

Abstract
Targeted therapy against epidermal growth factor receptor (EGFR) represents a major therapeutic advance in lung cancer treatment. Somatic mutations of the EGFR gene, most commonly L858R (exon 21) and short in-frame exon 19 deletions, have been found to confer enhanced sensitivity towards the inhibitors gefitinib and erlotinib. We have recently identified an EGFR mutation E884K, in combination with L858R, in a patient with advanced lung cancer who progressed on erlotinib maintenance therapy, and subsequently had leptomeningeal metastases that responded to gefitinib. The somatic E884K substitution appears to be relatively infrequent, and resulted in a mutant lysine residue that disrupts an ion pair with residue R958 in the EGFR kinase domain C-lobe, an interaction that is highly conserved within the human kinome as demonstrated by our sequence analysis and structure analysis. Our studies here, using COS-7 transfection model system, show that E884K works in concert with L858R in-cis, in a dominant fashion, to change downstream signaling, differentially induce MAPK-ERK1/2 signaling and associated cell proliferation, and differentially alter sensitivity of EGFR phosphorylation inhibition by ERBB family inhibitors in an inhibitor-specific fashion. Mutations of the conserved ion pair E884-R958 may result in conformational changes that alter kinase substrate recognition. The analogous E1271K-MET mutation conferred differential sensitivity towards preclinical MET inhibitors SU11274 (unchanged), and PHA665752 (more sensitive). Systematic bioinformatics analysis of the mutation catalog in the human kinome (COSMIC) revealed the presence of cancer-associated mutations involving the conserved E884 homologous residue, and adjacent residues at the ion pair, in known proto-oncogenes (KIT, RET, MET, FAK) and tumor suppressor gene (LKB1). Targeted therapy using small molecule inhibitors should take into account potential cooperative effects of multiple kinase mutations, and their specific effects on downstream signaling and inhibitor sensitivity. Improved efficacy of targeted kinase inhibitors may be achieved by targeting the dominant activating mutations present.
Keywords: EGFR, MET, mutation, tyrosine kinase inhibitor, sensitivity, resistance, structure, kinome
Introduction
Targeted therapy using epidermal growth factor receptor (EGFR) kinase inhibitors represents a major therapeutic advance in lung cancer treatment. Somatic mutations of the EGFR gene, most commonly L858R (exon 21) and short in-frame deletions in exon 19, have recently been identified as catalytic domain mutation hotspots (Shigematsu et al., 2006). These mutations confer enhanced sensitivity towards the anilinoquinazoline kinase inhibitors gefitinib and erlotinib (Lynch et al., 2004; Paez et al., 2004). A mutation conferring resistance to these two kinase inhibitors, T790M (exon 20), has also been found in the EGFR kinase domain and can account for about half of the cases of acquired resistance (Kobayashi et al., 2005). There are a number of other kinase domain mutations of EGFR that occur at lower frequencies, most often in combination with L858R (Tam et al., 2006). However, how these mutations might interact when present together in-cis is unknown.
We recently identified a novel EGFR kinase domain somatic mutation, E884K (Glu884Lys, exon 22) in a patient with stage IV non-small-cell lung cancer (NSCLC), in combination with the L858R mutation (L858R+E884K) (Choong et al., 2006). The patient initially received carboplatin/paclitaxel and erlotinib and then developed brain metastasis on maintenance erlotinib. In spite of further treatment with whole brain radiation, temozolomide, and irinotecan, the patient's disease progressed to symptomatic leptomeningeal carcinomatosis, which responded to gefitinib, a year after being off an EGFR kinase inhibitor. The L858R+E884K double mutation was found both in her pretreatment diagnostic thoracic lymph node biopsy specimen as well as the tumor cells (extracted by laser microdissection) within the cerebrospinal fluid during the course of leptomeningeal metastases (Choong et al., 2006). The E884K mutation represents the first mutation reported to show an apparent differential response to the two EGFR kinase inhibitors erlotinib and gefitinib, while L858R was known to be sensitizing to both. These findings led to our hypothesis that EGFR kinase mutations can work in concert to differentially alter inhibitor sensitivity and downstream signaling. Further biochemical analysis in our current study indicates that the double mutant EGFR (L858R+E884K) responds differently to gefitinib and erlotinib. We now show that E884K works in concert with L858R, and in a dominant fashion, to mediate differential sensitivity to kinase inhibitors via altered phosphorylation of AKT and STAT3 and were correlated with differential cellular cytotoxicity and induction of the apoptotic marker cleaved-PARP(Asp214) by EGFR inhibitors. Using a combination of bioinformatics and structural analyses, we further characterized the role of the E884 residue in EGFR kinase function. Our results further demonstrate that the ion pair formed by residues E884 and R958 in the EGFR kinase domain is a highly conserved feature of protein kinases in the human kinome, including many “druggable” targets such as MET. Disruption of the conserved ion pair in EGFR modulates downstream signal transduction and differentially alters kinase inhibitor sensitivity in an inhibitor-specific manner.
Results
E884K works in concert with L858R mutation to confer differential inhibitor sensitivity through inhibition of AKT and STAT3 downstream signaling
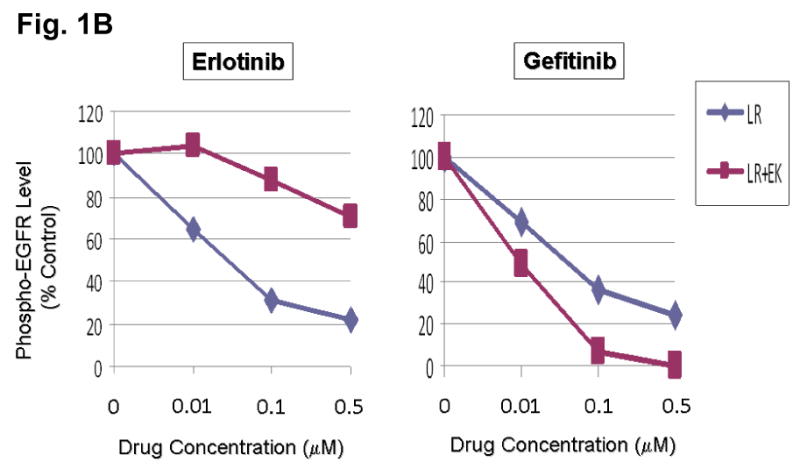
We hypothesize that EGFR kinase mutations can work in concert to differentially alter inhibitor sensitivity. To test this hypothesis, EGFR expression constructs engineered with L858R (LR) or dual mutations of L858R+E884K (LR+EK) were stably transfected into COS-7 cells. Cells were treated with increasing concentrations of either erlotinib or gefitinib in the presence of EGF stimulation (Fig. 1A). Compared to L858R alone, the L858R+E884K dual mutant was less sensitive to erlotinib in the inhibition of tyrosine phosphorylation of EGFR. Conversely, E884K worked in concert with L858R in-cis to further enhance the sensitivity of the mutant receptor to gefitinib inhibition (Fig. 1A, B). These findings correlated with the clinical course of the patient's response profile (Choong et al., 2006), and highlight the potential for EGFR kinase mutations to exert concerted effects in-cis to impact targeted inhibition.
Figure 1. E884K mutation of EGFR worked in concert with L858R to differentially alter sensitivity to EGFR kinase inhibitors erlotinib and gefitinib.
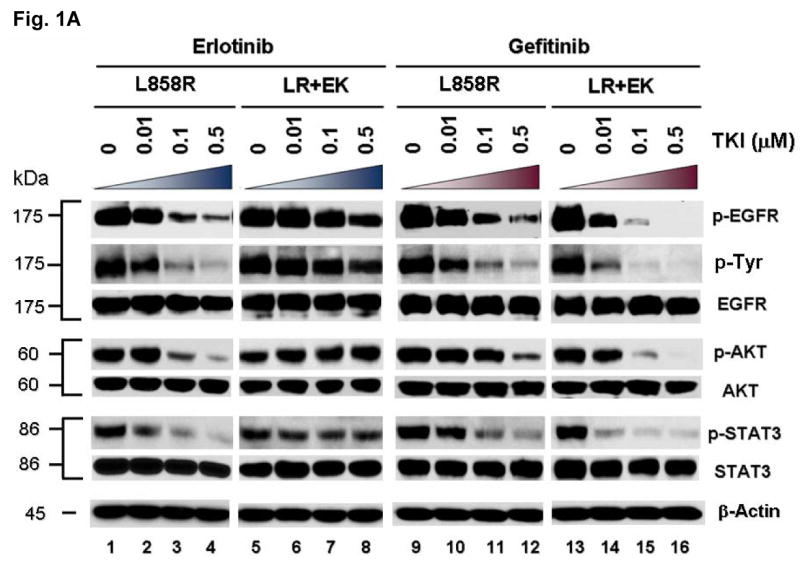
(A) Stable COS-7 transfects expressing the L858R and double mutant L858R+E884K variants of EGFR were used in the experiment. The endogenous wild type EGFR expression of parental COS-7 cells is negligible (data not shown). Cells were cultured in 0.5% BSA-containing serum-free media for 16 hours and then incubated with increasing concentrations of either erlotinib or gefitinib in the presence of EGF (100 ng/ml). Whole cell lysates were extracted for SDS-PAGE and immunoblotting using antibodies against: p-EGFR [Y1068], phosphotyrosine (p-Tyr), EGFR, p-AKT [S473], AKT, p-STAT3 [Y705], STAT3, c-PARP [cleaved-PARP(Asp214)], and β-actin. The experiment was performed in duplicate with reproducible results. The E884K mutation negatively modulated the effect of L858R to erlotinib inhibition in a dominant fashion but enhanced sensitivity of the mutant receptor to gefitinib inhibition.
(B) Densitometric quantitation of the p-EGFR [Y1068] levels showing differential alteration of sensitivity to erlotinib (more resistant) and gefitinib (more sensitive) by the E884K mutation when in-cis with L858R. The densitometric scanning of the p-EGFR immunoblot bands was performed digitally using the NIH ImageJ software program, and was normalized to the total EGFR expression levels.
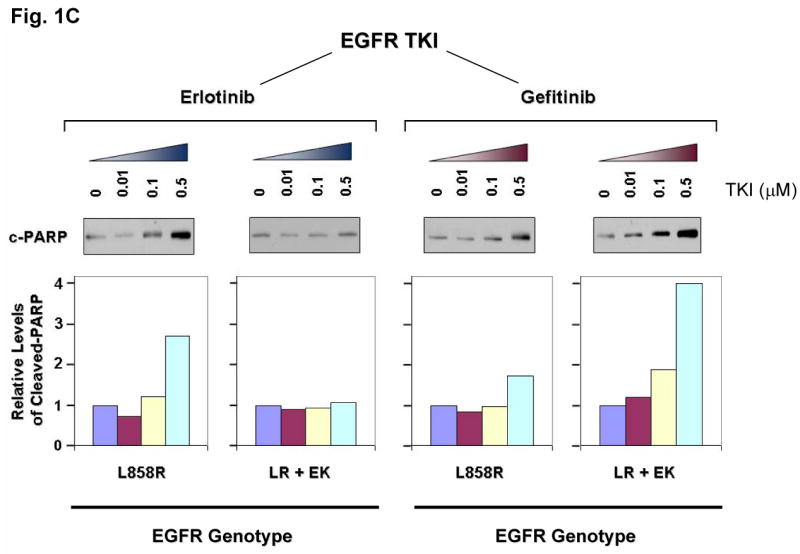
(C) Relative expression of the apoptotic marker, cleaved-PARP(Asp214) in L858R and L858R+E884K EGFR variants treated with increasing concentrations of erlotinib (left) and gefitinib (right). The immunoblot using anti-cleaved-PARP(Asp214) antibody is shown here (above) together with the densitometric quantitation (below) adjusted to β-actin loading control using the NIH ImageJ software program.
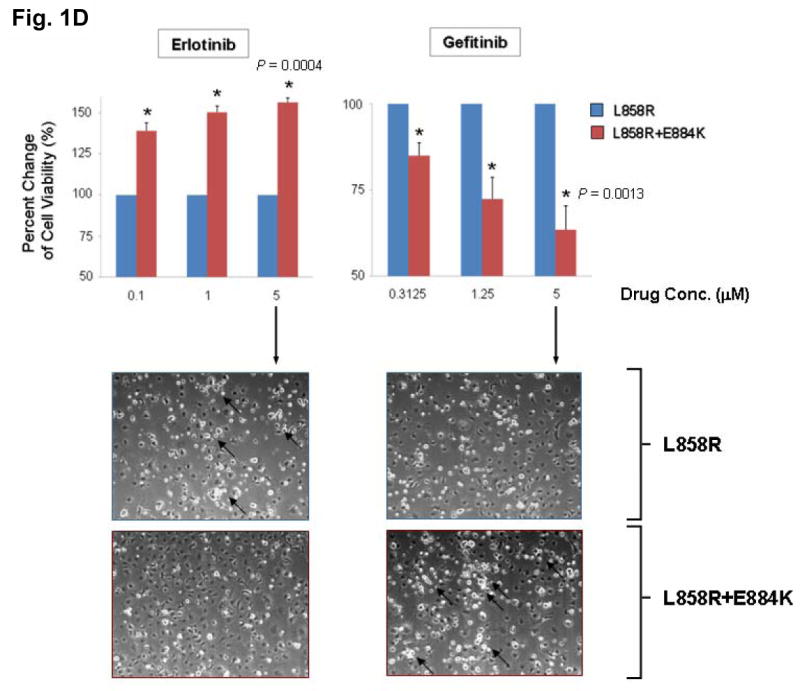
(D) COS-7 cells with stable transduced expression of L858R or L858R+E884K mutant EGFR were tested in cellular cytotoxicity assay in vitro under drug treatment with either erlotinib or gefitinib at indicated concentrations. Results are shown in percent change of cell viability of L858R+E884K EGFR-COS-7 compared to the control L858R EGFR-COS-7 cells at each concentration of TKI tested. E884K mutation, when in-cis with L858R, significantly decreased the sensitivity of cell viability inhibition by erlotinib compared with L858R alone; however, it significantly increased the sensitivity of cell viability inhibition by gefitinib compared with L858R alone. In the case of erlotinib, E884K was desensitizing to L858R, leading to lower cytotoxicity (56.3 ±2.68% increased viable cells after inhibition at 5 μM, P=0.0004) compared with L858R alone. Conversely, in gefitinib inhibition, E884K further sensitized L858R in-cis, leading to significantly higher cytotoxicity (63.5 ±6.86% decreased viable cells after inhibition at 5 μM, P=0.0013) compared with L858R alone. Error bar, S.D. (N=3). *, P<0.05, compared with L858R alone. Representative photomicrographs of cells after 48 hours of indicated inhibitor treatment (5 μM) in vitro were included to illustrate the presence of differential cytotoxicity as seen with the non-viable detached cells/cell fragments (10 x). Examples of increased floating non-viable cells are highlighted with arrows.
To gain insight into the mechanism of E884K modulation of EGFR tyrosine kinase inhibitor (TKI) sensitivity, we further studied its effect on downstream AKT and STAT3 signaling pathways with TKI inhibition. The effect on the downstream signal mediators p-AKT [S473] and p-STAT3 [Y705] correlated well with the inhibition of EGFR phosphorylation (Figure 1A); E884K in-cis with L858R decreased erlotinib inhibition of AKT and STAT3 phosphorylation but increased inhibition by gefitinib. The differential inhibition exerted by E884K on EGFR, AKT and STAT3 signaling also corresponded to the inhibitor induced expression pattern of the apoptotic marker, cleaved-PARP(Asp214) (Figure 1C). Similarly, there was an opposite effect of the E884K mutation over L858R in-cis in inducing cellular cytotoxicity by erlotinib and gefitinib (Figure 1D). Hence, E884K in-cis with L858R differentially altered inhibitor sensitivity when compared to L858R alone, through differential inhibition of the pro-survival AKT and STAT3 signaling pathways associated with altered induction of cleaved-PARP(Asp214).
E884K-EGFR modulates inhibitor sensitivity effects in an inhibitor-specific fashion
In order to further examine the hypothesis that EGFR mutations exert effects in combination that are unique to a specific kinase inhibitor, we further tested the mutant EGFR expressing L858R alone or L858R+E884K in-cis, against several other ERBB family tyrosine kinase inhibitors, including both reversible inhibitors (4557W, Lapatinib, GW583340, Tyrphostin, AG1478) and irreversible inhibitor (CL-387,785) (Figure 2, and Supplementary Figure 2). We focused on the effects of these inhibitors on the sensitivity of inhibition of the EGFR kinase phosphorylation in the mutant EGFR. Since the tyrosine phosphorylation of the EGFR has been shown to correlate well with its catalytic enzymatic activity, we used the tyrosine phosphorylation of the pY1068 (GRB1 binding site) epitope of EGFR as the surrogate measurement of the extent of inhibition by the TKIs. For 4557W (reversible dual TKI of EGFR/ERBB2), the E884K mutation modulated the L858R mutation in-cis, again in a dominant fashion, rendering the double mutant receptor more sensitive to the dual inhibitor (Figure 2). Hence, E884K mutation can work in concert with L858R to modulate mutant receptor sensitivity to different targeted inhibitors. Similarly, E884K further enhanced the sensitivity of L858R to the inhibition by the irreversible EGFR/ERBB2 inhibitor, CL-387,785. On the other hand, the sensitivity of EGFR phosphorylation between the L858R and L858R+E884K EGFR receptors in Tyrphostin AG1478 (reversible EGFR-TKI), GW583340 (reversible dual EGFR/ERBB2-TKI), and lapatinib (reversible dual EGFR/ERBB2-TKI) did not significantly differ. Hence, the E884K mutation, when in-cis with L858R, modulates the sensitivity of the mutant receptor towards ERBB family kinase inhibitors in an inhibitor-specific fashion.
Figure 2. Effects of L858R/E884K-EGFR on other EGFR kinase inhibitors.
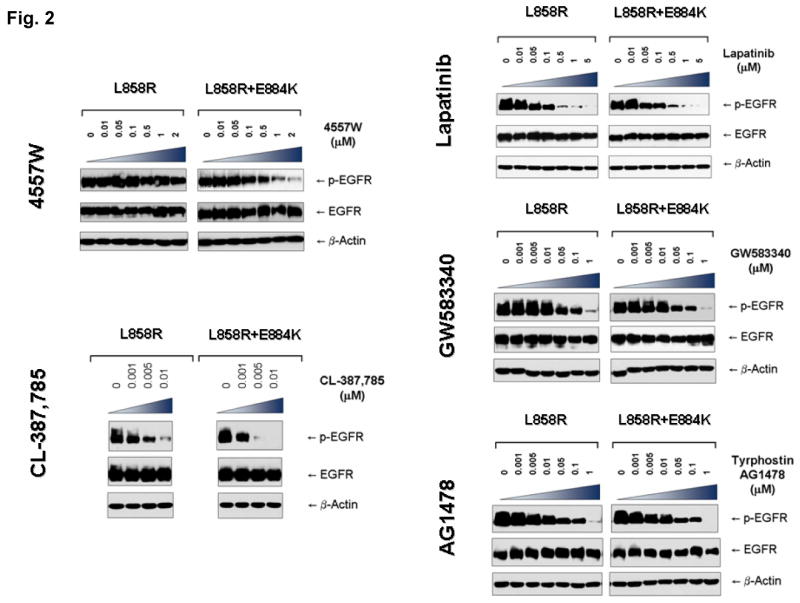
The EGFR mutation E884K modulated L858R mutation in-cis with inhibitor specific effects on the sensitivities to EGFR phosphorylation inhibition by the inhibitors (4557W, GW583340, AG1478, lapatinib, and CL-387,785). Stable COS-7 transfectant cells expressing equivalent levels of the following EGFR variants were used: L858R (LR), and L858R+E994K (LR+EK). Cells were cultured in 0.5% BSA-containing serum-free media for 16 hours, and then treated without or with increasing concentrations of the EGFR TKIs as indicated, in the presence of EGF stimulation (100 ng/ml). Whole cell lysates were extracted for SDS-PAGE and immunoblotting using the following antibodies: p-EGFR [Y1068], EGFR and β-actin. E884K mutation worked in concert with L858R in-cis to enhance the sensitivity of the mutant receptor to inhibition by the kinase inhibitor 4557W, and CL-387,785. On the other hand, it has little effects on the inhibition by lapatinib, GW583340, and AG1478.
E884K is activating, and can work cooperatively with L858R to differentially modulate downstream signal transduction
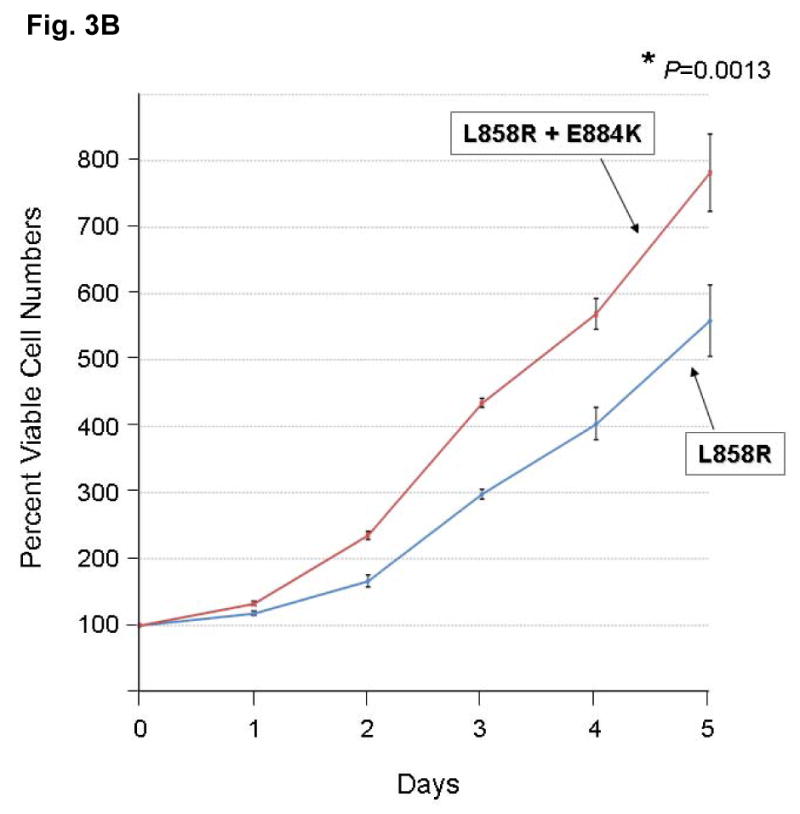
To address the question whether there are other downstream phosphoproteins that can be differentially activated by the E884K mutation compared to the activating L858R mutation, the global phosphotyrosine profiles of the cellular proteins induced by the mutant EGFR were examined. The E884K alone and L858R+E884K double mutant EGFR remained sensitive to EGF, and the E884K mutation cooperates with L858R when in-cis to further enhance the mutational effects on downstream phosphoprotein activation (data not shown). To date, essentially all mutational combinations involving L858R studied thus far were found to exist in-cis, suggesting potential cis mutation-to-mutation cooperation in EGFR signaling and possibly tumorigenesis (Tam et al., 2006). To determine the effect of E884K on mutant EGFR signaling, we next studied the EGFR activation of the downstream PI3K-AKT-MAPK(ERK1/2)-STAT pathway. E884K mutant (alone or in-cis with L858R) receptor exhibited constitutive activation of the tyrosine phosphorylated EGFR comparable to L858R (Figure 3A). E884K and L858R+E884K mutants remained sensitive to EGF and were activated by the ligand to a level comparable to L858R (Figure 3A). L858R was associated with downstream activation of p-AKT signaling, which was inducible by EGF stimulation. When in-cis with L858R, E884K mutation (L858R+E884K) downregulated constitutive AKT phosphorylation. E884K, alone or in-cis with L858R, can also mediate constitutive induction of p-STAT3 [pY705] (important for STAT3 dimerization and transcriptional activation of target genes) (Figure 3A). Interestingly, the double mutation, L858R+E884K conferred a distinctly more sensitive response to EGF stimulation selectively in the MAPK-ERK1/2 cell proliferation pathway compared to either wild type, E884K alone or L858R alone. Consistent with this differential signaling effect, the L858R+E884K-COS-7 cells had a significantly higher cell proliferation rate than that of the L858R-COS-7 cells in the MTS cell proliferation assay for 5 days (Figure 3B). At Days 3 and 5, the cell proliferation rate as determined by % viable cells increase during the assay period, was 1.46-fold (Day 3) and 1.40-fold (P=0.0013) higher (Day 5) in L858R+E884K than L858R alone. L858R+E884K also conferred a higher induction of p-CBL as well. Hence, the double mutation L858R+E884K modulated basal and stimulated downstream EGFR signaling differentially with differential effects on the AKT (down-regulated), CBL and MAPK-ERK1/2 phosphorylation (up-regulated). Moreover, E884K had a dominant effect over L858R, when in-cis, in these signaling modulatory effects.
Figure 3. Effects of mutational disruption of the Glu(E)884-Arg(R)958 ion pair in EGFR signaling.
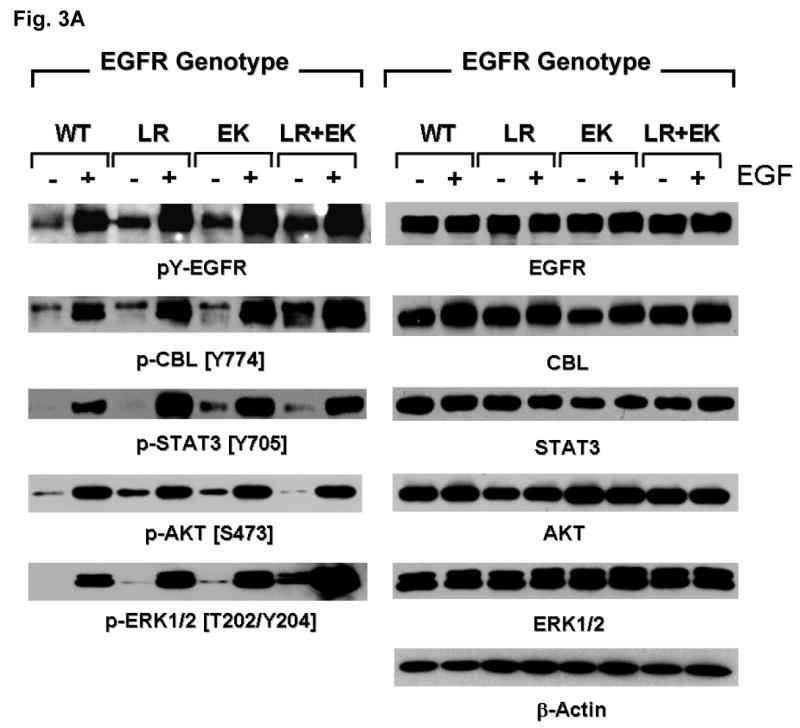
(A) Stable COS-7 transfectant cells expressing the various EGFR variants were cultured in 0.5% BSA-containing serum-free media, followed by EGF stimulation (100 ng/ml, 10 min). Whole cell lysates were prepared for SDS-PAGE and immunoblotted using the antibodies against: phosphotyrosine (pY-EGFR), EGFR, p-CBL[Y774], CBL, p-AKT [S473], AKT, p-ERK1/2 [T202/Y204], ERK1/2, p-STAT3 [Y705], STAT3, and β-actin. E884K, either alone or in-cis with L858R, modulated differential activation of downstream mutant EGFR signaling.
(B) The EGFR double mutations L858R+E884K conferred a significantly higher cell proliferation rate than L858R alone in the COS-7 cells stably expressing the transduced mutant EGFR. Cellular viability assay was performed with the cells growing in regular growth media (10% FBS) up to 5 days as described in Materials and Methods. The MTS viability assay was performed in triplicate. Error bar, S.D. *, P=0.0013.
Disruption of a conserved ion pair, Glu(E)884-Arg(R)958, in EGFR differentially alters kinase inhibitor sensitivity
Next, bioinformatics analysis of the E884 residue was performed by multiple kinase domain amino acid sequence alignments of the human kinome, using the AliBee multiple sequence alignment program (GeneBee) (Supplementary Figure 1). Amino acid alignments of the kinase domains of phylogenetically diverse groups of kinases such as among the ERBB family, the VEGFR family and the TRK family show that the E884 residue is highly conserved (Figure 4A). In addition, a second residue was also found to be highly conserved (R958) (Figure 4A). Further multiple sequence alignments of 321 human kinase domains show high conservation of both E884 and R958 residues of the EGFR kinase domain (Supplementary Figure 1). The glutamic acid residue (E884) is conserved in >77% and the arginine residue (R958) conserved in >55% of human kinases in the kinome.
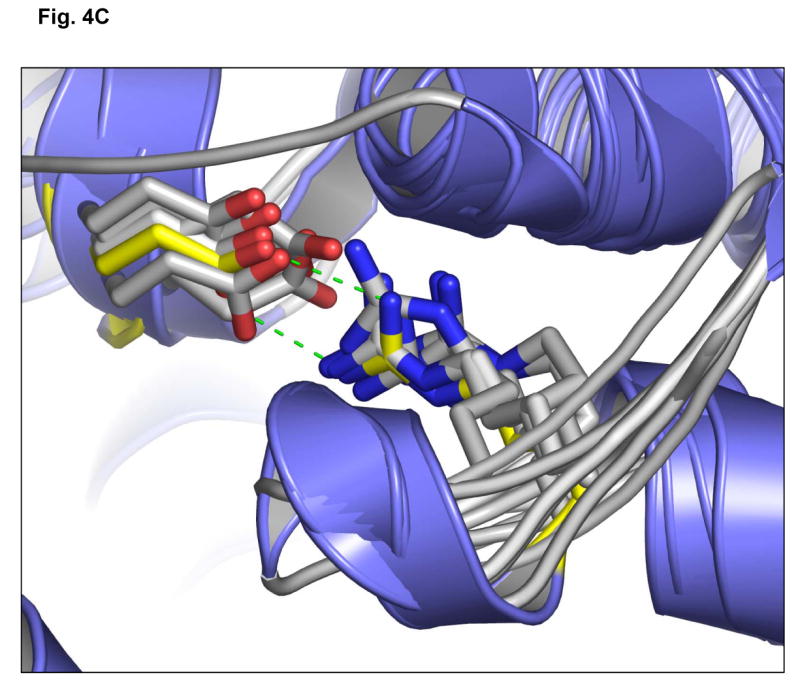
Figure 4. The ion pair Glu(E)884-Arg(R)958 in the EGFR kinase domain is a highly conserved feature in the human kinome.

(A) A selected list of 32 diverse human protein kinases with known importance as validated or potential cancer therapeutic targets, was included here with bioinformatics alignment analysis of the kinase domain amino acid sequences. Glu884 (E884) and Arg958 (R958) of EGFR are both highly conserved residues among these kinases. Left: Amino acid alignment of the kinase domains of 32 diverse members of human protein kinases showing E884-EGFR is highly conserved: EGFR, ERBB2, ERBB3, ERBB4, RON, MET, TYRO3, MER, AXL, RYK, RET, FGFR1, FGFR2, FGFR3, FLT3, KIT, PDGFR-A, PDGFR-B, VEGFR-1, VEGFR-2, VEGFR-3, SRC, EPHA2, EPHB2, FAK, ZAP70, TRK-A, TRK-B, TRK-C, IGFR1, ALK and c-ABL. Right: R958-EGFR is also highly conserved among diverse members of kinases. For the complete alignment analysis of kinase domains of the human kinome, see Supplementary Fig. 1.
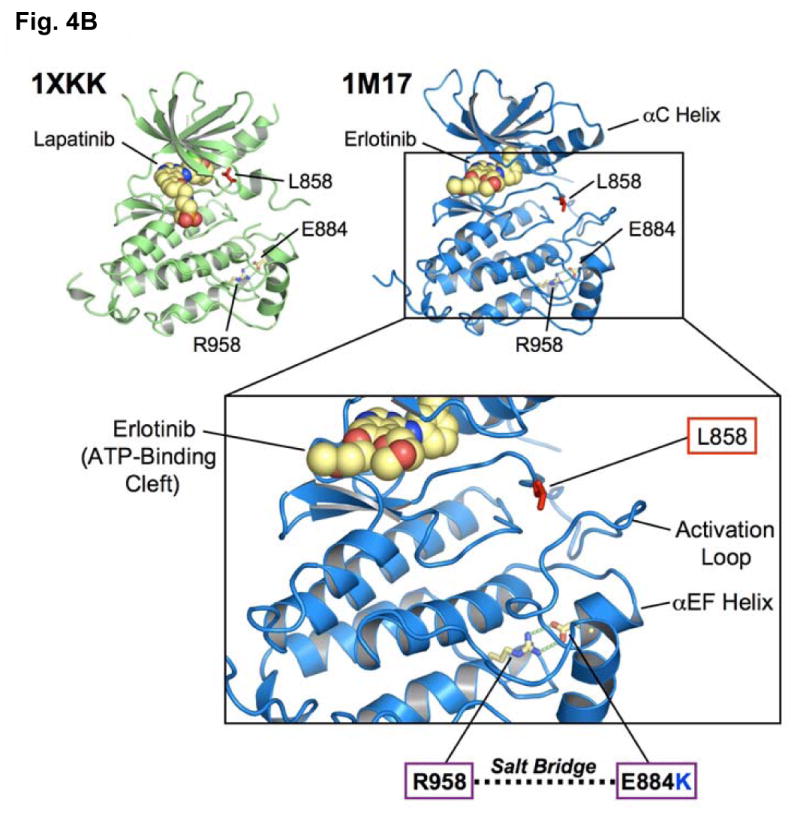
(B) EGFR kinase domain crystal structures [PDB accession codes 1M17 (Stamos et al., 2002) and 1XKK (Wood et al., 2004)] when in complex with erlotinib (blue) and lapatinib (green) are shown. The locations of L858 (exon 21) and E884 (exon 22) are highlighted. E884 (acidic) and R958 (basic) residues form an ion pair in wild type EGFR that would be disrupted by the E884K substitution from the acidic glutamic acid (E) to the basic lysine (K). The E884-R958 salt bridge is present in the kinase domain crystal structures complexed to either erlotinib or lapatinib.
(C) Superposition of the EGFR kinase domain with the catalytic domains of diverse kinases shows structural conservation of a buried Glu(E)-Arg(R) ion pair. The crystal structure of EGFR tyrosine kinase (PDB accession code: 1M17) (Stamos et al., 2002) was superimposed with the catalytic kinase domains of human CDK2 (PDB accession code: 1VYW) (Pevarello et al., 2004), human JNK3 (PDB accession code: 1PMQ) (Scapin et al., 2003), human insulin receptor kinase (PDB accession code: 1IR3) (Hubbard, 1997), ZAP-70 tyrosine kinase (PDB accession code: 1U59) (Jin et al., 2004), LCK kinase (PDB accession code: 1QPD) (Zhu et al., 1999) and MET (PDB accession code: 2RFS) (Bellon et al., 2008) using Cα atoms in the program DeepView/Swiss-PdbViewer v3.7. The conserved Glu-Arg ion pair is shown in stick format, with oxygen atoms colored red and nitrogens, blue. The EGFR side-chains are shown in yellow. The structural location of the ion pair is conserved in these crystal structures and helps orientate helix αEF. Figures 4B and 4C were prepared using the program PYMOL (www.pymol.org).
Finally, we mapped the locations of the L858R and E884K mutations onto the three-dimensional structure of the EGFR kinase domain complexed with erlotinib and with lapatinib [PDB accession codes 1M17 (Stamos et al., 2002) and 1XKK (Wood et al., 2004)] (Figure 4B). We also generated a superposition of the EGFR kinase domain with multiple diverse kinase catalytic domains (Figure 4C). These analyses show the structural conservation of the buried Glu(E)-Arg(R) ion pair and that the exon 22 residue, E884, is physically distant from L858 in exon 21. Furthermore, unlike L858, E884 is not proximal to the ATP-binding cleft of the kinase domain, making it difficult to predict its effects on kinase inhibitor interactions. Mutation of the acidic glutamate residue at codon 884 to a basic lysine will disrupt the highly conserved ion pair through charge-charge repulsion with the basic residue R958 (Figure 4B, C).
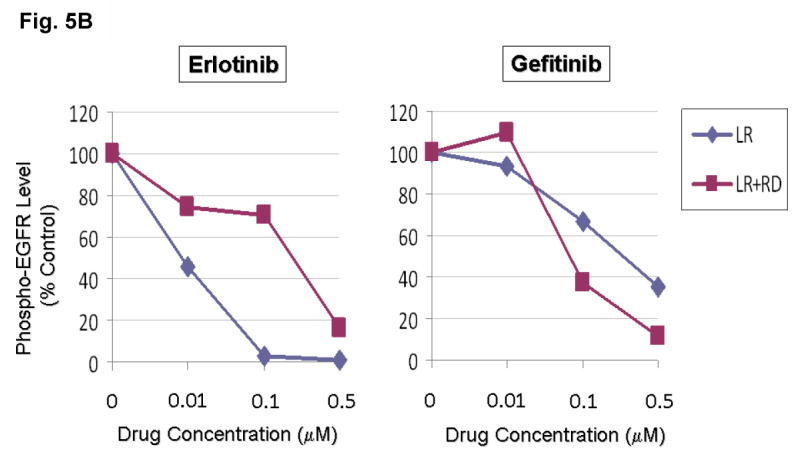
To further test the hypothesis of the disruption of the conserved E884-R958 salt bridge as a mechanism underlying the differential response of the mutant EGFR to kinase inhibitors, we tested the double mutant L858R+R958D against erlotinib and gefitinib (Figure 5). Substitution of the wild type Arg(R)958 with Asp(D)958 was created using site-directed mutagenesis. We hypothesized that the R958D substitution would disrupt the ion pair with E884 through electrostatic repulsion, in a way similar to the effect of the E884K substitution. COS-7 cells transfected to express the indicated mutant EGFR receptors were inhibited using either erlotinib or gefitinib in vitro with increasing concentrations. Similar to E884K, R958D modulated the sensitizing effect of L858R differentially to reversible EGFR inhibitors when in-cis (with L858R). R958D mutation, when in-cis with L858R, decreased the sensitivity of the mutant receptor to erlotinib inhibition, while increasing the sensitivity to gefitinib in a dominant fashion (Figure 5A, B).
Figure 5. Disruption of the conserved Glu(E)884-Arg(R)958 salt bridge by a R958D substitution differentially altered L858R mutant receptor sensitivity to EGFR inhibitors.
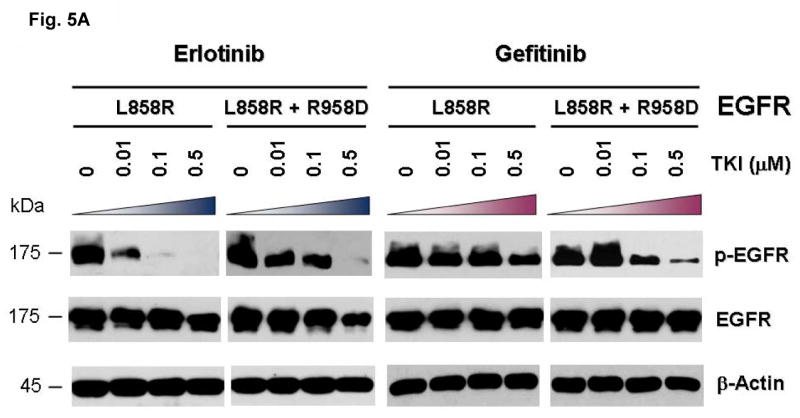
(A) Stable COS-7 transfects expressing the sensitizing L858R and double mutant L858R+R958D variants of EGFR were cultured in 0.5% BSA-containing serum-free media for 16 hours, and then incubated with increasing concentrations of either erlotinib or gefitinib, in the presence of EGF stimulation (100 ng/ml). Whole cell lysates were extracted for SDS-PAGE and immunoblotted using antibodies against the followings: p-EGFR [Y1068], EGFR and β-actin. R958D mutation modulated the effect of L858R on inhibitor sensitivity resulting in desensitization of the mutant receptor to erlotinib inhibition but modestly enhanced sensitivity to gefitinib inhibition.
(B) Densitometric quantitation of the p-EGFR [Y1068] levels showing that R958D mutation differentially altered L858R mutant receptor sensitivity to erlotinib (more resistant) and gefitinib (more sensitive). Densitometric scanning of the immunoblot signals shown in (A) was performed using NIH ImageJ software program, with normalization to total EGFR expression levels.
Mutational disruption of the conserved kinase ion pair in MET kinase by E1271K-MET also differentially alters the sensitivity of phosphorylation inhibition by MET inhibitors
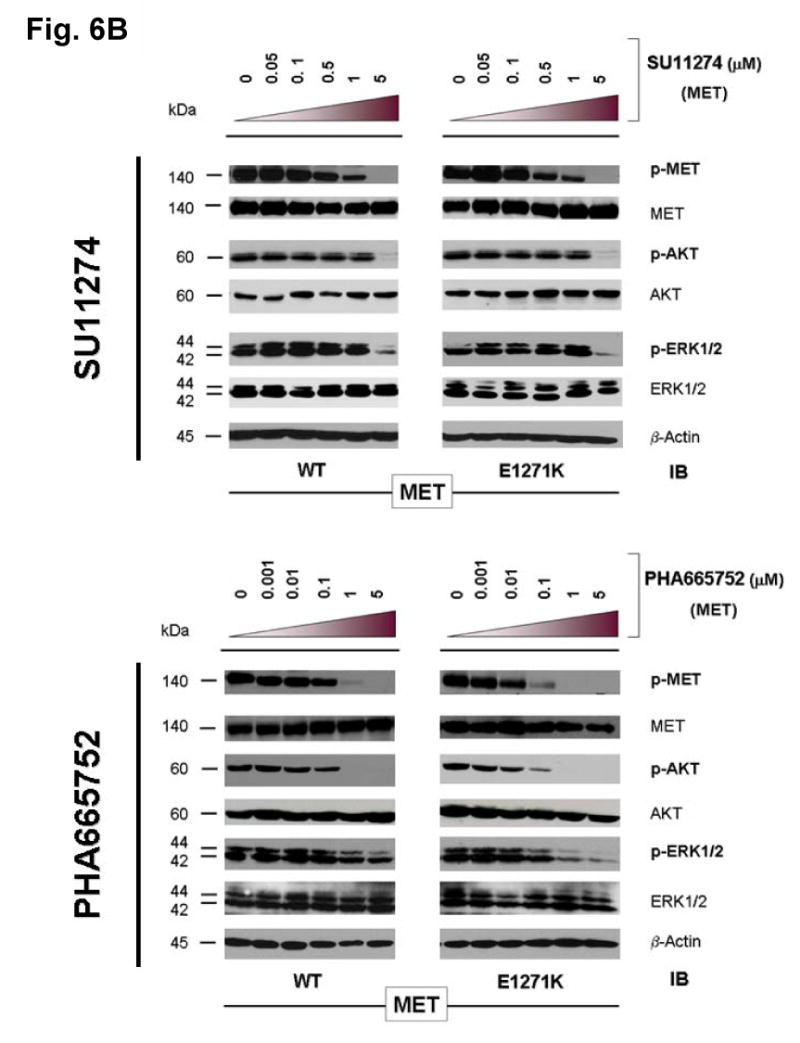
MET has been shown to play key role in the development of many human malignancies. A number of mutations have been identified in MET from various cancers. Recently, it has been shown that MET represents a key oncogenic signaling in lung cancer alongside with EGFR signaling (Guo et al., 2008; Rikova et al., 2007; Tang et al., 2008). Moreover, MET can cross-activate with EGFR when they are co-expressed, which happens rather frequently (Rikova et al., 2007; Tang et al., 2008). MET has also been shown to be an attractive therapeutic molecular target (Ma et al., 2003b; Mazzone et al., 2006; Peruzzi et al., 2006; Shinomiya et al., 2004; Smolen et al., 2006). Here, we test the hypothesis that E1271K mutation of MET, analogous to E884K-EGFR, can also differentially alter inhibitory sensitivity towards selective MET inhibitors (Figure 6). The Glu(E)1271-Arg(R)1345 constitutes the conversed ion-pair in MET kinase (Figure 4). The location of the E1271-R1234 ion pair in MET kinase is illustrated in the recently reported crystallographic structure of the MET kinase domain complexed with SU11274 (Bellon et al., 2008). Stable COS-7 transfectant cells expressing similar levels of wild type and E1271K-MET were used in this experiment using the two reversible preclinical MET inhibitors SU11274 (Ma et al., 2005a) and PHA665752 (Ma et al., 2005a; Ma et al., 2005b). We did not find any significant modulation of sensitivity to SU11274 inhibition in the E1271K-MET cells (Figure 6B). On the other hand, the E1271K mutation of MET enhanced the sensitivity of inhibition by PHA665752 in the phosphorylation of the mutant MET at its major autophosphorylation sites [pY1234/1235] (equivalent to pY1252/1253 phosphosites as in the full length MET transcript with a difference of 18 amino acids in the exon 10 with the common alternatively spliced variant) in the kinase domain, and its downstream signaling proteins AKT and ERK1/2 (Figure 6B). Hence, disrupting the MET kinase salt bridge by the E1271K mutation also differentially alters sensitivity to MET kinase inhibitors in an inhibitor-specific fashion.
Figure 6. Mutational disruption of the conserved E1271-R1345 ion pair in MET kinase salt bridge causes inhibitor specific modulation of sensitivity to SU11274 (unchanged) and PHA665752 (more sensitive).
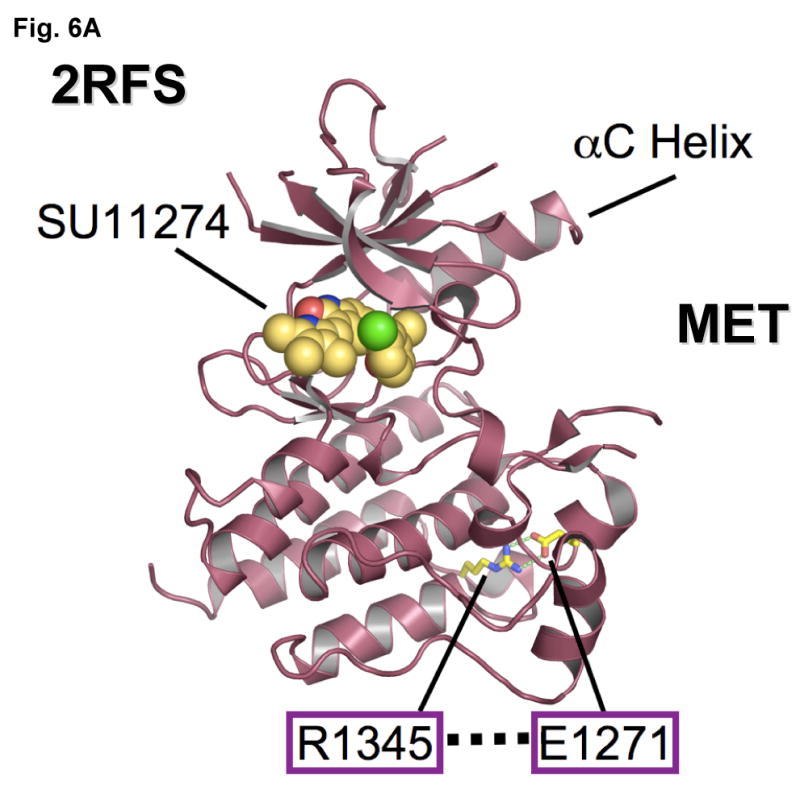
(A) MET kinase domain crystal structure (PDB accession code: 2RFS) (Bellon et al., 2008) highlighting the salt bridge between E1271 and R1345. Crystal structure was solved in complex with SU11274, shown. The conserved Glu-Arg ion pair is shown in stick format, with oxygen atoms colored red, nitrogens colored blue and carbon colored yellow. Figure was prepared using the program PYMOL (www.pymol.org).
(B) Stable COS-7 transfects expressing E1271K mutant MET were cultured in 0.5% BSA-containing serum-free media for 16 hours, then incubated with increasing concentrations of the MET inhibitors SU11274 (top) and PHA665752 (bottom) as indicated, in the presence of HGF stimulation (50ng/ml). Whole cell lysates were extracted for immunoblotting using antibodies against p-MET [Y1234/Y1235], MET, p-AKT, AKT, p-ERK1/2, ERK1/2 and β-actin. Wild-type MET expressing COS-7 tranfectant cells were included as control. E1271K mutation of MET increased the sensitivity of MET kinase phosphorylation inhibition by PHA665752.
Mutations at the conserved Glu(E)-Arg(R) ion pair in the human kinome
Since the E884K somatic mutation was originally identified in a never-smoker woman of Japanese descent, we performed mutational screening for the presence of mutation at the E884 and R958 residues of EGFR among a cohort of 67 lung tumor genomic DNA specimens from Japanese NSCLC patients (including sixty-six transbronchial biopsies and one surgical specimen). Non-synonymous mutations were not present in either residue location in this patient cohort. Based on our results suggesting the conserved structure and function of the Glu(E)-Arg(R) ion pair in EGFR and among other kinases in the kinome, we hypothesized that there would be other cancer-associated mutations at the conserved ion pair within the human kinome in kinases other than EGFR. Here, we performed bioinformatics survey of the updated Catalog of Somatic Mutations In Cancer (COSMIC) database (http://www.sanger.ac.uk/genetics/CGP/cosmic/) containing somatic mutations identified in kinases among human cancers (Figure 7). We have conducted a complete and comprehensive survey throughout the entire human kinome for mutations identified at the conserved Glu(E)-Arg(R) ion pair in COSMIC. We also documented here the hits identifying mutations clustered in the vicinity of the ion pair, 30 amino acids proximal or distal to the Glu(E) or Arg(R). Interestingly, several kinases within the kinome were found to have mutations occurred at the Glu(E) residue, homologous to the E884-EGFR residue. These include KIT (E839K), RET (E921K), STK11/LKB1 (E223*). These are all known cancer associated kinases that have dysregulated signaling in various human cancers, including GIST and hematological malignancies (KIT), papillary thyroid cancer [MEN2] (RET) and lung cancer (RET, LKB1).
Figure 7. Survey of identified mutations at the conserved salt bridge ion pair in the human kinome (COSMIC).




The COSMIC database for the human cancer genome re-sequencing was surveyed and screened for potential mutations identified at or near the conserved Glu(E)-Arg(R) salt bridge ion pair in the human kinome. A, (a) Glu(E)884-EGFR and analogous alignment in the kinome. (b) List of the mutations identified in the kinome is included as reference. B, (a) Arg(R)958-EGFR and analogous alignment in the kinome. (b) List of the mutations identified in the kinome is included as reference. The color code for the amino acids is included here.
We have identified several kinases within the kinome that have mutations occurred at the Glu (E) residue, homologous to the E884-EGFR. These include KIT (E839K), RET (E921K), and LKB1 (E223*). These are all known oncogenic kinases that have dysregulated signaling in various human cancers, including GIST and hematological malignancies (KIT), papillary thyroid cancer [MEN2] (RET) and lung adenocarcinoma (RET, LKB1). While KIT and RET are oncogenes, LKB1 has been shown to be tumor suppressor gene in lung cancer and here we showed clustering of truncation mutations at and near the salt bridge ion pair as a result of a number of mostly nonsense mutations among some missense mutations. While no mutation at E1271-MET was found, there are frequent clustered hotspots of mutations at its close vicinity, 3 amino acid residues proximally at M1268 (M1268T/I). This is a known activating mutation of MET frequently associated with metastatic lesions promoting tumor motility and progression. The selected kinases with positive “mutational hits” in our kinome bioinformatics screen are shown here for illustration.
Discussion
In the era of molecularly-targeted therapeutics in cancer therapy, the impact of cancer-associated mutations on kinase inhibitor sensitivity-resistance has increasingly important implications in the success of novel targeted inhibitors such as erlotinib (EGFR-TKI). Furthermore, knowledge of mutational correlation with inhibitor sensitivity-resistance would likely facilitate more effective and “personalized” targeted therapeutics development in cancer therapy. The clinical course of the patient where the somatic E884K mutation was identified (Choong et al., 2006) suggested that different mutations of a target kinase, such as EGFR, may lead to differential responses to targeted kinase inhibitors. Alternatively, one may postulate that there might be differences in cerebrospinal fluid penentrance by TKIs that could potentially account for central nervous system failure with disease progression in the compartment on therapy (Jackman et al., 2006). Our biochemical studies here now show that E884K mutation in-cis with L858R differentially altered inhibitor sensitivity when compared to L858R alone, through differential inhibition of the pro-survival AKT and STAT3 signaling pathways associated with altered induction of cleaved-PARP(Asp214). This is also shown to occur in an inhibitor-specific manner within the class of various ERBB family small molecule inhibitors, including reversible single EGFR or dual inhibitors (gefitinib, erlotinib, lapatinib, 4557W, GW583340, and AG1478) and irreversible EGFR inhibitor (CL-387,785).
Moreover, the E884K alone and L858R+E884K double mutant EGFR remained sensitive to EGF, and the E884K mutation cooperates with L858R when in-cis to enhance the mutational effects on downstream phosphoprotein activation. To date, essentially all mutational combinations involving L858R studied were found to exist in-cis, suggesting potential cis mutation-to-mutation cooperation in EGFR signaling and possibly tumorigenesis (Tam et al., 2006). Interestingly, the double mutation, L858R+E884K conferred a distinctly more sensitive response to EGF stimulation selectively in the MAPK-ERK1/2 cell proliferation pathway compared to either wild type, E884K alone or L858R alone. Hence, the double mutation L858R+E884K modulated downstream EGFR signaling differentially with distinctly different effects on the AKT (downregulated) and MAPK-ERK1/2 phosphorylation (upregulated). Moreover, E884K had a dominant effect over L858R, when in-cis, in these signaling modulatory effects. E884K, alone or in-cis with L858R, can also mediate induction of p-STAT3 [pY705] (important for STAT3 dimerization and transcriptional activation of target genes), and may have a role in differential regulation of STAT3 activation and thus nuclear translocation for transcriptional activity (Lo et al., 2005). Our data also share some similarities to the recent findings that various activating “gain-of-function” mutations of FLT3, while all induced FLT3 kinase activation constitutively, they showed differential downstream signaling activation along the STAT3, STAT5, AKT, MAPK-ERK1/2 paths (Frohling et al., 2007). EGFR somatic doublet mutations are potentially more frequent than previously understood, with majority of them representing driver/driver mutations rather than driver/passenger mutations (Chen et al., 2008). Future kinome targeted therapies should take into account of oncogenic effects of doublet mutations in the targets and detailed analysis of the identified doublet mutations would be warranted.
Through sequence bioinformatics and structural analysis, we identified the highly conserved E884-R958 ion pair in EGFR kinase domain that is conserved, both by sequence homology and by structural salt-bridge formation, across the entire human kinome. Many of the protein kinases in the human kinome are “druggable” therapeutic targets for various human cancers (Krause et al., 2005; Ma et al., 2005a). This striking finding provides a structural basis for the potential mechanism of alteration of substrate specificity. This hypothesis is substantiated by our study using mutational disruption of the E884-R958 ion pair via a R958D substitution resulting in an opposite electrostatic charge between the wild type and the mutant residue at codon 958. Similar differential sensitivity towards gefitinib (more sensitive) and erlotinib (more resistant) was observed in our in vitro EGFR inhibition study here. It is interesting to note that this salt bridge is located directly between two regions critical for normal EGFR activation, the intermolecular EGFR activation interface and the activation loop. Residue R958 falls between helices αH and αI and is proximal to the intermolecular EGFR activation interface recently revealed by structure-directed studies (Zhang et al., 2006). Residue E884 is the conserved glutamate of the MALE motif (MAPE in PKA) and falls within helix αEF at the C-terminus of the activation loop. This salt bridge helps orientate helix αEF. In the recent EGFR kinase domain crystal structure bound to a peptide substrate analogue (PDB accession code: 2GS6) (Zhang et al., 2006), helix αEF packs against the substrate analogue suggesting that disruption of the salt bridge by an acquired E884K mutation could influence substrate recognition and binding. The acquisition of a lysine at codon 884 may therefore bring about local conformation disruptions that alter EGFR interactions with downstream substrates. Although we did not identify further E884K mutation (or any mutations involving R958 residue) in EGFR from the Japanese patients tumor sample cohort, the results of our study may have implication on the potential impact of cancer-associated mutations that may interrupt the integrity of the salt bridge of a kinase. Since the human kinome is a rich source of “druggable” targets, we extended our search through bioinformatics data-mining from the COSMIC human cancer genome re-sequencing project. To this end, we identified several proximal ion pair residue substitutions recorded in the COSMIC database at the E884 (EGFR) homologous residue, in the oncogenic kinases KIT, and RET; as well as in the tumor suppressor gene LKB1 (also known as STK11). Mutations at the neighboring residues of the conserved motif MAPE(884), as exemplified in FAK-A612V, MET-M1268I/T, RET-M918T and RET-A919V, as well as the truncational nonsense mutation in LKB1-Q220* were also identified from the COSMIC database. Furthermore, the juxtaposing proximal region to the MAPE(884) conserved motif in the kinome also appears to harbor mutational hotspots in the human cancer genome. Nonetheless, the significance of these mutations with respect to the kinase structure and signaling function is not clear. While KIT has been extensively characterized with an established oncogenic role in some hematologic malignancies and GIST, it has not been found to play key role in lung cancer. However, recent studies have implicated interesting oncogenic role of RET (Thomas et al., 2007), FAK (Ma et al., 2007; Rikova et al., 2007), MET (Ma et al., 2005a), and tumor suppressor role of LKB1 (Ji et al., 2007) in lung cancer.
Recently, better understanding of signaling network interactions between EGFR and MET is beginning to emerge (Guo et al., 2008; Tang et al., 2008). MET genomic oncogenic amplification has also been identified to correlate with acquired resistance to EGFR inhibitors (gefitinib/erlotinib) with or without T790M-EGFR mutation (Bean et al., 2007; Engelman et al., 2007). Numerous kinase domain mutations of MET have been identified in previous studies, many of them shown to be activating and most frequently found in metastatic tumor lesions compared with the primaries (Di Renzo et al., 2000). The E1271-MET conserved ion pair residue occurs within the conserved MALE motif, where M1268 is a mutational hotspot frequently found substituted in human cancers (M1268T/I). This is a known activating mutation of MET frequently associated with metastatic lesions promoting tumor motility and progression. Our results here demonstrate that E1271K-MET, effectuated differential effect on sensitivity towards the two preclinical MET inhibitors, SU11274 (unchanged) and PHA665752 (sensitizing). Hence, mutations in the kinase domain of MET may play a role in modulating the inhibitory spectrum of MET inhibitors, similar to what is established in EGFR targeted therapy using gefitinib/erlotinib. Whether these mutationally-specific differences in inhibitor sensitivity would eventually be clinical relevant is not clear at present and should be a focus of future research. MET is emerging as an important therapeutic target in cancer therapy beyond EGFR. More detailed studies to better define the relative role of kinase mutations in MET and how they can modulate inhibitor sensitivity would be warranted. Furthermore, non-kinase mutations of MET, in the extracellular sema domain and the short cytoplasmic juxtamembrane domain, have been identified to be important in lung cancer and mesothelioma (Jagadeeswaran et al., 2006; Ma et al., 2005a; Ma et al., 2003a). Little is known about the correlation of inhibitor sensitivity with these non-kinase mutations, and they should be included in future studies. Bellon et al. recently compared the crystal structures of a novel MET inhibitor AM7, and that of SU11274 when bound to the unphosphorylated form of MET kinase (Bellon et al., 2008). They identified a novel binding mode of a MET inhibitor AM7 compared with SU11274 and raised the possibility of designing TKIs that have improved specific activity and specificity towards different mutant profiles in different cancers; hence “mutationally-targeted inhibitors”.
While the role of kinase domain mutations in modulating the sensitivity-resistance to small molecule inhibitors, in the case of BCR/ABL, KIT and EGFR, has been quite extensively studied, in-depth understanding of the relative role of mutations in other target kinases such as MET, RET, FAK in determining specific inhibitor sensitivity is still largely lacking. The ion pair formed by residues E884 and R958 in the EGFR kinase domain is a highly conserved feature in the human kinome, and mutations of this conserved ion pair may result in conformational changes that alter kinase substrate recognition. The discovery that disruption of the conserved E884-R958 ion pair affects EGFR signal transduction and inhibitor sensitivity indicates the clinical importance of in vitro and biochemical analysis for all documented resistance mutations. Our analysis also suggests that targeted therapy using small molecule inhibitors should take into account potential cooperative effects of multiple intramolecular kinase mutations. As the number of targeted TKIs available increases, it is anticipated that a “personalized” approach to cancer therapy based on knowledge of the activating mutations present should improve the efficacy of these treatments.
Materials and Methods
Plasmid Constructs and Site-Directed Mutagenesis
The plasmids pcDNA3.1 containing the full-length wild-type EGFR and the L858R-EGFR cDNA insert was a generous gift from Dr. Stanley Lipkowitz (NIH/NCI). The generation of the kinase domain missense mutations of EGFR, E884K, L858R+E884K and L858R+R958D, were performed using the QuikChange Site-Directed Mutagenesis XL II kit (Stratagene, La Jolla, CA) as previously described (Choong et al., 2006). The E1271K mutation of MET was introduced into the wild type MET plasmid (Ma et al., 2003a). Incorporation of the correct mutations was confirmed by direct DNA sequencing of the constructs.
Cell Culture and Transfection
COS-7 cells were grown as previously described (Choong et al., 2006). Transfection method was described in Supplemental Materials and Methods.
Cell Proliferation and Cytotoxicity Assays
Cell proliferation and cytotoxicity assays were performed using tetrazolium compound based CellTiter 96® AQueous One Solution Cell Proliferation (MTS) assay (Promega) (see Supplemental Materials and Methods).
Preparation of Cell Lysates and Immunoblotting
Whole cell lysates were extracted, separated by 7.5% SDS-PAGE, immunoblotted using the various primary antibodies indicated, and developed with SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL) as previously described (Choong et al., 2006). The following primary antibodies were used: phosphotyrosine (4G10, Upstate Biotechnology, Lake Placid, NY), phospho-EGFR [Y1068] (BioSource International, Camarillo, CA), EGFR (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-STAT3 [Y705] (Cell Signaling), STAT3 (Zymed, South San Francisco, CA), phospho-AKT [S473] (Cell Signaling), AKT (Biosource), phospho-ERK1/2 [T202/Y204] (Cell Signaling), ERK1/2 (Biosource International), cleaved-PARP(Asp214) [c-PARP(Asp214)] (Cell Signaling), and β-actin (Santa Cruz).
Chemicals
The details regarding the EGFR TKIs and MET TKIs used in this study are described in the Supplemental Materials and Methods.
Lung Tumor Genomic DNA Extraction and DNA Sequencing
Genomics DNA was extracted using standard techniques from sixty-seven NSCLC patients treated from July 1995 to March 2003 at Osaka Prefectural Medical Center for Respiratory and Allergic Disease (Osaka, Japan). All tumor samples were utilized in accordance with Institutional Review Board protocol, with patients' informed consent wherever necessary. Screening for mutations within exon 22 (harboring E884) and exon 23 (harboring R958) was performed using standard single-strand conformational polymorphism (SSCP) analysis, followed by direct DNA sequencing when indicated (details see Supplemental Materials and Methods).
Bioinformatics Sequence Analysis
Multiple sequence alignments of kinase domains in the human kinome were performed for 321 human kinase domains. The positions of the conserved glutamate (E) and arginine (R) residues are colored purple and EGFR indicated in red. FASTA files for human kinase domains were obtained from the kinase database at Sugen/Salk (Kinbase) and aligned with the AliBee multiple sequence alignment program (GeneBee) (Brodsky et al., 1992) using Clustal format. Resulting alignments were colored using JalView 2.2 (Clamp et al., 2004) according to sequence conservation (BLOSUM62). In addition, the amino acid sequences from a selected list of 32 diverse human protein kinases were obtained from the ENSEMBL database (www.ensembl.org). The amino acid sequences of these kinase domains were analyzed and aligned using the EMBL-EBI online CLUSTALW software (www.ebi.ac.uk/clustalw).
Structural Analysis
EGFR crystal structures (PDB accession codes 1M17, 1XKK and 2GS6) (Stamos et al., 2002; Wood et al., 2004; Zhang et al., 2006) were analyzed using the program O (Jones et al., 1991). Superposition of the EGFR kinase domain with the catalytic domains of diverse kinases was performed to study the structural conservation of a buried Glu(E)-Arg(R) ion pair. The crystal structure of EGFR tyrosine kinase (PDB accession code: 1M17) (Stamos et al., 2002) was superimposed with the catalytic kinase domains of human CDK2 (PDB accession code: 1VYW) (Pevarello et al., 2004), human JNK3 (PDB accession code: 1PMQ) (Scapin et al., 2003), human insulin receptor kinase (PDB accession code: 1IR3) (Hubbard, 1997), ZAP-70 tyrosine kinase (PDB accession code: 1U59) (Jin et al., 2004), LCK kinase (PDB accession code: 1QPD) (Zhu et al., 1999), and MET (PDB accession code: 2RFS) (Bellon et al., 2008) using Cα atoms in the program DeepView/Swiss-PdbViewer v3.7. Figures were prepared using the program PYMOL (www.pymol.org).
Supplementary Material
Acknowledgments
Patrick C. Ma is supported by NIH/National Cancer Institute-K08 Career Development Award (5K08CA102545-04), American Cancer Society (Ohio)-Institutional Research Grant (IRG-91-022, Case Comprehensive Cancer Center), and Ohio Cancer Research Associates (Give New Ideas A Chance) Grant Award. Titus J. Boggon is an American Society of Hematology Junior Faculty Scholar. Edward T. Petri is supported by a NIH/National Cancer Institute T32 training grant (5T32CA009085-32). We thank Dr. Zhenghe J. Wang (Department of Genetics, Case Western Reserve University) for critically reading the manuscript and helpful suggestions.
Abbreviations
- ATP
adenosine triphosphate
- COSMIC
Catalogue of Somatic Mutations In Cancer
- EGFR
epidermal growth factor receptor
- p44/42 MAPK (ERK1/2)
p44/42 mitogen-activated protein kinase (extracellular signaling-regulated kinase 1/2)
- FBS
fetal bovine serum
- MEN2
multiple endocrine neoplasm syndrome type 2
- NSCLC
non-small cell lung cancer
- PCR
polymerase chain reaction
- p-Tyr
phosphotyrosine
- c-PARP
cleaved-poly (ADP-ribose) polymerase
- RTK
receptor tyrosine kinase
- SSCP
single strand conformation polymorphism
- STAT3
signal transducer and activator of transcription 3
- TKI
tyrosine kinase inhibitor
- VEGFR
vascular endothelial growth factor receptor
References
- Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellon SF, Kaplan-Lefko P, Yang Y, Zhang Y, Moriguchi J, Rex K, et al. c-Met inhibitors with novel binding mode show activity against several hereditary papillary renal cell carcinoma related mutations. J Biol Chem. 2008;283:2675–2683. doi: 10.1074/jbc.M705774200. [DOI] [PubMed] [Google Scholar]
- Brodsky LI, Vasiliev AV, Kalaidzidis YL, Osipov YS, Tatuzov RL, Feranchuk SI. GeneBee: the program package for biopolymer structure analysis. Dimacs. 1992;8:127–139. [Google Scholar]
- Choong NW, Dietrich S, Seiwert TY, Tretiakova MS, Nallasura V, Davies GC, et al. Gefitinib response of erlotinib-refractory lung cancer involving meninges--role of EGFR mutation. Nat Clin Pract Oncol. 2006;3:50–57. doi: 10.1038/ncponc0400. quiz 51 p following 57. [DOI] [PubMed] [Google Scholar]
- Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–427. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- Di Renzo MF, Olivero M, Martone T, Maffe A, Maggiora P, Stefani AD, et al. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene. 2000;19:1547–1555. doi: 10.1038/sj.onc.1203455. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- Frohling S, Scholl C, Levine RL, Loriaux M, Boggon TJ, Bernard OA, et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell. 2007;12:501–513. doi: 10.1016/j.ccr.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A. 2008;105:692–697. doi: 10.1073/pnas.0707270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. Embo J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman DM, Holmes AJ, Lindeman N, Wen PY, Kesari S, Borras AM, et al. Response and resistance in a non-small-cell lung cancer patient with an epidermal growth factor receptor mutation and leptomeningeal metastases treated with high-dose gefitinib. J Clin Oncol. 2006;24:4517–4520. doi: 10.1200/JCO.2006.06.6126. [DOI] [PubMed] [Google Scholar]
- Jagadeeswaran R, Ma PC, Seiwert TY, Jagadeeswaran S, Zumba O, Nallasura V, et al. Functional analysis of c-Met/hepatocyte growth factor pathway in malignant pleural mesothelioma. Cancer Res. 2006;66:352–361. doi: 10.1158/0008-5472.CAN-04-4567. [DOI] [PubMed] [Google Scholar]
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- Jin L, Pluskey S, Petrella EC, Cantin SM, Gorga JC, Rynkiewicz MJ, et al. The three-dimensional structure of the ZAP-70 kinase domain in complex with staurosporine: implications for the design of selective inhibitors. J Biol Chem. 2004;279:42818–42825. doi: 10.1074/jbc.M407096200. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005a;65:1479–1488. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- Ma PC, Kijima T, Maulik G, Fox EA, Sattler M, Griffin JD, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003a;63:6272–6281. [PubMed] [Google Scholar]
- Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003b;22:309–325. doi: 10.1023/a:1023768811842. [DOI] [PubMed] [Google Scholar]
- Ma PC, Schaefer E, Christensen JG, Salgia R. A selective small molecule c-MET Inhibitor, PHA665752, cooperates with rapamycin. Clin Cancer Res. 2005b;11:2312–2319. doi: 10.1158/1078-0432.CCR-04-1708. [DOI] [PubMed] [Google Scholar]
- Ma PC, Tretiakova MS, Nallasura V, Jagadeeswaran R, Husain AN, Salgia R. Downstream signalling and specific inhibition of c-MET/HGF pathway in small cell lung cancer: implications for tumour invasion. Br J Cancer. 2007;97:368–377. doi: 10.1038/sj.bjc.6603884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzone M, Comoglio PM. The Met pathway: master switch and drug target in cancer progression. FASEB J. 2006;20:1611–1621. doi: 10.1096/fj.06-5947rev. [DOI] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Peruzzi B, Bottaro DP. Targeting the c-Met signaling pathway in cancer. Clin Cancer Res. 2006;12:3657–3660. doi: 10.1158/1078-0432.CCR-06-0818. [DOI] [PubMed] [Google Scholar]
- Pevarello P, Brasca MG, Amici R, Orsini P, Traquandi G, Corti L, et al. 3-Aminopyrazole inhibitors of CDK2/cyclin A as antitumor agents. 1. Lead finding. J Med Chem. 2004;47:3367–3380. doi: 10.1021/jm031145u. [DOI] [PubMed] [Google Scholar]
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Scapin G, Patel SB, Lisnock J, Becker JW, LoGrasso PV. The structure of JNK3 in complex with small molecule inhibitors: structural basis for potency and selectivity. Chem Biol. 2003;10:705–712. doi: 10.1016/s1074-5521(03)00159-5. [DOI] [PubMed] [Google Scholar]
- Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118:257–262. doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- Shinomiya N, Gao CF, Xie Q, Gustafson M, Waters DJ, Zhang YW, et al. RNA interference reveals that ligand-independent met activity is required for tumor cell signaling and survival. Cancer Res. 2004;64:7962–7970. doi: 10.1158/0008-5472.CAN-04-1043. [DOI] [PubMed] [Google Scholar]
- Smolen GA, Sordella R, Muir B, Mohapatra G, Barmettler A, Archibald H, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci U S A. 2006;103:2316–2321. doi: 10.1073/pnas.0508776103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- Tam IY, Chung LP, Suen WS, Wang E, Wong MC, Ho KK, et al. Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin Cancer Res. 2006;12:1647–1653. doi: 10.1158/1078-0432.CCR-05-1981. [DOI] [PubMed] [Google Scholar]
- Tang Z, Du R, Jiang S, Wu C, Barkauskas DS, Richey J, et al. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br J Cancer. 2008;99:911–922. doi: 10.1038/sj.bjc.6604559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–351. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Zhu X, Kim JL, Newcomb JR, Rose PE, Stover DR, Toledo LM, et al. Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Structure. 1999;7:651–661. doi: 10.1016/s0969-2126(99)80086-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.