Data represent the mean ± SEM; statistical significance was assessed by a two-tailed Student’s T-test.
Results
Senescent BJ fibroblasts stimulate the growth of preneoplastic cells in vitro and in vivo
The tumor microenvironment is an important contributor to neoplastic progression and an increasing body of evidence suggests that it also plays a pivotal role in the early stages of the transformation process. One factor that impacts the stromal compartment is age. Indeed, the presence of senescent cells increases with age (15-17) and these cells possess the capacity to promote preneoplastic cell growth in several models (19, 20, 25, 26). To address the molecular mechanism by which senescent fibroblasts mediate preneoplastic cell growth, we first established a culture system that allowed us to quantitate cell growth in vitro. Because the presence of senescent cells within the skin of aging individuals increases with age (15-17) and our microarray analysis (see below) was carried on human skin fibroblasts, we choose to utilize preneoplastic immortalized keratinocytes (HaCaT and N.p.c.T.) for our studies (21, 22). To facilitate quantification of HaCaT and N.p.c.T. cell growth, we transduced them with click beetle red luciferase (CBR) and grew them in the presence of young or senescent BJ fibroblasts. Senescence can be induced by continued passage leading to replicative senescence (RS), which is believed to be telomere based or by any number of other methods collectively referred to as stress-induced premature senescence (SIPS) (19, 27, 28), all of which have been shown to stimulate the growth of preneoplastic cells (19, 25, 26, 29). Therefore, we chose to utilize bleomycin treatment, which reliably induces senescence of BJ skin fibroblasts (Fig. 1A). To confirm that cells undergoing RS and SIPS equivalently stimulated preneoplastic growth, we compared their ability to stimulate the growth of HaCaT cells. As shown in Fig. 1B, RS and SIPS fibroblasts were equally proficient at inducing the growth of HaCaT cells compared to HaCaT cells grown in the presence of young BJ fibroblasts. In addition, live animal luciferase imaging revealed that BJ fibroblasts undergoing RS or SIPS efficiently stimulated the growth of HaCaT xenografts in vivo (Fig. 1C and data not shown). Histological analysis of xenografts confirmed that observed by our live animal imaging. Injection of HaCAT cells and senescent fibroblasts resulted in large benign lesions. In contrast, injection of HaCAT cells alone or in the presence of young fibroblasts resulted in small, nearly undetectable lesions (Fig. 1D).
Figure 1. Senescent fibroblasts stimulate the growth of preneoplastic cells.

A, continued passage (RS) and treatment with 100 ug/ml bleomycin (SIPS) induces robust senescence characterized by induction of senescence associated β–galactosidase (SA-βGal) staining. Magnification 20X. B, fibroblasts undergoing RS or SIPS stimulate the in vitro growth of HaCaT keratinocytes expressing click beetle red luciferase. HaCaT cells were plated on lawns of young, RS, or SIPS fibroblasts and their growth was measured by relative luciferase activity. Wells containing HaCAT cells and young fibroblasts were set to 1. Fold increase in growth over wells with young fibroblasts is plotted. p<0.05. C, HaCAT cells were injected alone or with young or senescent fibroblasts (RS) into NOD/SCID mice. For each injection, 2.5×105 HaCaT cells were coinjected with 7.5×105 fibroblasts. In vivo imaging of luciferase activity was used to assess HaCaT cell growth weekly. Each line represents the average photons/sec for 6 independent injection sites. Student’s t test at day 28, p < 0.05. D, Hematoxylin and eosin stains of histological sections obtained from xenografts injected with HaCaT cells alone or in the presence of young or senescent fibroblasts (top to bottom). Magnification 10X.
Analysis of the senescent transcriptome
Given the observed stimulation of HaCaT cell growth after coculture with both RS and SIPS fibroblasts, we next carried out microarray analysis to identify putative senescent-associated candidate genes. Given that fibroblasts undergoing RS or SIPS similarly induced the growth of preneoplastic cells (20) (Fig. 1B), we hypothesized that a common core of genes was responsible for their growth-promoting activities. Therefore, we conducted microarray analysis under conditions that captured the cell culture conditions utilized in our coculture experiments (i.e. serum-free and 3% O2). Using these conditions, RNA was isolated from BJ skin fibroblasts that had undergone RS or SIPS following bleomycin treatment and compared the expression profile to young BJ fibroblasts.
Analysis of the resultant microarray data revealed a significant alteration in gene expression in cells undergoing senescence versus their younger counterparts. As expected, gene expression differences were observed between cells undergoing RS versus SIPS. However, a significant overlap in the gene expression patterns of RS and SIPS fibroblasts was apparent (Fig. 2A). Indeed, when comparing genes differentially expressed in each senescent population compared to young fibroblasts, 354 were coordinately regulated in RS and SIPS fibroblasts (Fig. 2B). GO analysis of the data revealed that the coordinately regulated gene groups included proliferative and mitogenic genes, genes involved in extracellular matrix (ECM) functions and inflammation (Fig. 2C). Many of these genes code for secreted growth factors, chemokines, wound repair proteins, and matrix remodeling proteins, which have been implicated in tumor stroma-epithelial interactions (10, 11). We also found that senescent fibroblasts increased expression of AREG and MMP3, which impact preneoplastic cell growth and morphology, respectively (19, 27). These observations demonstrate that senescent fibroblasts possess a significantly altered gene expression pattern compared to young fibroblasts and supports the hypothesis that activation of senescence is likely to alter the microenvironment, leading to enhanced preneoplastic cell growth and tumor formation in a manner analogous to cancer associated fibroblasts (CAFs).
Figure 2. Analysis of the senescence transriptome reveals significant overlap between cells undergoing replicative and stress-induced premature senescence.
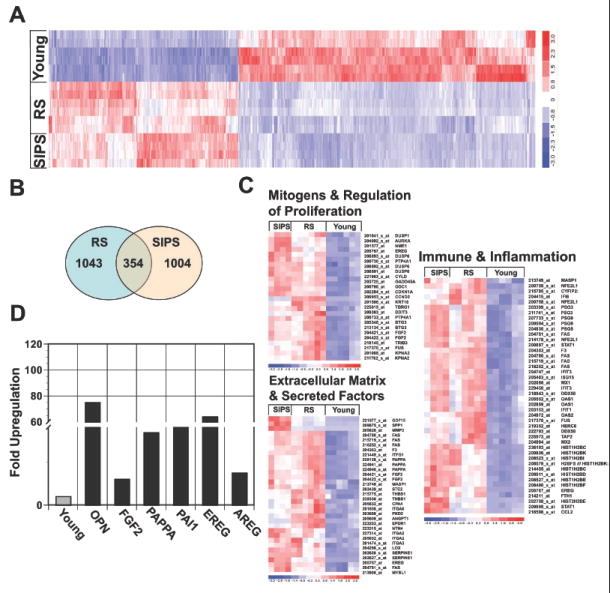
A, microarray analysis of young BJ fibroblasts and BJ fibroblasts undergoing RS or bleomycin induced SIPS. Hierarchical clustering of consistent transcript alterations across all conditions and replicas are plotted. B, Venn diagram plots genes that significantly overlap in fibroblasts undergoing RS and SIPS versus young. C, Gene ontology analysis (GO) of upregulated classes in cells undergoing RS and SIPS compared to their younger counterparts (i.e. Immune & Inflammation, Mitogens & Regulation of Proliferation, Extracellular Matrix & Secreted Factors). D, Fold upregulation of select genes in senescent fibroblasts (SIPS) was confirmed by qRT-PCR. Expression in young fibroblasts was set as one for all genes.
To validate our microarray results, we chose a subset of target genes based on their reported secretory and mitogenic properties, reasoning that they would impact preneoplastic cell growth. Pregnancy-associated plasma protein A (PAPPA) is a metalloprotease that cleaves insulin-like growth factor binding protein 4 (IGFBP4) – an inhibitor of insulin growth factor (IGF) (30). The direct result of PAPPA’s activity is increased bioavailability of IGF, which is a potent growth-regulatory protein. Amphiregulin (AREG) is a member of the epidermal growth factor family that stimulates the growth and proliferation of epithelial cells. It was previously documented that AREG is expressed in senescent fibroblasts and functions in a paracrine fashion to stimulate the growth of prostate epithelial cells (19). PAI-1, an inhibitor of tissue plasminogen activator (tPA) and urokinase (uPA) was previously shown to mediate induction of senescence (31). Finally, we examined the expression of osteopontin (OPN), a multifunctional secreted glycoprotein that has been implicated in diverse stages of carcinogenesis (reviewed in (32)). qRT-PCR analysis verified the upregulation pattern observed in the microarray for all of the selected genes (Fig. 2D).
Identification of stromal-derived OPN in preneoplastic lesions coincident with senescent stroma
Our microarray data indicated that OPN was significantly upregulated in senescent fibroblasts, suggesting that it impacts preneoplastic cell growth. OPN was originally identified as a transformation-associated matricellular protein produced by cancer cells (33). OPN is a secreted protein that is extensively modified by phosphorylation and glycosylation (34) and is targeted by several metalloproteinases (35). While OPN contributes to mineralization and ECM integrity, it also functions as a potent signaling cytokine (34). Intriguingly, OPN-null mice display a significant delay in the appearance and incidence of papillomas in a classical two-stage skin chemical carcinogenesis model (36), suggesting that OPN plays a key role in skin tumorigenesis. To date, studies have focused on the function of epithelial-derived OPN in tumorigenesis and its value as a prognostic marker (37). However, given the high levels of OPN expressed in senescent fibroblasts (Fig. 2) and the delayed cancer phenotype observed in OPN-null mice (36), we postulated that OPN expressed within the stromal compartment contributes to the transformation process.
Given the possibility that stromal-derived OPN contributes to transformation, we next sought to determine whether senescent stroma was present within a preneoplastic lesion and whether it was coincident with OPN expressing cells. To address this possibility, we took advantage of the two-stage skin carcinogenesis model. We choose to focus on this model because previous studies demonstrated that 12-O-tetradecanoylphorbol-13-acetate (TPA) treatment of murine skin leads to OPN expression (38). In addition, OPN null mice display a delayed tumor phenotype when treated with this protocol (36). To determine whether 7,12-dimethylbenz(a)anthracene (DMBA) and/or TPA were sufficient to induce senescence, we first examined skin sections obtained from mice that were treated with a single dose of DMBA followed by three treatments of TPA. We utilized SA-βGal staining to assess the presence of senescent cells. We observed the appearance of SA-βGal positive cells within the stromal compartment of mice treated with DMBA and TPA as well as TPA alone (Fig. 3A). Remarkably, the appearance of senescent stroma precedes the presence of overt hyperplasia as evidenced by the positive staining in TPA only-treated mice (AT) within 7 days of treatment (Fig. 3A). In addition, the same stromal area displayed p16-positive cells, another senescent marker (Supplemental Fig. 1). Using this model, previous work demonstrated the presence of SA-βGal within the epithelial compartment of papillomas that arose several months after the initial DMBA/TPA exposure (39). While we see very limited SA-βGal staining within the epithelial compartment of the skin, we postulate that the differences observed between our studies is due to the time at which the tissue was analyzed. Indeed, in our hands SA-βGal staining of papillomas arising several months after initial DMBA treatment did reveal limited staining within the epithelial compartment (Supplemental Fig. 2).
Figure 3. Senescent stroma is present in preneoplastic lesions following DMBA-TPA treatment.
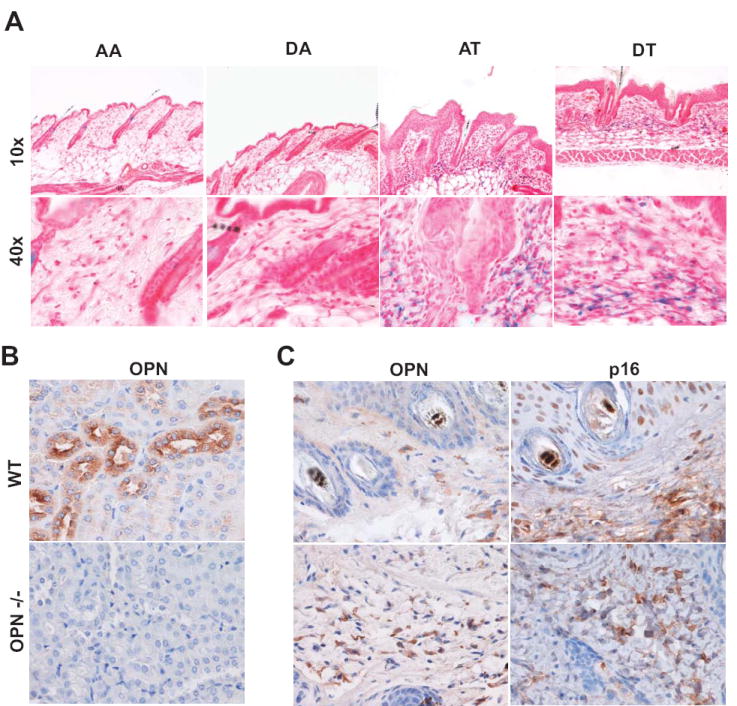
A, Representative SA-βGal staining of frozen tissue sections reveal robust staining within the stromal compartment (n=3). Skin treatments are as follows: (1) AA, acetone followed by weekly acetone treatment (2) DA, DMBA followed by weekly acetone treatment; (3) AT, acetone followed by weekly TPA treatment; (4) DT, DMBA followed by weekly TPA treatments. B, Immunohistochemical analysis of osteopontin (OPN) in kidney sections from wild type (C57/B6) and OPN deficient mice. Magnification upper panel 10X and lower panel 40X. C, OPN and p16 expression in DMBA/TPA induced papillomas (upper panel) and associated skin (lower panel). Serial paraffin embedded tissue sections were stained with antibodies against OPN and the cell cycle inhibitor p16. Magnification 40X. (n=2 of 6).
We next analyzed tissue from papillomas obtained from mice treated with a single dose of DMBA followed by repeated twice-weekly treatments with TPA, which produces papillomas at predictable latency(40). As shown in Fig. 3C (upper panel), OPN staining within the stromal compartment of a papilloma coincided with p16 staining while limited staining was observed within the epithelial compartment. In addition, we also observed OPN positive cells in the stromal compartment of DMBA-TPA treated skin adjacent to papillomas (Fig. 3C lower panel). The presence of p16 expression is consistent with the presence of senescent cells (16, 41). SA-βGal staining was also evident within the stromal compartment of the papillomas, further supporting the possibility that the OPN expression was within senescent stromal cells (Supplemental Fig. 2).
Stromal-derived OPN stimulates the growth of preneoplastic cells
Senescent fibroblasts possess growth-promoting activities that stimulate preneoplastic cell growth (19, 29) and our data suggested that OPN functions as an important mediator of this activity. Therefore, to directly assess whether stromal-derived OPN impacted preneoplastic cell growth, BJ fibroblasts were stably transduced with virally encoded RNAi hairpin constructs targeting OPN or a control hairpin construct. We found that knockdown of OPN in senescent fibroblasts by three independent hairpins (shOPN-7, shOPN-8, and shOPN-9) resulted in dramatically reduced mRNA levels (Fig. 4A) and undetectable protein levels (Fig. 4B) when compared to cells transduced with a control hairpin (shSCR). Fibroblasts transduced with hairpins targeting OPN entered senescence upon bleomycin treatment at the same rate as observed in control cells, indicating that OPN expression is not necessary for induction of senescence (Fig. 4C).
Figure 4. Senescent-derived OPN is necessary for preneoplastic cell growth.
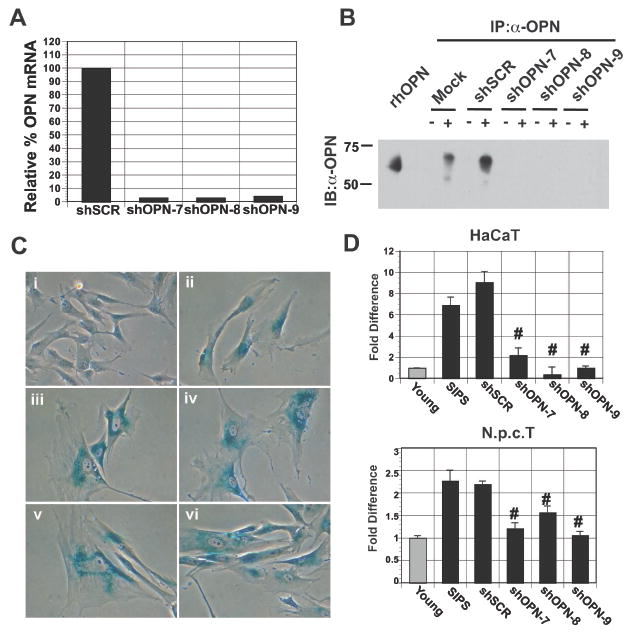
A, Stable knockdown of OPN expression by viral-based RNAi is revealed by qRT-PCR. OPN mRNA expression in senescent fibroblasts transduced with a control hairpin (shSCR) was set to 100%. Three independent short hairpins targeting OPN (shOPN-7, -8, -9) resulted in robust knockdown of OPN mRNA. B, Western blot analysis (IB) of OPN immunoprecipitations (IP) from cultured supernatants obtained from young (-) or senescent (+) fibroblasts mock transduced, transduced with a control hairpin (shSCR), or transduced with hairpins targeting OPN (shOPN-7, -8, -9). Recombinant human OPN (rhOPN) is included as a reference. C, RNAi directed loss of OPN does not affect the induction of senescence upon bleomycin treatment. SA-βGal expression in i) young, ii) SIPS, iii) shSCR SIPS, iv) shOPN-7 SIPS, v) shOPN-8 SIPS, and vi) shOPN-9 SIPS BJ fibroblasts. Magnification 40X. D, Upper Panel: Quantification of HaCAT cell growth when plated on lawns of fibroblasts. Preneoplastic cell growth was measured by relative luciferase activity and wells where HaCAT cells were plated with young BJ fibroblasts were set to 1. OPN expression was depleted in senescent BJ fibroblasts by one of three independent hairpins (shOPN-7, -8, -9) to greater than 95% in all cases. A control hairpin (shSCR) was used as a negative control. Depletion of OPN expression reduced preneoplastic cell growth to levels observed in cells plated with young fibroblasts. #p<0.05. Lower Panel: Quantification of N.p.c.T cell growth when plated on lawns of fibroblasts. Loss of OPN expression (shOPN-7, -8, -9) resulted in reduced growth similar to that observed with HaCaT cells shown in upper panel. #p<0.05.
Having established that our RNAi constructs efficiently reduced OPN to undetectable levels without impacting the induction of senescence, we next investigated how loss of OPN impacted the growth-promoting properties of senescent fibroblasts. Senescent fibroblasts mock transduced or transduced with a control hairpin (shSCR) increased the growth of HaCaT and N.p.c.T cells compared to cells grown on young fibroblasts (Fig. 4D). In contrast, RNAi directed knockdown of OPN in senescent fibroblasts resulted in a significant reduction in HaCaT and N.p.c.T cell growth (Fig. 4D). Indeed, we found that loss of OPN resulted in HaCaT and N.p.c.T cell growth similar to that observed when these cells were plated on young fibroblasts, indicating that OPN is necessary for stimulation of preneoplastic cell growth by senescent fibroblasts.
Stromal-derived OPN stimulates the growth of preneoplastic cells in xenografts
Loss of OPN significantly reduced the growth promoting properties of senescent fibroblasts in vitro. Therefore, we next addressed the impact of loss of stromal derived OPN on the growth of HaCaT cells in xenografts. Utilizing live in vivo luciferase imaging, we found that injection of HaCaT cells alone or in combination with young fibroblasts resulted in little growth of xenografts. In contrast, coinjection of senescent fibroblasts and HaCaT cells resulted in robust growth in vivo (Fig. 5A). Similarly, senescent fibroblasts expressing a control hairpin (shSCR) significantly stimulated HaCaT cell growth. In contrast, OPN depletion abolished HaCaT cell growth indicating that OPN derived from senescent fibroblasts is necessary in vivo (Fig. 5A). Immunohistological analysis of CD45 and F4/80 in xenografts revealed a decrease in immune infiltration in lesions resulting from injection of senescent cells expressing a hairpin targeting OPN versus a control hairpin (Fig. 5B). Together, these experiments indicate that stromal-derived OPN impacts the microenvironment and significantly stimulates preneoplastic cell growth.
Figure 5. Loss of OPN expression in senescent fibroblasts results in reduced preneoplastic cell growth in vivo.
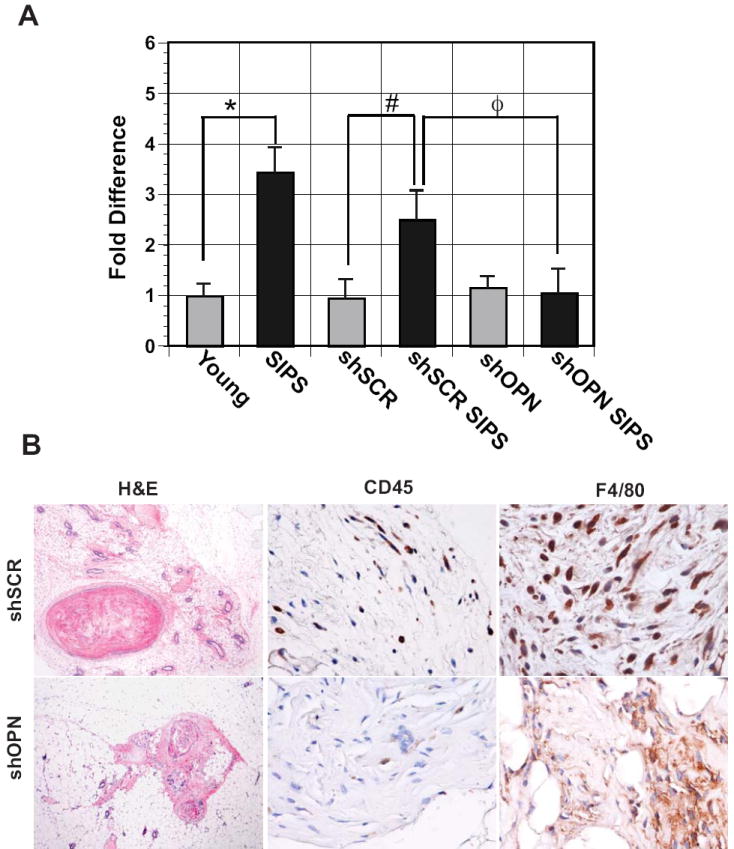
Fibroblasts were uninfected (young and SIPS), infected with a control virus (shSCR, sSCR SIPS are young and senescent fibroblasts, respectively), or a virus that knocks down OPN expression (shOPN, shOPN SIPS are young and senescent fibroblasts, respectively) greater than 95%. Relative to SIPS fibroblasts, knockdown of OPN resulted in a significant reduction in growth of HaCaT xenografts in NOD/SCID mice (n=3). *p=0.003, #p=0.04, and Φp= 0.06. B, Immunohistochemical analysis of xenografts obtained from mice injected with HaCaT cells and senescent fibroblasts expressing a control hairpin (shSCR) (upper panel) or a hairpin targeting OPN (shOPN) (lower panel). Shown are Hematoxylin and Eosin (H&E) and staining for the immune markers CD45 and F4/80, which identify leukocytes and macrophages, respectively. Photos focus on stromal sections showing CD45 or F4/80 positive staining. Magnification for H&E, 10X and for CD45 and F4/80, 40X.
rhOPN is sufficient to stimulate the growth of preneoplastic cells
The above experiments demonstrate that stromal-derived OPN significantly impacts preneoplastic cell growth. We next sought to determine whether OPN is sufficient to promote preneoplastic cell growth. To address this question, we treated preneoplastic cells (HaCaT/N.p.c.T) with recombinant human OPN (rhOPN) and measured its impact on cell growth. We found that addition of rhOPN to both HaCaT and N.p.c.T cells conferred a modest but significant growth advantage compared to mock-treated cells, indicating that OPN acts as a paracrine factor that is sufficient for preneoplastic cell growth (Fig. 6). The increased growth observed upon rhOPN treatment was less robust then when HaCaT or N.p.c.T cells are plated directly on senescent fibroblasts expressing OPN (Fig. 4). Because senescent fibroblasts express numerous secretory proteins it is possible that the reduced response observed upon rhOPN treatment reveals a requirement for additional factors. Alternatively, the reduced response could be attributed to a lack of adequate protein modifications on the rhOPN protein. As stated earlier, OPN is a highly modified protein (42) and careful inspection of a longer exposure of our western blot (Supplemental Fig. 3) revealed the presence of additional OPN species in supernatants obtained from senescent fibroblasts that were not present in the rhOPN preparations.
Figure 6. OPN is sufficient to stimulate the growth of preneoplastic cells.
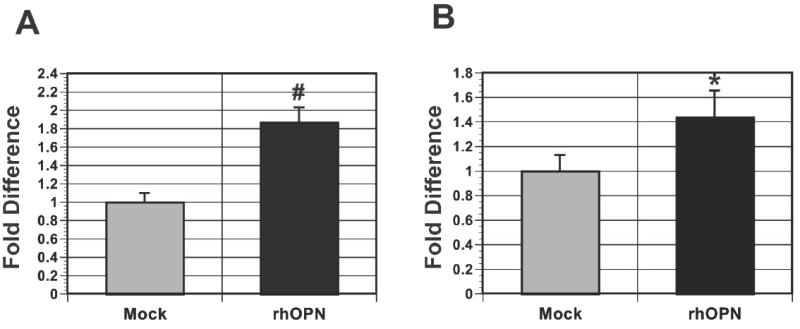
Cell growth was measured by relative luciferase activity and the growth of cells receiving BSA alone was set to 1. A, addition of 100 ng/ml of rhOPN was sufficient to stimulate the growth of HaCaT cells. #p<0.05. B, addition of 100 ng/ml of rhOPN was sufficient to stimulate the growth of N.p.c.T. cells. *p=0.05.
Discussion
Alterations in the tissue microenvironment in combination with cell autonomous mutations collaborate to drive the transformation process. Here, we identify osteopontin (OPN) as a critical stromal-derived mediator of preneoplastic cell growth. Utilizing microarray analysis we found that OPN is upregulated in senescent fibroblasts while being undetectable in their younger counterparts. We found that senescent fibroblast-derived OPN was necessary for preneoplastic cell growth in vitro and in vivo. In addition, recombinant OPN was sufficient to stimulate the growth of preneoplastic cells. Finally, examination of papillomas that arose following treatment of mice with a DMBA/TPA protocol revealed the presence of senescent stromal cells and OPN expression within the stromal compartment. Taken together, our data argue that senescent fibroblast-derived OPN contributes to tumorigenesis and reveal a previously uncharacterized stromal function for OPN that may represent a novel therapeutic target and/or biomarker for the early stages of cancer development.
Investigations into the cell types within the microenvironment that impact tumorigenesis have revealed that macrophages, B and T cells, mast cells, adipocytes, endothelial cells, and fibroblasts can affect primary growth and metastasis. Cancer associated fibroblasts (CAF) exert their influence on neoplastic progression through several mechanisms such as secretion of paracrine factors that directly stimulate cancer cells and impact proliferation and survival. While senescent fibroblasts differ from CAFs in regards to their proliferative capacity, they possess the ability to stimulate preneoplastic and neoplastic cell growth in a manner analogous to CAFs (19, 20, 43). This latter observation raises the possibility that the accumulation of senescent cells within aged individuals alters the tissue microenvironment that in turn collaborates with cell autonomous mutations to drive the transformation process. Intriguingly, we show that senescent stroma appears before the emergence of a papilloma, arguing that stromal changes arise early in the transformation process. In support of this hypothesis, our microarray data indicate that in addition to OPN, senescent fibroblasts express numerous factors capable of remodeling the microenvironment and modulating cell growth in a paracrine fashion. We propose that these early changes, which include the appearance of senescent stroma and concomitant upregulation of OPN, provide a rich microenvironment for tumorigenesis.
OPN is a pleiotropic protein to which numerous functions have been ascribed including mineralization and calcification, osteoclast activation, endothelial cell survival, wound healing, migration, lymphocyte proliferation and immunosurveillance (32). Interestingly, in human cancer, OPN expression levels correlate with tumor grade and elevated OPN serum levels is a poor prognostic indicator in a wide variety of tumors (32). Murine studies revealed that OPN null mice display a significant delay in tumorigenesis when treated with the classic two-step skin carcinogenesis model of DMBA/TPA treatment (36), suggesting that OPN plays a key role in cellular transformation.
To date, investigation into OPN’s role in tumorigenesis has been limited to the impact of epithelial-derived OPN on transformation and progression. However, a functional role for stromal-derived OPN in tumorigenesis has not been investigated even though other cell types including macrophages and neutrophils express OPN under both normal and pathological conditions (44). Notably, a recent microarray analysis of the stromal compartment of human breast cancers identified osteopontin as one of twenty-six genes that when overexpressed is a strong predictor of clinical outcome (45). Our results showing increased OPN in the stromal compartment of DMBA/TPA treated mice suggest an even more general role for OPN in epithelial tumorigenesis. Together, these observations indicate that non-epithelial derived OPN is present at various stages of tumorigenesis in human cancer as well as mouse models, and support a physiologic role for this source of OPN in transformation.
OPN is clearly important for the growth promoting effects of senescent fibroblasts as OPN depletion from senescent fibroblasts drastically reduces growth of preneoplastic cells. However, our data indicate that recombinant human OPN (rhOPN) does not fully mimic the enhanced preneoplastic cell growth observed when cells are grown in the presence of senescent fibroblasts expressing OPN. The inability of rhOPN to recapitulate the magnitude of preneoplastic cell growth observed in our coculture conditions raises the possibility that other factors cooperate with fibroblast-derived OPN. Alternatively, posttranslational modifications of stromal-derived OPN may be important. OPN is a heavily modified protein that possesses metalloproteinases (MMP) cleavage sites (35). Both protein modifications and cleavage impact OPN function (46, 47). Interestingly, analysis of our microarray data indicated that several MMPs are expressed at high levels in senescent fibroblasts. Western blot analysis of conditioned medium obtained from senescent fibroblasts revealed the presence of OPN species that were not present in our rhOPN preparation, raising the possibility that one or more of these species mediates the growth-promoting activities of senescent fibroblasts. In addition, despite the high levels of OPN mRNA present within senescent fibroblasts, we did not detect the protein in the cell pellets but instead were only able to detect OPN in the conditioned media of senescent fibroblasts, indicating that a significant amount of the protein is present as a soluble factor within the extracellular space. Such data suggests that the presence of OPN in the stroma of premalignant lesions such as papillomas may be underappreciated.
Our data and that of others (19, 20) support a role for senescent stroma in preneoplastic lesions but does not preclude its involvement in the later stages of transformation and cancer development. Interrogation of our senescent microarray data revealed a prominent inflammatory profile, which could facilitate recruitment of other components of the microenvironment in addition to impacting the ECM, bone marrow cell recruitment, and angiogenesis. Intriguingly, CAFs also express many of these factors, some of which are able to promote vascularization by recruiting endothelial cells (48). Furthermore, a recent report demonstrates that CAFs secrete extracellular matrix tracks on which tumor cells can migrate, thus facilitating metastasis (49). While OPN has been shown to be important for metastasis and is a validated prognostic marker in human neoplasias, it was recently shown to function as a systemic instigator by mobilizing and recruiting bone marrow-derived cells to the tumor stroma (50). Interestingly, immunohistochemical analysis of xenografts arising from injections with stromal cells in the absence of OPN revealed a decrease in immune infiltration compared to those arising in the presence of OPN expressing stromal cells. This observation raises the possibility that senescent stromal-derived OPN affects recruitment of cells of the innate immune system potentially through bone marrow mobilization. These results underscore the importance of paracrine signaling and the interplay between different components of the tumor microenvironment at multiple stages of transformation.
Supplementary Material
Immunohistochemistry in skin sections from mice treated with DMBA or TPA alone, or with DMBA-TPA. Mice were treated with DMBA followed by weekly acetone treatment (DA), acetone followed by weekly TPA treatment (AT), or DMBA followed by weekly TPA treatments (TA). Frozen skin sections (exposed to the same treatment as shown on Fig. 3) were stained for p16 – a senescent marker. Magnification 40X.
SA-βGal staining of frozen tissue sections obtained from papillomas reveals the presence of senescent cells within the stromal compartment. Magnification 20X.
The gel is a longer exposure of the gel pictured in Fig. 4 that reveals the presence of at least two unique OPN species that are not present in the rhOPN preparation.
Acknowledgments
We thank members of the Stewart Laboratory for valuable discussions; Charlotte Kuperwasser for advice and inspiration; Cynthia Hernandez~DeLuca for technical assistance; Abhishek Saharia for graphical assistance, Loren Michel and Julien Duxin for critical reading; Lisa Coussens and Cecilia Giachelli for the CD45 and OPN staining protocol. This work was supported in part by Philip Morris USA Inc. and Philip Morris International, Molecular Imaging Center Grant, P50-CA94056. E.P. was support by the Lucille P. Markey and the DOD Breast Cancer Research Program Predoctoral Traineeship Award W81XWH-06-1-0691, L. S. was supported by a P30-CA91842 grant, S.B. was supported by the Howard Hughes Medical Institute through the Undergraduate Biological Sciences Education Program and a Hoopes Award. S.D was supported by RO1-GM055479 awarded to Dr. Kopan. This work is dedicated to the memory of Allan Thomas Stewart.
References
- 1.DePinho RA. The age of cancer. Nature. 2000;408(6809):248–54. doi: 10.1038/35041694. [DOI] [PubMed] [Google Scholar]
- 2.Sakr WA, Haas GP, Cassin BF, Pontes JE, Crissman JD. The frequency of carcinoma and intraepithelial neoplasia of the prostate in young male patients. J Urol. 1993;150(2 Pt 1):379–85. doi: 10.1016/s0022-5347(17)35487-3. [DOI] [PubMed] [Google Scholar]
- 3.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumor cells with defined genetic elements. Nature. 1999;400:464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 4.Hahn WC, Meyerson M. Telomerase activation, cellular immortalization and cancer. Ann Med. 2001;33(2):123–9. doi: 10.3109/07853890109002067. [DOI] [PubMed] [Google Scholar]
- 5.Zhao JJ, Roberts TM, Hahn WC. Functional genetics and experimental models of human cancer. Trends Mol Med. 2004;10(7):344–50. doi: 10.1016/j.molmed.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Martins-Green M, Boudreau N, Bissell MJ. Inflammation is responsible for the development of wound-induced tumors in chickens infected with Rous sarcoma virus. Cancer Res. 1994;54(16):4334–41. [PubMed] [Google Scholar]
- 7.de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer cell. 2005;7(5):411–23. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Peek RM, Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006;208(2):233–48. doi: 10.1002/path.1868. [DOI] [PubMed] [Google Scholar]
- 9.Greenson JK. Dysplasia in inflammatory bowel disease. Seminars in diagnostic pathology. 2002;19(1):31–7. [PubMed] [Google Scholar]
- 10.Tuxhorn JA, Ayala GE, Rowley DR. Reactive stroma in prostate cancer progression. J Urol. 2001;166(6):2472–83. [PubMed] [Google Scholar]
- 11.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1(1):46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59(19):5002–11. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nielsen M, Thomsen JL, Primdahl S, Dyreborg U, Andersen JA. Breast cancer and atypia among young and middle-aged women: a study of 110 medicolegal autopsies. Br J Cancer. 1987;56(6):814–9. doi: 10.1038/bjc.1987.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhathal PS, Brown RW, Lesueur GC, Russell IS. Frequency of benign and malignant breast lesions in 207 consecutive autopsies in Australian women. Br J Cancer. 1985;51(2):271–8. doi: 10.1038/bjc.1985.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castro P, Giri D, Lamb D, Ittmann M. Cellular senescence in the pathogenesis of benign prostatic hyperplasia. Prostate. 2003;55(1):30–8. doi: 10.1002/pros.10204. [DOI] [PubMed] [Google Scholar]
- 16.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311(5765):1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 18.Trougakos IP, Saridaki A, Panayotou G, Gonos ES. Identification of differentially expressed proteins in senescent human embryonic fibroblasts. Mech Ageing Dev. 2006;127(1):88–92. doi: 10.1016/j.mad.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 19.Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, Nelson PS. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006;66(2):794–802. doi: 10.1158/0008-5472.CAN-05-1716. [DOI] [PubMed] [Google Scholar]
- 20.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98(21):12072–7. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106(3):761–71. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rheinwald JG, Hahn WC, Ramsey MR, et al. A two-stage, p16(INK4A)- and p53-dependent keratinocyte senescence mechanism that limits replicative potential independent of telomere status. Mol Cell Biol. 2002;22(14):5157–72. doi: 10.1128/MCB.22.14.5157-5172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98(1):31–6. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci U S A. 2004;101(33):12288–93. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang G, Rosen DG, Zhang Z, et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A. 2006;103(44):16472–7. doi: 10.1073/pnas.0605752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang J, Chen W, Xia J, et al. Extracellular matrix secreted by senescent fibroblasts induced by UVB promotes cell proliferation in HaCaT cells through PI3K/AKT and ERK signaling pathways. Int J Mol Med. 2008;21(6):777–84. [PubMed] [Google Scholar]
- 27.Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118(Pt 3):485–96. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aoshiba K, Tsuji T, Nagai A. Bleomycin induces cellular senescence in alveolar epithelial cells. Eur Respir J. 2003;22(3):436–43. doi: 10.1183/09031936.03.00011903. [DOI] [PubMed] [Google Scholar]
- 29.Krtolica A, Campisi J. Cancer and aging: a model for the cancer promoting effects of the aging stroma. Int J Biochem Cell Biol. 2002;34(11):1401–14. doi: 10.1016/s1357-2725(02)00053-5. [DOI] [PubMed] [Google Scholar]
- 30.Conover CA, Bale LK, Overgaard MT, et al. Metalloproteinase pregnancy-associated plasma protein A is a critical growth regulatory factor during fetal development. Development (Cambridge, England) 2004;131(5):1187–94. doi: 10.1242/dev.00997. [DOI] [PubMed] [Google Scholar]
- 31.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8(8):877–84. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rangaswami H, Bulbule A, Kundu GC. Osteopontin: role in cell signaling and cancer progression. Trends Cell Biol. 2006;16(2):79–87. doi: 10.1016/j.tcb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 33.Senger DR, Wirth DF, Hynes RO. Transformed mammalian cells secrete specific proteins and phosphoproteins. Cell. 1979;16(4):885–93. doi: 10.1016/0092-8674(79)90103-x. [DOI] [PubMed] [Google Scholar]
- 34.Wai PY, Kuo PC. The role of Osteopontin in tumor metastasis. J Surg Res. 2004;121(2):228–41. doi: 10.1016/j.jss.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 35.Agnihotri R, Crawford HC, Haro H, Matrisian LM, Havrda MC, Liaw L. Osteopontin, a novel substrate for matrix metalloproteinase-3 (stromelysin-1) and matrix metalloproteinase-7 (matrilysin) J Biol Chem. 2001;276(30):28261–7. doi: 10.1074/jbc.M103608200. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh YH, Juliana MM, Hicks PH, et al. Papilloma development is delayed in osteopontin-null mice: implicating an antiapoptosis role for osteopontin. Cancer Res. 2006;66(14):7119–27. doi: 10.1158/0008-5472.CAN-06-1002. [DOI] [PubMed] [Google Scholar]
- 37.Fedarko NS, Jain A, Karadag A, Van Eman MR, Fisher LW. Elevated serum bone sialoprotein and osteopontin in colon, breast, prostate, and lung cancer. Clin Cancer Res. 2001;7(12):4060–6. [PubMed] [Google Scholar]
- 38.Craig AM, Smith JH, Denhardt DT. Osteopontin, a transformation-associated cell adhesion phosphoprotein, is induced by 12-O-tetradecanoylphorbol 13-acetate in mouse epidermis. J Biol Chem. 1989;264(16):9682–9. [PubMed] [Google Scholar]
- 39.Sun P, Yoshizuka N, New L, et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 2007;128(2):295–308. doi: 10.1016/j.cell.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 40.Yuspa SH. The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis. J Dermatol Sci. 1998;17(1):1–7. doi: 10.1016/s0923-1811(97)00071-6. [DOI] [PubMed] [Google Scholar]
- 41.Collado M, Gil J, Efeyan A, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:7051–642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 42.Rittling SR, Chambers AF. Role of osteopontin in tumour progression. Br J Cancer. 2004;90(10):1877–81. doi: 10.1038/sj.bjc.6601839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JL, Wang MJ, Sudhir PR, Chen GD, Chi CW, Chen JY. Osteopontin promotes integrin activation through outside-in and inside-out mechanisms: OPN-CD44V interaction enhances survival in gastrointestinal cancer cells. Cancer Res. 2007;67(5):2089–97. doi: 10.1158/0008-5472.CAN-06-3625. [DOI] [PubMed] [Google Scholar]
- 44.Denhardt DT, Noda M, O’Regan AW, Pavlin D, Berman JS. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest. 2001;107(9):1055–61. doi: 10.1172/JCI12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finak G, Bertos N, Pepin F, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14(5):518–27. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 46.Christensen B, Kazanecki CC, Petersen TE, Rittling SR, Denhardt DT, Sorensen ES. Cell type-specific post-translational modifications of mouse osteopontin are associated with different adhesive properties. J Biol Chem. 2007;282(27):19463–72. doi: 10.1074/jbc.M703055200. [DOI] [PubMed] [Google Scholar]
- 47.Kazanecki CC, Uzwiak DJ, Denhardt DT. Control of osteopontin signaling and function by post-translational phosphorylation and protein folding. J Cell Biochem. 2007;102(4):912–24. doi: 10.1002/jcb.21558. [DOI] [PubMed] [Google Scholar]
- 48.Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 49.Gaggioli C, Hooper S, Hidalgo-Carcedo C, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9(12):1392–400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 50.McAllister SS, Gifford AM, Greiner AL, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008;133(6):994–1005. doi: 10.1016/j.cell.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Immunohistochemistry in skin sections from mice treated with DMBA or TPA alone, or with DMBA-TPA. Mice were treated with DMBA followed by weekly acetone treatment (DA), acetone followed by weekly TPA treatment (AT), or DMBA followed by weekly TPA treatments (TA). Frozen skin sections (exposed to the same treatment as shown on Fig. 3) were stained for p16 – a senescent marker. Magnification 40X.
SA-βGal staining of frozen tissue sections obtained from papillomas reveals the presence of senescent cells within the stromal compartment. Magnification 20X.
The gel is a longer exposure of the gel pictured in Fig. 4 that reveals the presence of at least two unique OPN species that are not present in the rhOPN preparation.