

Abstract
Murine melanomas produce site-specific experimental brain metastases that reflect clinical reality. When injected into the internal carotid artery of mice, the K-1735 melanoma cells produce metastatic lesions only in the brain parenchyma, whereas the B16 melanoma cells and the somatic hybrid cells of the B16 x K-1735 melanoma cells produce metastatic lesions only in the leptomeninges and ventricles. In the present study, we identified TGF-β2, an isoform of the TGF-β family, as a molecular determinant of melanoma cell growth in the brain parenchyma. We found that the TGF-β2 mRNA was highly expressed by the K-1735 cells, whereas the B16 cells or any B16 x K-1735 somatic cell-cell fusion hybrids have low expression. Transfection of the TGF-β2 gene into B16 cells resulted in the production of microscopic metastatic lesions in the brain parenchyma, without a decrease in metastasis to the leptomeninges or ventricles. TGF-β2 knockdown in the K-1735 melanoma cells significantly reduced metastasis to the brain parenchyma but did not induce metastasis to the leptomeninges or ventricles. These data demonstrate that TGF-β2 expression by murine melanoma cells is necessary for the establishment and growth of metastases in the brain parenchyma.
Keywords: TGF-β2, melanoma, brain metastasis
Introduction
Brain metastases are the most common intracranial tumors, outnumbering primary brain tumors by at least 10-fold (1). Melanoma is the third most common cancer producing brain metastases after carcinomas of the lung and breast (2). The prognosis of patients with melanoma brain metastasis is poor, and even the best available treatments provide a median survival of only 3.7 months for these patients (3). Previously, we developed an in vivo model of experimental brain metastasis by injecting tumor cells into the internal carotid artery of anesthetized mice (4) and reported that two widely used melanoma cells, B16 syngeneic to the C57BL/6 mouse and K-1735 syngeneic to the C3H/HeN mouse, produce site-specific brain metastasis. The K-1735 cells produced metastasis only in the brain parenchyma, whereas B16 cells and somatic hybrid cells from cell-cell fusion between B16 x K-1735 melanoma cells produced metastatic lesions only in the leptomeninges and ventricles (5, 6). This difference in site specificity of metastasis was attributed to the in vitro response of melanoma cells to transforming growth factor-beta (TGF-β), which stimulated the proliferation of K-1735 cells (6).
TGF-β is a prominent family of cytokines essential for the development and maintenance of a multicellular organism. TGF-β has been recognized to play a wide range of roles in cancer as well (7–9). In glioblastoma multiforme (GBM), the most common and most aggressive form of primary brain tumor, TGF-β2 has been shown to play essential role in its pathogenesis. Particularly, TGF-β2 in culture supernatants of human GBM cells has been identified as an immunosuppressive factor (10, 11). Later studies of GBM clinical specimens revealed that TGF-β2 is overexpressed (12, 13). Antisense therapy to reduce the production of TGF-β2 improved survival of rats with experimental glioma (14–17).
To identify the molecular determinants of site-specific melanoma brain metastasis, we analyzed the expression of growth factors VEGF and TGF-β by murine B16 and K-1735 melanoma cells. We found that the K-1735 cells express high level of TGF-β2 mRNA, whereas the B16 cells express low level of TGF-β2 mRNA. Further studies with the somatic hybrid cells from cell-cell fusion between B16 x K-1735 melanoma cells revealed a correlation between the expression level of TGF-β2 and the ability of these melanoma cells to produce lesions in the brain parenchyma of mice. Only K-1735 cells produce the TGF-β2 cytokine whereas the B16 or B16 x K-1735 hybrid cells do not. In addition, B16-BL6 melanoma cells overexpressing the TGF-β2 gene produced microscopic metastatic lesions in the brain parenchyma, as well as unchanged metastasis to the leptomeninges and ventricles. Short hairpin RNA (shRNA)-mediated knockdown of TGF-β2 expression in K-1735 C4 cells resulted in decreased production of experimental parenchymal brain metastasis without growth in the leptomeninges and brain ventricles. These data demonstrate a direct relationship between TGF-β2 expression and tumor cell growth in the brain parenchyma.
Materials and Methods
Mice
Specific pathogen-free female mice of the inbred strains C57BL/6, C3H/HeN, and C57BL/6 x C3H/HeN F1 (hereafter designated B6C3F1) were purchased from the Animal Production Area, NCI-Frederick Cancer Research Facility (Frederick, MD). At the time of the experiments, the mice were 6 to 8 weeks old. The animals were maintained in facilities approved by the American Association for Accreditation of Laboratory Animal Care in accordance with current U. S. Department of Agriculture, Department of Health and Human Services, and NIH regulations and standards.
Murine melanoma cell lines
The highly invasive and metastatic B16-BL6 melanoma (18, 19) was originally derived from the B16-F1 line syngeneic to C57BL/6 mice (20). The K-1735 melanoma, induced in a C3H/HeN mouse by chronic exposure to UV light followed by painting with croton oil (21), was the gift of Dr. Margaret L. Kripke (The University of Texas M. D. Anderson Cancer Center, Houston, TX). The parental tumor was cloned in vitro by a double-cloning method (22). Of the large number of clones thus isolated, clone 4 (designated C4) is highly metastatic and produces melanotic tumor foci in the lungs and brain of syngeneic mice (23, 24).
All tumor cell lines were maintained on plastic in modified Eagle’s medium supplemented with 10% fetal bovine serum, sodium pyruvate, nonessential amino acids, L-glutamine, and 2-fold vitamin solution (Invitrogen, Carlsbad, CA). The cultures were free of Mycoplasma and the following murine viruses: reovirus type 3; pneumonia virus; K virus; Theiler’s encephalitis virus; Sendai virus; minute virus; mouse adenovirus; mouse hepatitis virus; lymphocytic choriomeningitis virus; ectromelia virus; and lactate dehydrogenase virus (assayed by M. A. Bioproducts, Walkersville, MD). Cultures were maintained for no longer than 12 weeks after recovery from frozen stock, and all in vitro studies were carried out with cultures of passages 6 to 10.
For in vivo studies, tumor cells in exponential growth phase were harvested by a 1-min treatment with 0.25% trypsin-0.02% EDTA solution (w/v). The flask was tapped to detach the cells, supplemented with medium, and the cell suspension gently agitated to produce a single-cell suspension. The cells were washed and resuspended in Ca2+-free and Mg2+-free Hanks’ balanced salt solution (HBSS). Only suspensions of single cells with viability exceeding 90% were used.
Injection of tumor cells into the carotid artery to produce experimental brain metastasis
Mice were anesthetized by intraperitoneal injection of pentobarbital sodium, restrained on a cork board in a supine position, and placed under a dissecting microscope. The preparation and exposure of the carotid artery has been described in detail previously (4–6). Briefly, the mouse’s head was stabilized on a cork board with a rubber band placed between the teeth of the upper jaw. The hair over the trachea was shaved, the neck prepared for surgery with alcohol-iodine, and the skin cut by a mediolateral incision. After blunt dissection, the trachea was exposed. The muscles were separated to expose the right common artery, which was then separated from the vagal nerve. The artery was prepared for injection distal to the point of division into the internal and external carotid arteries. A ligature of 5-0 silk suture was placed in the distal part of the common carotid artery. A second ligature was placed and tied loosely proximal to the injection site. To control bleeding from the carotid artery by regurgitation from distal vessels, a sterile cotton tip applicator was inserted under the artery just distal to the injection site to elevate the carotid artery. The artery was nicked with a pair of microscissors, and a <30-gauge glass cannula was inserted into the lumen of the blood vessel. To assure proper delivery, cells (1 × 105/0.1 ml HBSS) were injected slowly, and then the cannula was removed. The second ligature was tightened, and the skin was closed with staples.
When the tumor cells are properly injected into the internal carotid artery and the animals recover from deep anesthesia and the microsurgical procedure, the incidence of experimental brain metastasis in C57BL/6, C3H/HeN and B6C3F1 mice is 100%.
Subcutaneous tumor inoculation to measure in vivo tumorigenicity
The K-1735 C4 melanoma cells and the shRNA-transduced cells (1 × 106/0.1 ml) were inoculated subcutaneously into syngeneic C3H/HeN mice. The tumor size in two perpendicular diameters was measured with calipers every 3 days beginning on day 5. The tumor volume was calculated by the formula V = (π/6) × (L × W2).
Semi-quantitative RT-PCR analysis of TGF-β ligand expressions
The expression of TGF-β isoforms was determined by reverse-transcription polymerase chain reaction (RT-PCR). Total RNA of cultured tumor cells was isolated using TRIzol Reagent according to the manufacturer’s recommendations (Invitrogen, Carlsbad, CA). The first-strand cDNA was synthesized using a commercial kit according to the manufacturer’s protocol (Promega, Madison, WI) and was used as a template for subsequent PCR reactions. PCR was conducted under routine conditions for 25~28 cycles. The following primers were used to amplify the specific murine TGF-β isoforms: 5′-GCTACTGCCGCTTCTGCT-3′ (forward) and 5′-GCCCTGTATTCCGTCTCC-3′ (reverse) for TGF-β1, 5′-TACTACGCCAAGGAGGTT-3′ (forward) and 5′-AATTATTAGACGGCACGAA-3′ (reverse) for TGF-β2, and 5′-GCAAAGGGCTCTGGTAGT-3′ (forward) and 5′GTCTCCATTGGGCTGAAA-3′ (reverse) for TGF-β3.
Determination of TGF-β1 and TGF-β2 protein levels
The level of TGF-β1 and TGF-β2 proteins in the culture supernatants was determined by ELISA kits according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). All cells subjected to ELISA assays were cultured for 48 h in minimal essential medium supplemented with 1% fetal bovine serum.
Transfection of B16-BL6 melanoma cells with mTGF-β2
Full-length murine TGF-β2 cDNA (IMAGE #4035366) was acquired from the Mammalian Gene Collection (Open Biosystems, Huntsville, AL). The full-length cDNA insert was cloned from the pCMV-SPORT6 vector by EcoRV/NotI double digestion and was subsequently cloned into EcoRV/NotI linearized mammalian expression vector pIRESneo (Clontech, Mountain View, CA). The resulting plasmid was transfected into B16-BL6 cells using Lipofectamine 2000 Transfection Reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). Stable cell lines were selected for 3 wk under gradually elevated concentrations of G418 from 0.5–1.0 mg/ml.
Knockdown expression of mTGF-β2 in K-1735 C4 melanoma mediated by shRNA
Short hairpin RNA (shRNA) specific for the murine TGF-β2 gene was acquired from the Elledge-Hannon library (Open Biosystems, Huntsville, AL). Clone V2LMM_40830 was chosen as the target of the murine TGF-β2 transcript that is distinct from the TGF-β1 and TGF-β3 isoforms. The target sequence is: TGCTGTTGACAGTGAGCGCGGTGTATAAATCGAGACCAAATTAGTGAAGCCACAGATGTATTTGGTCTCGATTTATACACCTTGCCTACTGCCTCGGA. The mTGF-β2-specific shRNA was cloned in the pGIPZ lentiviral expression vector. The shRNA-containing lentiviral vector was cotransfected with lentiviral packaging and pseudotyping vectors psPAX2/pMD2G into HEK-293FT cells (Invitrogen, Carlsbad, CA) to produce shRNA-carrying lentivirus particles. Culture supernatants were collected at 24 h and 48 h after transfection and filtered through 0.45-μm membranes to generate cell-free virus supernatant. K-1735 C4 cells were transduced by the resulting viral particles, and positive clones were selected and maintained in puromycin (3–5 μg/ml). pGIPZ lentivirus of a non-silencing shRNA control (RHS4346, Open Biosystems) with no homology to known mammalian genes was used as the negative control for the knockdown experiment.
Immunohistochemistry
Sections of formalin-fixed, paraffin-embedded brains were deparaffinized in xylene, rehydrated in graded alcohol, and transferred to PBS. Heat induced epitope retrieval was performed by heating the slides in Target Retrieval Solution (S1699, Dako USA) for 25 min in a rice cooker. After being cooled down to room temperature, the slides were rinsed twice with PBS. Endogenous peroxidase was blocked by the use of 3% hydrogen peroxide in PBS for 12 min. The treated slides were blocked in PBS containing 5% normal horse serum/1% normal goat serum and incubated overnight at 4°C in a humidified chamber with a 1:100 dilution of rabbit polyclonal antibody to TGF-β2 antigen (sc-90, Santa Cruz Biotechnology). The sections were then rinsed, reblocked for 10 min (see above) and incubated for 60 min with 1:500 dilution of peroxidase-conjugated anti-rabbit secondary antibody (111-036-047, Jackson ImmunoResearch Laboratories). The slides were then rinsed with PBS and incubated with either diaminobenzidine/DAB (Research Genetics, Huntsville, AL) or Romulin AEC Chromogen kit (RAEC810L, Biocare Medical, CA). The sections were then washed with distilled water and counterstained with Gill’s hematoxylin.
Histological studies
At various time points, the mice injected with melanoma cells were euthanized. The brain parenchyma and skull with leptomeninges were collected and fixed in 10% buffered formalin. To assure representative sampling and adequate comparison between the right and left hemispheres, each brain was cut in 4 coronal sections of equal width. Four level sections from each sample (16 sections/brain: 5 μm thickness) were stained with hematoxylin and eosin and examined microscopically for the presence of lesions.
Statistical analysis
The significance of differences in TGF-β2 ELISA assay was analyzed by two-tailed Student’s t test with a cutoff of 0.05. Survival estimates and median survivals were determined using the method of Kaplan and Meier. The survival data were tested for significance using a log-rank (Mantel-Cox) test.
Results
Murine melanoma cells produce site-specific brain metastasis
Murine B16-BL6 and K-1735 C4 melanoma cells were injected into the internal carotid artery of syngeneic mice C57BL/6 and C3H/HeN, respectively, as well as of the B6C3F1 mice (F1 of C57BL/6 x C3H/HeN). Three to four weeks later, mice with neurological symptoms (e.g., circling, unbalanced, immobile) were euthanized. Their brains were collected for analysis of experimental metastasis. The K-1735 C4 melanoma cells produced grossly visible tumor growths only in the brain parenchyma (Fig. 1A) and exhibited diffuse-infiltrative lesions upon microscopic examination (Fig. 1C). The B16-BL6 melanoma cells produced growths only in the leptomeninges and ventricles (Fig. 1B, 1D).
Figure 1.
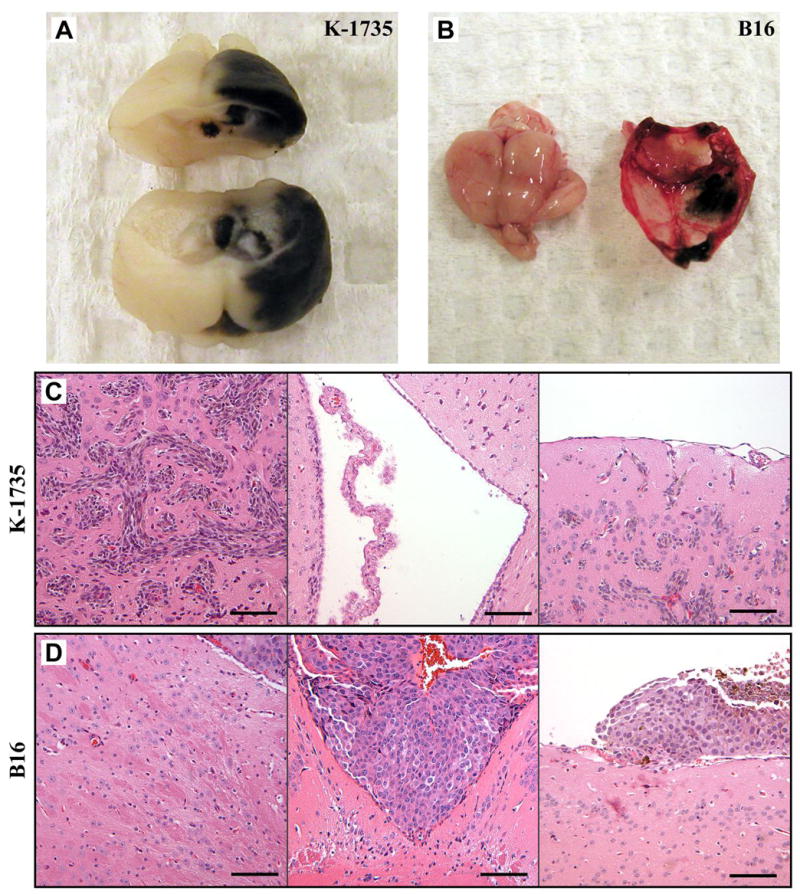
Site-specific brain metastasis produced by murine K-1735 C4 (A, C) and B16-BL6 (B, D) melanoma cells. A, grossly visible parenchymal brain lesions produced by K-1735 C4 cells. B, leptomeningeal lesions produced by B16-BL6 cells. C, K-1735 C4 cells produce diffuse-infiltrative lesions in the brain parenchyma. The ventricle is not invaded by K-1735 C4 melanoma cells; choroid plexus is shown. K-1735 C4 cells do not grow in the leptomeninges, but only in the brain parenchyma. D, B16-BL6 cells do not produce lesions in the parenchyma; the small tumor lesion (shown in right upper corner of panel) is growing in the ventricle. B16-BL6 melanoma cells produce metastatic lesions in the ventricle. B16-BL6 melanoma cells produce large lesions in the leptomeninges.
To rule out that the site-specific melanoma brain metastasis is due to different genetic background of recipient mice (B16-BL6 into C57BL/6 mice and K-1735 C4 into C3H/HeN mice), we injected both melanoma lines into the internal carotid artery of B6C3F1 mice. As clearly shown in Table 1, the site specificity is not associated with the genetic background of the injected mice.
Table 1.
Site-specific brain metastasis produced by B16-BL6 and K-1735 C4 melanoma cells in syngeneic and B6C3F1 cross mice
| C57BL/6
|
B6C3F1 |
|||
|---|---|---|---|---|
| Cell line | Leptomeninges, ventricles | Parenchyma | Leptomeninges, ventricles | Parenchyma |
| B16-BL6 | 14/14 | 0/14 | 4/4 | 0/4 |
| B16-BL6/vector control | 15/15 | 0/15 | 5/5 | 0/5 |
| B16-BL6/TGF-β2 OE | 19/19 | 5/19* | 5/5 | 1/5* |
| C3H/HeN
|
B6C3F1
|
|||
| Cell line | Leptomeninges, ventricles | Parenchyma† | Leptomeninges, ventricles | Parenchyma |
|
| ||||
| K-1735 C4 | 0/20 | 20/20 | 0/5 | 5/5 |
| K-1735 C4/shRNA control | 0/19 | 19/19 | 0/5 | 5/5 |
| K-1735 C4/TGF-β2 shRNA | 0/20 | 17/20‡ | 0/4 | 4/4 |
B16-BL6 cells overexpressing the TGF-β2 cytokine produced microscopic tumor lesions in the brain parenchyma.
K-1735 C4 cells and derivative cells produced grossly visible tumors in the brain parenchyma. The production of parenchymal lesions was significantly delayed in mice injected with K-1735 C4/TGF-β2 shRNA cells.
Three mice killed 24 d after tumor injection had no visible brain parenchymal lesions.
TGF-β2 is differentially expressed in the two melanoma lines
Semi-quantitative RT-PCR was used to analyze the expression of VEGF and TGF-β growth factors in cultured melanoma cells. In both sparse and dense cultures, the TGF-β2 mRNA, an isoform of the TGF-β family, was highly expressed by the K-1735 C4 cells, but lowly expressed by the B16-BL6 cells (Fig. 2A). Both lines also expressed VEGF to similar levels.
Figure 2.
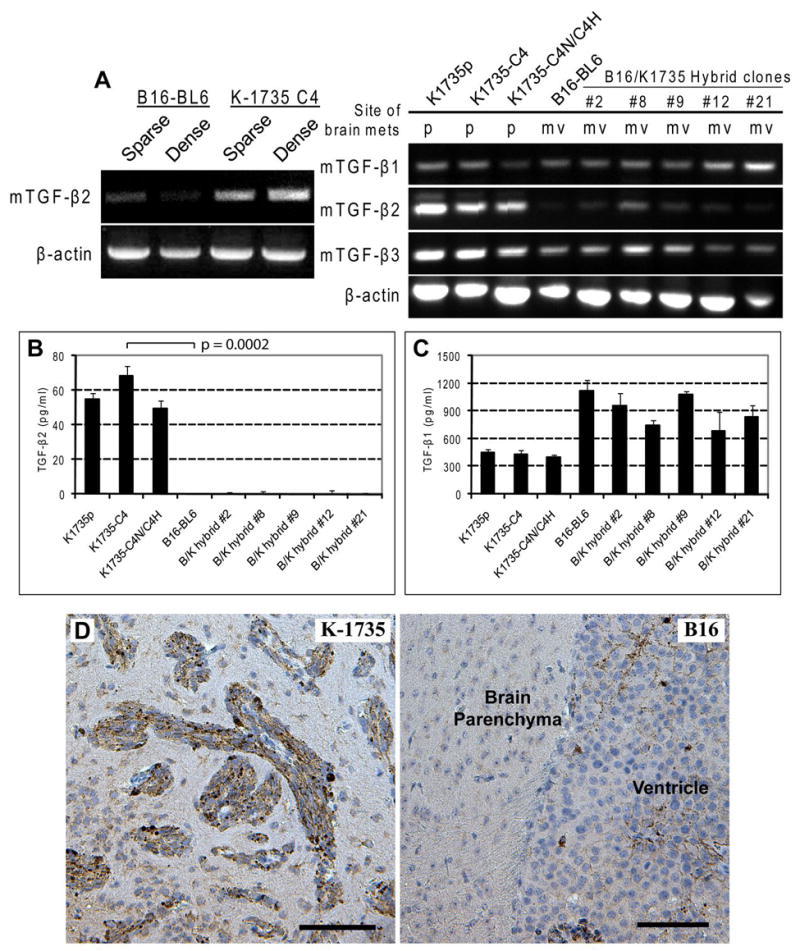
TGF-β2 expression is correlated with site specificity of melanoma brain metastasis. A, semi-quantitative RT-PCR analysis of murine TGF-β2 expression. Under both sparse and dense conditions, K-1735 C4 melanoma cells express high level of TGF-β2, whereas B16-BL6 melanoma cells express low level of the cytokine. Expression of all 3 TGF-β isoforms were analyzed for K-1735 parental cells, K-1735 clone 4 cells, K-1735 C4 self-fusion hybrid cells, and 5 individual clones of somatic hybrid cells from cell-cell fusion between B16-BL6 and K-1735 C4 melanoma cells. The expression of TGF-β2 is correlated with site specificity of brain metastasis (p, parenchyma; mv, leptomeninges and ventricle). B, ELISA assay was used to determine the secreted TGF-β2 cytokine in the culture supernatant. Only K-1735 melanoma cells produce TGF-β2. C, all melanoma cells produce significant levels of TGF-β1 (ELISA of culture supernatant). D, Diffuse-infiltrative brain parenchymal lesions produced by K-1735 C4 melanoma cells stained positive for TGF-β2. Brain ventricle metastasis produced by B16-BL6 melanoma cells negative for TGF-β2. (Note melanin deposits within the metastasis.) Scale bar: 100 μm.
TGF-β2 expression is correlated with site-specific brain metastasis
To determine whether the production of TGF-β2 is correlated with the site-specificity of melanoma brain metastasis, we expanded the expression analysis to somatic hybrid cells from cell-cell fusion between the B16-BL6 x K-1735 C4 melanoma cells. RT-PCR results showed that only K-1735 C4 cells and its self-fusion hybrid cells expressed high level of TGF-β2 mRNA, whereas the B16-BL6 cells and the B16 x K-1735 cross-hybrid cells expressed low level of the TGF-β2 transcript. In contrast, all of the cells in our analysis expressed similar levels of TGF-β1 and TGF-β3 mRNA (Fig. 2A). The RT-PCR findings were enhanced by ELISA measurements of the protein level of TGF-β2 in cell culture supernatants (Fig. 2B) where TGF-β2 ligand production was not detected for B16-BL6 cells or any of the B16 x K-1735 cross-hybrid cells. The differences in production of TGF-β2 were maintained, whereas all cells produced significant amounts of TGF-β1 (Fig. 2C).
To test whether the differential expression of TGF-β2 from in vitro cultures of K-1735 C4 and B16-BL6 cells is still maintained in vivo, we did immunohistochemical studies of brain sections with either melanoma tumor. Brain parenchymal tumors produced by K-1735 C4 melanoma cells were stained positive for TGF-β2 expression, whereas the ventricle tumor produced by B16-BL6 cells were stained negative for TGF-β2 expression (Fig. 2D). The immunohistochemistry results corroborate the in vitro findings.
Overexpression of murine TGF-β2 in B16-BL6 cells
To study the causal role that TGF-β2 plays in site-specific brain metastasis, full-length murine TGF-β2 cDNA was cloned into mammalian expression vector pIRESneo under the control of the early CMV promoter. We transfected the expression vector into the B16-BL6 cells. Because of the presence of internal ribosome entry site (IRES) between the TGF-β2 gene and the neo cassette, stably transfected cells were selected over 3 wk by increasing concentrations (0.5–1 mg/ml) of G418. TGF-β2 ELISA of the culture supernatant of the resulting cell population revealed overexpression of TGF-β2 in transfected B16-BL6 melanoma cells (Fig. 3A). No changes in growth rate or gross morphological differences were found between the parental B16-BL6 cells, vector control cells, and cells overexpressing TGF-β2.
Figure 3.
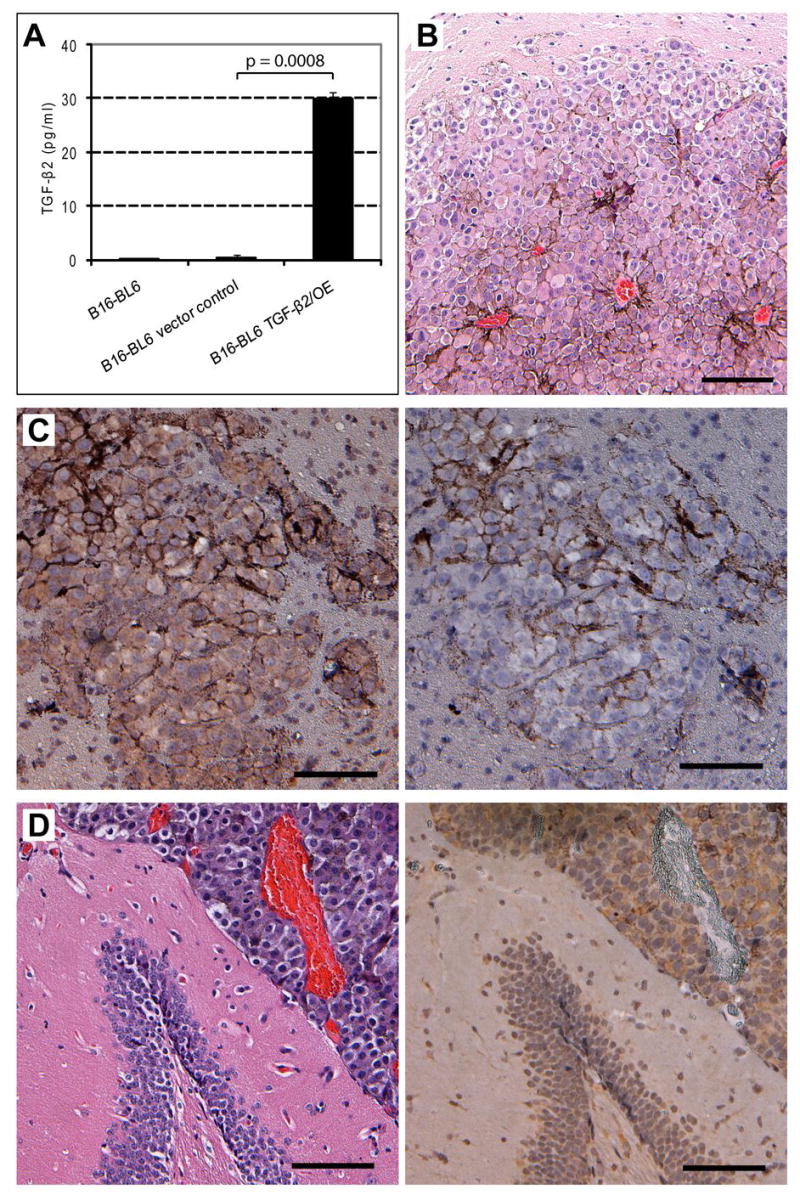
B16 melanoma cells overexpressing TGF-β2 produce microscopic metastatic lesions in the brain parenchyma. A, full-length murine TGF-β2 cDNA was cloned into pIRESneo expression vector and transfected into B16-BL6 cells. Stable clones were selected and maintained for 3 wk in gradually elevated concentrations of G418 (0.5–1 mg/ml). Production of TGF-β2 was measured by ELISA, p<0.001. B, (H&E staining) after injection into the internal carotid artery, TGF-β2-overexpressing B16-BL6 melanoma cells produced microscopic lesions in the brain parenchyma. Note the enlarged blood vessels in the parenchymal tumor lesion. C, parenchymal lesion produced by TGF-β2 overexpressing B16-BL6 cells positive for TGF-β2. Antibody control negative for TGF-β2. D, (H&E staining) overexpression of TGF-β2 did not inhibit growth of B16-BL6 cells in the brain ventricle. Ventricle tumor lesion is also TGF-β2 positive. Scale bar: 100 μm.
B16-BL6 cells overexpressing TGF-β2 produce tumor lesions in the brain parenchyma
B16-BL6 cells that overexpress TGF-β2 were injected into the internal carotid artery (5 × 104 cells/0.1 ml HBSS) of syngeneic C57BL/6 and B6C3F1 mice. The mice were euthanized when they became moribund. Mice in groups of 5 or 10 for each cell line were injected with the genetically engineered cell lines including the proper controls. Cells were also injected into the B6C3F1 mice (n=5 per group) to rule out the possible interference from the host genetic background. The experiments were repeated twice in syngeneic C57BL/6 mice and once in the B6C3F1 mice (Table 1).
TGF-β2-overexpressing B16-BL6 cells produced microscopic lesions in the brain parenchyma (Fig. 3B), whereas the parental B16-BL6 cells or B16-BL6 cells transfected with the pIRESneo vector did not. Overexpression of TGF-β2 by B16-BL6 melanoma cells did not inhibit their growth in the leptomeninges or ventricles (Figs. 3D). Both lesions in the brain parenchyma and ventricles expressed TGF-β2 (Figs. 3C, 3D). The tumor growth in the leptomeninges and ventricles was undiminished. Because the major cause for mortality was the leptomeningeal growth, no difference in survival was found between mice injected with TGF-β2-expressing B16-BL6 cells or parental B16-BL6 cells or B16-BL6 cells transfected with the pIRESneo vector control. These data indicate that TGF-β2 is essential for the growth of melanoma cells in the brain parenchyma. Further evidence for this conclusion is provided by the next experiment.
Short hairpin RNA (shRNA)-mediated knockdown of TGF-β2 expression in K-1735 C4 melanoma
To provide causal evidence for the role of TGF-β2 in the production of metastasis to the brain parenchyma, we knocked down the TGF-β2 expression in K-1735 C4 cells using shRNA-mediated interference. Lentiviral vector pGIPZ carrying the TGF-β2-specific shRNA sequence was co-transfected with packaging plasmids into HEK-293FT cells to produce lentiviral particles to transduce the K-1735 C4 melanoma cells. Cells with shRNA vector incorporated into the genome were selected and maintained in puromycin (3–5 μg/ml). TGF-β2 ELISA of the culture supernatant of the resulting cells showed that its production was suppressed in transduced K-1735 C4 melanoma cells (Fig. 4A). To show the specificity of the shRNA mediated knockdown, TGF-β1 production was measured by ELISA. No differences in production of TGF-β1 were found among K-1735 C4, control shRNA, and TGF-β2 knockdown cells (Fig. 4A).
Figure 4.
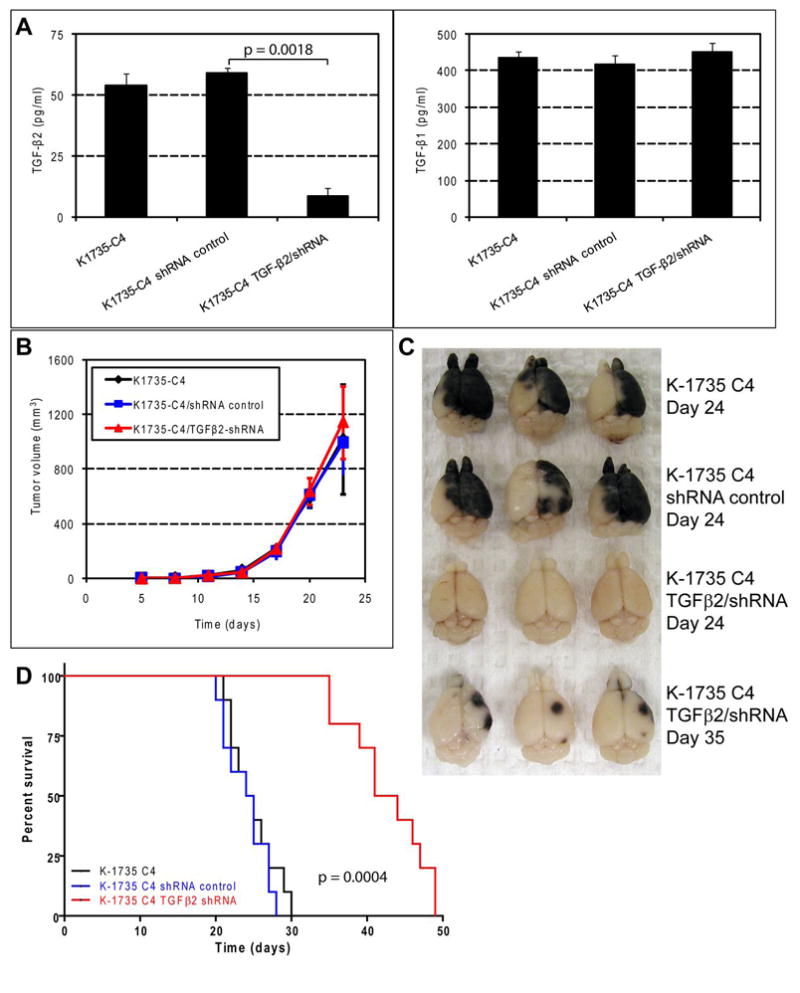
Short hairpin RNA-mediated TGF-β2 knockdown in K-1735 cells decreases metastasis to the brain parenchyma and prolongs the survival times of tumor-bearing mice. A, lentiviral particles carrying the TGF-β2-specific shRNA sequence were used to infect K-1735 C4 cells. Cells were selected and maintained for 3 wk in puromycin (3–5 μg/ml). The resulting cell cultures were analyzed by ELISA for production of TGF-β2. TGF-β2 shRNA-mediated knockdown had no effect on the production of TGF-β1. B, tumorigenicity was measured by injection of 106/0.1 ml of K-1735 C4, K-1735 C4/shRNA-control, and K-1735 C4/TGF-β2 shRNA cells into the subcutis. Tumor volume was calculated using the formula, V = (π/6) × (L×W2). C, gross brain metastasis lesions produced by K-1735 C4 cells (24 d after injection), K-1735 C4/shRNA control cells (24 d after injection), and K-1735 C4/TGF-β2-shRNA (24 d and 35 d after injection). D, survival mice were injected in the carotid artery with 105/0.1 ml of K-1735 C4, shRNA control, and TGF-β2 knockdown cells. Short hairpin RNA-mediated knockdown of TGF-β2 expression significantly prolongs the survival of tumor-bearing mice (median survival 24.5 d vs. 42.5 d, p<0.001).
Knockdown of TGF-β2 expression decreased growth of K-1735 cells in the brain parenchyma
K-1735 C4 cells, K-1735 C4 cells transduced with control shRNA with no homology to any known mammalian genes, and K-1735 C4 cells with shRNA-mediated lower TGF-β2 production (1 × 105 cells/0.1 ml HBSS) were injected into the internal carotid artery of syngeneic C3H/HeN and B6C3F1 mice (Table 1). Twenty four days later, all mice injected with K-1735 C4 tumor cells or K-1735 C4 cells transduced with control shRNA became moribund and were euthanized. Their brains contained grossly visible large melanoma lesions (Fig. 4C). In contrast, mice injected with TGF-β2 knockdown cells were asymptomatic. Three of these asymptomatic mice were euthanized and autopsied. No obvious lesions were detected upon visual inspection (Fig. 4C). At 35 d after injection, 3 mice from the TGF-β2 knockdown group were euthanized. The mice exhibited few small grossly visible melanoma lesions in the brain parenchyma (Fig. 4C). Six weeks after injection, the remaining mice bearing the TGF-β2 knockdown tumors became moribund and were euthanized. To repeat the observed effects of TGF-β2 knockdown on the brain parenchymal growth of the K-1735 C4 melanoma cells, we injected the tumor cells into another group of 10 mice for each cell line and studied their survival. Survival time was significantly prolonged in mice with knockdown expression of TGF-β2, as compared to control mice (Fig. 4D). Upon autopsy, we did not find lesions in the leptomeninges or brain ventricles, indicating that low or high expression of TGF-β2 is not correlated with growth of melanoma cells in these areas of the brain. This experiment was repeated twice in the syngeneic C3H/HeN mice with 10 animals for each group of tumor cell injection and once in the B6C3F1 mice with 5 animals for each group of tumor cell injection (Table 1).
To eliminate the possibility that TGF-β2 knockdown decreased in vivo tumorigenicity of K-1735 C4 cells, we inoculated the parental K-1735 C4 cells, control shRNA cells, and TGF-β2 knockdown cells into the subcutis of syngeneic mice (n=5). No differences in tumor take and volume were found among these groups (Fig. 4B).
Discussion
In the brain, the leptomeninges and ventricles form a contiguous compartment filled with cerebrospinal fluid produced by the choroid plexus in the ventricles. The brain parenchyma is composed of neurons, various glial cells, and endothelial cells that perform all central nervous system functions. Therefore, the brain parenchyma and the leptomeninges/ventricles system represent two distinct microenvironments in the central nervous system. Human melanoma often produces leptomeningeal or parenchymal brain metastasis (25). Metastatic tumor growth in the leptomeninges confers a particularly dismal prognosis for the patient due to its spreading distribution and inaccessibility to surgical resection (26). In experimental models of brain metastasis, tumor growth at either site results in quick morbidity and mortality of the host animals. Absolute site-specific brain metastasis was observed after two murine melanomas, B16-BL6 and K-1735 C4, were injected into the internal carotid artery of mice to produce experimental brain metastasis. We have previously reported that B16-BL6 melanoma cells produce metastasis in the leptomeninges and ventricles, whereas the K-1735 melanoma cells produce metastasis in the brain parenchyma (5). To rule out the possibility that the observed site-specificity is due to the distinct genetic backgrounds from which these two murine melanoma cells were derived, the site-specific brain metastasis was reproduced in both syngeneic mice (C57BL/6 for B16-BL6 and C3H/HeN for K-1735 C4, respectively) and B6C3F1 mice (C57BL/6 x C3H/HeN F1 cross) (6).
In 1889, Steven Paget first proposed the “seed and soil” hypothesis of cancer metastasis, stating that the pattern of distant organ metastasis is not random but dependent on multiple interactions between tumor cells and a congenial microenvironment (27). In the present study, we analyzed the VEGF and TGF-β growth factor expressions by the murine B16-BL6 and K-1735 C4 melanoma cells using semi-quantitative RT-PCR. Both cytokines have been implicated in tumor interactions with the brain microenvironment. Our experiments revealed that TGF-β2, an isoform of the TGF-β cytokine family, is only highly expressed by the K-1735 C4 melanoma cells. Interestingly, when the analyses were expanded to somatic hybrid cells from cell-cell fusion between B16-BL6 and K-1735 C4 melanoma cells (6), we found that the TGF-β2 expression was strictly correlated with the in vivo phenotype of site-specific brain metastasis. Melanoma cells that grew only in the brain parenchyma expressed high level of TGF-β2 (i.e., K-1735 C4 cells and K-1735 C4 self-fusion hybrid cells), whereas all other melanoma cells that grew only in the leptomeninges and ventricles (i.e., B16-BL6 cells and different clones of hybrid cells made from B16-BL6 x K-1735 C4 cells) did not produce detectable amount of the TGF-β2 ligand or expressed low level of the mRNA transcript.
To examine the causal evidence of the role of TGF-β2 in determining site-specific brain metastasis, we overexpressed the TGF-β2 gene in B16-BL6 cells and knocked down TGF-β2 expression in K-1735 C4 cells by shRNA-mediated interference. The resulting genetically engineered cells were injected into the internal carotid artery. Again, TGF-β2 expression was correlated with site-specific brain metastasis. B16-BL6 cells overexpressing the TGF-β2 gene produced microscopic lesions in the brain parenchyma as well as major tumors in the leptomeninges and ventricles. K-1735 C4 cells with shRNA-mediated knockdown of TGF-β2 expression produced few slow-growing lesions in the brain parenchyma but no lesions in the leptomeninges or ventricles. The overall survival of these mice was significantly prolonged as compared to the control mice. Collectively, these results suggest that expression of TGF-β2 is necessary for the growth of melanoma metastasis in the brain parenchyma. The number of metastatic lesions produced in the brain parenchyma by B16-BL6 cells expressing TGF-β2 was smaller than those produced by K-1735 C4 cells. This may be due to the lower level of TGF-β2 in the B16-BL6 TGF-β2-transfected cells as compared with that in the K-1735 C4 cells. Alternatively, the TGF-β2-transfected B16-BL6 cells may lack additional co-factors that are necessary for the growth of melanoma metastasis in the brain parenchyma. Short hairpin RNA-mediated TGF-β2 knockdown in K-1735 C4 cells did not completely block the production of this growth factor, which may account for the fact that K-1735 C4 cells with TGF-β2 knockdown produce few slow-growing lesions that eventually kill the host animals.
Early studies of GBM culture supernatants suggested the presence of an immunosuppressive factor (28, 29). Later, this factor was isolated and identified as the homologue to TGF-β1 and it was named TGF-β2 (10, 11). The immunosuppressive effect of the GBM culture supernatants is inhibited by anti-TGF-β2 but not by anti-TGF-β1 antibodies (13). This TGF-β2 cytokine was then found to be produced by a large variety of human GBM cell lines (30). In situ hybridization studies confirmed the expression of TGF-β2 by multiple clinical specimens of human GBM (12). Recent evidence suggested that TGF-β2 mediated immunosuppression involves regulatory T cells (31). TGF-β2 was also shown to increase the permeability of endothelial cell monolayer in an in vitro model of blood brain barrier (32). Antisense therapy of experimental glioma in rats (15–17) produced encouraging results.
Our present data demonstrate that growth of murine melanoma cells in the brain parenchyma is associated with expression of TGF-β2. These results corroborate the findings from studies of GBM cells and demonstrate that common microenvironmental factors in a specific organ/tissue may determine tumor take and growth. Whether anti-TGFβ2 therapies currently under trial for GBM patients (33–37) might be effective for melanoma brain metastasis should therefore be investigated.
Acknowledgments
Grant support: This work was supported in part by Cancer Center Support Core grant CA16672 and SPORE in Prostate Cancer grant CA90270 from the National Cancer Institute, National Institutes of Health.
The authors thank Lionel Santibañez for critical editorial review and Lola López for excellent assistance with the preparation of this manuscript.
References
- 1.Patchell RA. The management of brain metastases. Cancer Treat Rev. 2003;29:533–40. doi: 10.1016/s0305-7372(03)00105-1. [DOI] [PubMed] [Google Scholar]
- 2.Johnson JD, Young B. Demographics of brain metastasis. Neurosurg Clin N Am. 1996;7:337–44. [PubMed] [Google Scholar]
- 3.Sampson JH, Carter JH, Jr, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11–20. doi: 10.3171/jns.1998.88.1.0011. [DOI] [PubMed] [Google Scholar]
- 4.Schackert G, Fidler IJ. Development of in vivo models for studies of brain metastasis. Int J Cancer. 1988;41:589–94. doi: 10.1002/ijc.2910410419. [DOI] [PubMed] [Google Scholar]
- 5.Schackert G, Fidler IJ. Site-specific metastasis of mouse melanomas and a fibrosarcoma in the brain or meninges of syngeneic animals. Cancer Res. 1988;48:3478–84. [PubMed] [Google Scholar]
- 6.Fujimaki T, Fan D, Staroselsky A, Gohji K, Bucana C, Fidler I. Critical factors regulating site-specific brain metastasis of murine melanomas. Int J Oncol. 3:789–99. doi: 10.3892/ijo.3.5.789. [DOI] [PubMed] [Google Scholar]
- 7.Sporn MB. The early history of TGF-β, and a brief glimpse of its future. Cytokine Growth Factor Rev. 2006;17:3–7. doi: 10.1016/j.cytogfr.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 8.Massague J. TGF-β in cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bierie B, Moses HL. Tumour microenvironment: TGF-β: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–20. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 10.de Martin R, Haendler B, Hofer-Warbinek R, et al. Complementary DNA for human glioblastoma-derived T cell suppressor factor, a novel member of the transforming growth factor-β gene family. EMBO J. 1987;6:3673–7. doi: 10.1002/j.1460-2075.1987.tb02700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wrann M, Bodmer S, de Martin R, et al. T cell suppressor factor from human glioblastoma cells is a 12.5-kd protein closely related to transforming growth factor-β. EMBO J. 1987;6:1633–6. doi: 10.1002/j.1460-2075.1987.tb02411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maxwell M, Galanopoulos T, Neville-Golden J, Antoniades HN. Effect of the expression of transforming growth factor-β 2 in primary human glioblastomas on immunosuppression and loss of immune surveillance. J Neurosurg. 1992;76:799–804. doi: 10.3171/jns.1992.76.5.0799. [DOI] [PubMed] [Google Scholar]
- 13.Bodmer S, Strommer K, Frei K, et al. Immunosuppression and transforming growth factor-β in glioblastoma: preferential production of transforming growth factor-β 2. J Immunol. 1989;143:3222–9. [PubMed] [Google Scholar]
- 14.Jachimczak P, Bogdahn U, Schneider J, et al. The effect of transforming growth factor-β 2-specific phosphorothioate-anti-sense oligodeoxynucleotides in reversing cellular immunosuppression in malignant glioma. J Neurosurg. 1993;78:944–51. doi: 10.3171/jns.1993.78.6.0944. [DOI] [PubMed] [Google Scholar]
- 15.Liau LM, Fakhrai H, Black KL. Prolonged survival of rats with intracranial C6 gliomas by treatment with TGF-β antisense gene. Neurol Res. 1998;20:742–7. doi: 10.1080/01616412.1998.11740594. [DOI] [PubMed] [Google Scholar]
- 16.Fakhrai H, Dorigo O, Shawler DL, et al. Eradication of established intracranial rat gliomas by transforming growth factor β antisense gene therapy. Proc Natl Acad Sci USA. 1996;93:2909–14. doi: 10.1073/pnas.93.7.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Wang Q, Kleinschmidt-DeMasters BK, Franzusoff A, Ng KY, Lillehei KO. TGF-β2 inhibition augments the effect of tumor vaccine and improves the survival of animals with pre-established brain tumors. J Neurooncol. 2007;81:149–62. doi: 10.1007/s11060-006-9222-1. [DOI] [PubMed] [Google Scholar]
- 18.Hart IR. The selection and characterization of an invasive variant of the B16 melanoma. Am J Pathol. 1979;97:587–600. [PMC free article] [PubMed] [Google Scholar]
- 19.Poste G, Doll J, Hart IR, Fidler IJ. In vitro selection of murine B16 melanoma variants with enhanced tissue-invasive properties. Cancer Res. 1980;40:1636–44. [PubMed] [Google Scholar]
- 20.Fidler IJ. Selection of successive tumour lines for metastasis. Nat New Biol. 1973;242:148–9. doi: 10.1038/newbio242148a0. [DOI] [PubMed] [Google Scholar]
- 21.Kripke ML. Speculations on the role of ultraviolet radiation in the development of malignant melanoma. J Natl Cancer Inst. 1979;63:541–8. doi: 10.1093/jnci/63.3.541. [DOI] [PubMed] [Google Scholar]
- 22.Fidler IJ, Gruys E, Cifone MA, Barnes Z, Bucana C. Demonstration of multiple phenotypic diversity in a murine melanoma of recent origin. J Natl Cancer Inst. 1981;67:947–56. [PubMed] [Google Scholar]
- 23.Talmadge JE, Fidler IJ. Enhanced metastatic potential of tumor cells harvested from spontaneous metastases of heterogeneous murine tumors. J Natl Cancer Inst. 1982;69:975–80. [PubMed] [Google Scholar]
- 24.Radinsky R, Beltran PJ, Tsan R, Zhang R, Cone RD, Fidler IJ. Transcriptional induction of the melanocyte-stimulating hormone receptor in brain metastases of murine K-1735 melanoma. Cancer Res. 1995;55:141–8. [PubMed] [Google Scholar]
- 25.de la Monte SM, Moore GW, Hutchins GM. Patterned distribution of metastases from malignant melanoma in humans. Cancer Res. 1983;43:3427–33. [PubMed] [Google Scholar]
- 26.Raizer JJ, Hwu WJ, Panageas KS, et al. Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro-Oncol. 2008;10:199–207. doi: 10.1215/15228517-2007-058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 28.Fontana A, Hengartner H, de Tribolet N, Weber E. Glioblastoma cells release interleukin 1 and factors inhibiting interleukin 2-mediated effects. J Immunol. 1984;132:1837–44. [PubMed] [Google Scholar]
- 29.Schwyzer M, Fontana A. Partial purification and biochemical characterization of a T cell suppressor factor produced by human glioblastoma cells. J Immunol. 1985;134:1003–9. [PubMed] [Google Scholar]
- 30.Leitlein J, Aulwurm S, Waltereit R, et al. Processing of immunosuppressive pro-TGF-β 1,2 by human glioblastoma cells involves cytoplasmic and secreted furin-like proteases. J Immunol. 2001;166:7238–43. doi: 10.4049/jimmunol.166.12.7238. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Yang P, Zhou H, Meng Q, Huang X. Involvement of Foxp3-expressing CD4+ CD25+ regulatory T cells in the development of tolerance induced by transforming growth factor-β2-treated antigen-presenting cells. Immunology. 2008;124:304–14. doi: 10.1111/j.1365-2567.2007.02769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishihara H, Kubota H, Lindberg RL, et al. Endothelial cell barrier impairment induced by glioblastomas and transforming growth factor β2 involves matrix metalloproteinases and tight junction proteins. J Neuropathol Exp Neurol. 2008;67:435–48. doi: 10.1097/NEN.0b013e31816fd622. [DOI] [PubMed] [Google Scholar]
- 33.Fakhrai H, Mantil JC, Liu L, et al. Phase I clinical trial of a TGF-β antisense-modified tumor cell vaccine in patients with advanced glioma. Cancer Gene Ther. 2006;13:1052–60. doi: 10.1038/sj.cgt.7700975. [DOI] [PubMed] [Google Scholar]
- 34.Schlingensiepen R, Goldbrunner M, Szyrach MN, et al. Intracerebral and intrathecal infusion of the TGF-β 2-specific antisense phosphorothioate oligonucleotide AP 12009 in rabbits and primates: toxicology and safety. Oligonucleotides. 2005;15:94–104. doi: 10.1089/oli.2005.15.94. [DOI] [PubMed] [Google Scholar]
- 35.Schlingensiepen KH, Schlingensiepen R, Steinbrecher A, et al. Targeted tumor therapy with the TGF-β2 antisense compound AP 12009. Cytokine Growth Factor Rev. 2006;17:129–39. doi: 10.1016/j.cytogfr.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Hau P, Jachimczak P, Schlingensiepen R, et al. Inhibition of TGF-β2 with AP 12009 in recurrent malignant gliomas: from preclinical to phase I/II studies. Oligonucleotides. 2007;17:201–12. doi: 10.1089/oli.2006.0053. [DOI] [PubMed] [Google Scholar]
- 37.Schlingensiepen KH, Fischer-Blass B, Schmaus S, Ludwig S. Antisense therapeutics for tumor treatment: the TGF-β2 inhibitor AP 12009 in clinical development against malignant tumors. Recent Results Cancer Res. 2008;177:137–50. doi: 10.1007/978-3-540-71279-4_16. [DOI] [PubMed] [Google Scholar]