

Abstract
Interferon Regulatory Factor 8 (IRF8) has been shown to suppress tumor development at least partly through regulating apoptosis of tumor cells; however, the molecular mechanisms underlying IRF8 regulation of apoptosis are still not fully understood. Here, we demonstrated that disrupting IRF8 function resulted in inhibition of cytochrome C release, caspases 9 and 3 activation, and PARP cleavage in soft tissue sarcoma (STS) cells. Inhibition of the mitochondrion-dependent apoptosis signaling cascade is apparently due to blockage of caspase 8 and Bid activation. Analysis of signaling events upstream of caspsse 8 revealed that disrupting IRF8 function dramatically increases FLIP mRNA stability, resulting in increased IRF8 protein level. Furthermore, primary myeloid cells isolated from IRF8 null mice also exhibited increased FLIP protein level, suggesting that IRF8 might be a general repressor of FLIP. Nuclear IRF8 protein was absent in 92% (55/60) of human STS specimens, and 99% (59/60) human STS specimens exhibited FLIP expression, suggesting that the nuclear IRF8 protein level is inversely correlated with FLIP level in vivo. Silencing FLIP expression significantly increased human sarcoma cells to both FasL and TRAIL-induced apoptosis, and ectopic expression of IRF8 also significantly increased the sensitivity of these human sarcoma cells to FasL and TRAIL-induced apoptosis. Taken together, our data suggest that IRF8 mediates FLIP expression level to regulate apoptosis and targeting IRF8 expression is a potentially effective therapeutic strategy to sensitize apoptosis-resistant human STS to apoptosis, thereby possibly overcoming chemoresistance of STS, currently a major obstacle in human STS therapy.
Keywords: Soft tissue sarcoma, IRF8, FLIP, Caspase 8, Apoptosis, Mitochondria
Introduction
Soft tissue sarcoma (STS) is a heterogeneous group of mesenchymal malignancies. One of the most prominent characteristics of human STS is their resistance to apoptosis (1). Because the vast majority of cytotoxic modalities exert their anti-tumor effects by inducing tumor cell apoptosis, resistance to apoptosis is one of the most significant challenges in sarcoma chemotherapy. Consequently, identifying factors that regulate the anti-apoptotic phenotype and elucidating the underlying molecular mechanisms of these apoptosis regulatory factors are crucial for the success of molecular target-based therapeutics for human sarcoma.
IRF8 (also known as interferon consensus sequence-binding protein or ICSBP) is a transcription factor of the interferon regulatory factor family (2, 3). Mice with a null mutation of IRF8 exhibit two prominent phenotypes (4). The first is impaired immunity against virus infections. The second is deregulated hematopoiesis leading to development of a disease reminiscent of human chronic myelogeneous leukemia (CML). One of the important features of these IRF8-deficient myeloid cells is their increased resistance to apoptosis (4, 5). In humans, IRF8 expression is high in normal hematopoietic cells but impaired in myeloid leukemia (6). Recently, we have shown that IRF8 may also play a role as a tumor suppressor in non-hematopoietic (solid) tumors. Loss of IRF8 expression was observed in human carcinoma cells when compared to matched primary tumor cells (7, 8). We further demonstrated that loss of IRF8 expression or function significantly enhanced the metastatic potential of sarcoma cells in mouse models of experimental lung metastasis (8, 9). IRF8 tumor suppression function has also been demonstrated in lens, colon, esophageal and nasopharyngeal carcinoma cells (10, 11). More recently, it has been revealed that the chromosome region containing IRF8 gene is frequently deleted in nasopharyngeal carcinoma cells, which is characteristics of a tumor suppressor gene (11).
The molecular mechanisms underlying IRF8 modulation of apoptosis are not fully understood and are possibly tumor type or tumor differentiation stage-specific. Ectopic expression of IRF8 was shown to repress Bcl-xL expression in human monocytic leukemia cells (5). However, ectopic expression of IRF8 resulted in decreased Bcl-2 but not Bcl-xL expression in mouse myeloid leukemia cells (12). It has also been demonstrated that a decreased nuclear IRF8 protein level correlates with increased Bcl-2 protein level in splenocytes of leukemia-bearing 12/15-lipoxygenase (Alox15)-deficient mice (13). Moreover, it has recently been shown that IRF8 also mediates Fas-mediated apoptosis via regulating Fap-1 expression in monocytic leukemia cells (14). In addition, disruption of IRF8 function in human monocytic leukemia cells resulted in inhibition of activation of STAT1, a key mediator in the IFN-γ signaling pathway that is essential for Fas activation and Fas-mediated apoptosis (15). We have recently demonstrated that loss of IRF8 expression or function diminishes mouse sarcoma cell sensitivity to apoptosis induction (16) and IRF8 mediates IFN-γ-sensitized and Fas-mediated apoptosis by participating in STAT1 activation and Fas receptor expression in sarcoma cells (16). Taken together, it is clear that IRF8 regulates death receptor-initiated and mitochondrion-dependent apoptosis pathways in tumor cells, but it is not clear whether IRF8 mediates multiple apoptosis-related genes in the same tumor cells or different target genes of the same apoptosis pathway in different types of tumor cells. In the present study, we carried out a detailed analysis of the mechanistic link between IRF8 and the mitochondrion-dependent apoptosis pathway in mouse sarcoma cells and primary myeloid cells, identifying FLIP, a key regulator of the death receptor-initiated apoptosis pathways, as a target of IRF8.
Materials and Methods
Tumor cell lines
The mouse sarcoma cell line CMS4 was kindly provided by Dr. A. Deleo (University of Pittsburg, Pittsburg, PA). The human sarcoma cell line HT1080 was obtained from ATCC. CMS4 cells were transfected with pcDNA vector control plasmid or pcDNA containing a mouse IRF8 mutant as previously described (16).
Tumor specimens and immunohistochemistry
Human STS specimens were collected from surgical specimens at The University of Texas MD Anderson Cancer Center. For immunohistochemical staining of IRF8 protein, tissues were fixed with formalin and embedded in paraffin. Sections were cut from formalin-fixed, paraffin-embedded tissues and printed on glass slides as a tissue microarray. Specimens were blocked with normal goat serum (1.5%), and then incubated with a goat anti-IRF8 polyclonal antibody (Santa Cruz Biotech. Santa Cruz, CA) at 1:100 dilution or rabbit anti-FLIP polyclonal antibody (Abcam. Cambridge, MA) at 1:100 dilution for 30 min, followed by rinsing and staining with anti-goat or anti-rabbit biotinylated antibody (1:2,000) for another 30 min. Color was developed by incubation with 3’3-diaminobenzidine (DAB) solution (Sigma, St. Louis, MO), followed by rinsing and counterstaining with hematoxylin.
Cell surface marker analysis
For cell surface TRAIL receptor (DR4 and DR5) measurement, tumor cells were stained with FITC-conjugated mouse anti-human DR4 and DR5 monoclonal antibodies (Alexis Biochemicals, San Diago, CA) respectively, or with isotype-matched control for 30 min, then washed 3 times in PBS. The stained cells were then analyzed by flow cytometry FACS Calibur with CellQuest (BD Biosciences, San Diego, CA).
RT-PCR analysis
RT-PCR analysis was carried out as previously described (17). The PCR primer sequences are as follows: IRF8: forward: 5’-CCAGATTTTGAGGAAGTGACG-3’, reverse: 5’-TGGGAGAATGCTGAATGGTGC-3’; FLIPS: forward: 5’-ACTTAGCAATTGCCACCATGTCTGCTGAAGTCATCCATCAG-3’, reverse: 5’-ATTCACCAGTCACATGGAACAATTTCCAAGAAT-3’; and β-actin: forward: 5’-ATTGTTACCAACTGGGACGACATG-3’, reverse: 5’-CTTCATGAGGTAGTCTGTCAGGTC-3’. PCR products were visualized on 1-2% Agarose gel under UV light. Image was acquired by ImagQuant 300 with IQuest Capture 300 software (GE, Pittsburgh, PA).
FLIP mRNA half-live measurement
Tumor cells were seeded in 6-well plates overnight. Actinomycin-D (Invitrogen) was added to a final concentration of 10 μg/ml. Total RNA was isolated from the treated cells at different time points. RT-PCR was then used to analyze FLIPs and β-actin mRNA levels. The PCR band intensity was quantified using NIH image J program (National Institutes of Health, Bethesda, MD). The ratio of FLIPs vs β-actin was determined for each time point. The value of untreated cells was set at 100%. The FLIPs mRNA half-live is defined as the time when 50% of the FLIPs mRNA have degraded after actinomycin-D treatment.
Preparation of bone marrow-derived myeloid cells
Mouse bone marrow-derived dendritic cells were generated from marrow stem cells obtained from the femurs of IRF8 null mice and wt littermate control mice as described previously (18).
Preparation of cytosol and organelle-enriched mitochondrion fractions
Cells were resuspended in ice-cold cytosol extraction buffer [10 mM Hepes, pH 7.4, 250 mM Sucrose, 70 mM KCl, 1.5mM MgCl2, 1 mM EDTA, 1 mM EGTA, and 0.01% digitonin, protease and phosphatase inhibitors (Calbiochem, La Jolla, CA) for 5 minutes on ice (1×106 cells/25 μl buffer). The cell suspension was then centrifuged at 1,000g for 5 minutes in 4°C. The supernatant was transferred to a new tube and centrifuged at 20,000g for 10 min at 4°C. The supernatant was then collected as cytosol fraction. To isolate the mitochondrial fraction, the pellet from the first centrifuge was rinsed once with PBS, then resuspended in the same volume (1×106 cells/25 μl buffer) of ice-cold mitochondrion lysis buffer [50 mM Tris-HCl pH7.5, 100 mM NaCl, 10mMMgCl2, 2 mM EGTA, 2 mM EDTA, 1% NP40, 10% glycerol protease and phosphatase inhibitor cocktails (Calbiochem)] for 10 min on ice, followed by centrifugation at 10,000g for 10 min. The supernatant was collected as organelle-enriched mitochondrion fraction.
Fas DISC isolation
Biotin-anti-mouse mAb (clone Jo2, BD Biosciences) and isotype-matched IgG control mAb were incubated with Streptavidin T1 Dynabeads (Invitrogen). The antibody-conjugated beads were then incubated with IFN-γ-treated tumor cells for 10 min at room temperature. Bead-bound cells were collected using a magnetic stand and lysed on ice for 30 min. The beads were washed 5 times in BSA plus 0.1% BSA. The bead-bound proteins were eluted by resuspending the washed beads in SDS-protein gel loading buffer and heating at 95°C for 5 min. The eluted proteins were resolved by 4-20% SDS-polyacrylamide (PAGE) gradient gels and analyzed by Western blotting analysis.
Western blotting analysis
Western blotting analysis was carried out as previously described (19). The blots were probed with the following primary antibodies: anti-IRF8 antibody (C-19, Santa Cruz Biotech) at 1:200 (for tumor cells) or 1:1000 (for myeloid cells) dilution; anti-FLIP antibody (Santa Cruz) at 1:1000 for human FLIP or anti-FLIP (Cell Signaling, Danvers, MA) at 1:1000 for mouse FLIP; Anti-Cytochrome C (BD Biosciences) at 1:500; anti-cleaved caspase 3 (Cell Signaling) at 1:1000; anti-cleaved caspase 9 (Cell Signaling) at 1:1000; anti-cleaved PARP (Cell Signaling) at 1:1000; anti-cleaved caspase 8 (R&D System, Minneapolis, MN) at 0.5 μg/ml; anti-tBid (Millipore) at 1:200; and anti-β-actin (Santa Cruz) at 1:1000 for human β-actin or anti-β-actin (Sigma, St Louis, MO) at 1:5000 for mouse β-actin. Blots were detected using the ECL Plus (Amersham Pharmacia Biotech, NJ) or Super Signaling (Pierce, Rockford, IL) Western detection kit.
RNA interference of FLIP
FLIP-specific siRNA were designed and synthesized by Qiagen (Valencia, CA). Four different siRNA duplexes, recognizing the conserved region of both FLIPL and FLIPs, were tested in HT1080 cells for their effectiveness to silence FLIP expression. The most effective siRNA (5’-CACCTTGTTTCGGACTATAGA-3’) was used for silencing FLIP expression in HT1080 cells. A scramble siRNA (5’-ATAGCGACTAAACACATCAA-3’) was obtained from Dharmacon (Lafayette, CO) and used as control. Transfections were carried out using Lipofectamine (Invitrogen) according to the manufacturer’s instructions.
Cell cycle analysis
Cells were collected and fixed in 70% ethanol for 30 min. The fixed cells were washed in PBS and incubated with DNA extraction buffer (192 mM Na2HPO4, 4 mM citric acid, pH 7.8) for 5 min. RNase A and propidium iodide was added to the cells and incubated for 30 min. The cell cycle was analyzed by flow cytometry.
Measurement of apoptotic cell death
Apoptotic cell death was measured by PI staining as previously described (19). Briefly, tumor cells were seeded in 6-well culture plates (1 × 105 cells/well) and cultured in the absence or presence of recombinant human FasL or TRAIL (PeproTech; Rocky Hill, NJ). Adherent and suspended cells were then harvested and incubated with PI solution (Trivegin, Gathesburg, MD) for 5 min at room temperature, then immediately analyzed by flow cytometry. The percentage of cell death was calculated by the formula: % cell death=% PI-positive cells after FasL or TRAIL treatment - % PI-positive cells with FasL or TRAIl treatment.
Results
Disruption of IRF8 function inhibits mitochondrion-dependent apoptosis
A mouse sarcoma cell line CMS4 stably expressing a dominant-negative IRF8 mutant (CMS4.K79E) (15) was used to test the hypothesis that IRF8 mediates apoptosis through regulating target genes in the intrinsic apoptosis pathway. Because release of cytochrome C is the initial step of the mitochondrion-dependent apoptosis pathway, we first measured cytochrome C release by Western blotting analysis. Constitutive cytochrome C is detectable in the cytosol fractions of untreated CMS4 and CMS4.Vecttor cells, albeit at low levels, and IFN-γ treatment alone did not alter the cytochrome C release rate (Fig. 1A). FasL treatment dramatically increased the cytochrome C levels in both CMS4 and CMS4.Vector cells. In contrast, no constitutive or FasL-induced cytochrome C release was detected in CMS4.K79E cells (Fig 1A).
Figure 1. IRF8 regulates cytochrome C-mediated activation of caspases 3 and 9 in the mitochondria-dependent apoptosis pathway.
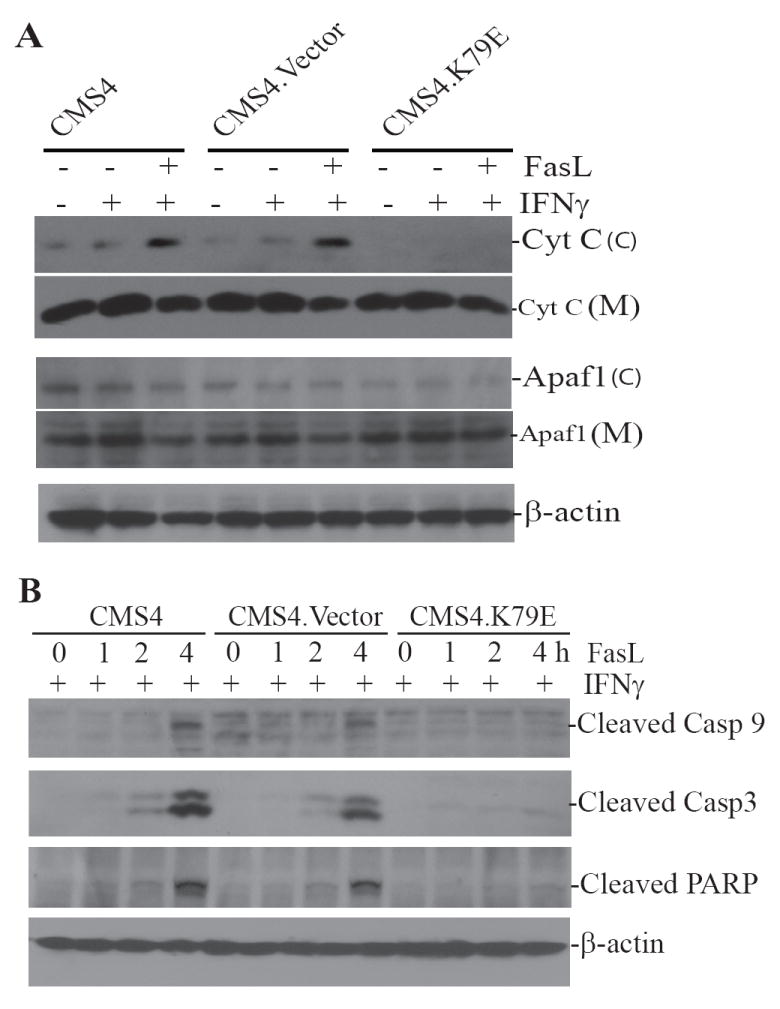
A. Cytochrome C release assay. Tumor cells were treated with IFN-γ overnight, following by incubation with recombinant FasL for 4 h. Cytosol (C) and organelle-enriched mitochondrion (M) fractions were then prepared. The fractions were analyzed by Western blotting analysis for cytochrome C and Apaf1 protein levels. The blots of cytosol fractions were stripped and reprobed with anti-mouse β-actin antibody. B. Caspases 3 and 9 activation. Tumor cells were treated with IFN-γ overnight, followed by incubation with recombinant FasL for various time as indicated. Cytosolic fractions were then prepared and analyzed for activated caspase 9 and caspase 3, as well as cleaved PARP. The blots were stripped and reprobed with anti-mouse β-actin antibody.
One of the consequences of cytochrome C release is formation of apoptosome containing cytochrome C, Apaf-1 and procaspase-9 in the cytosol of the cells, leading to activation of caspase 9 through an intrinsic autocatalytic cleavage of procaspase 9 (20). Western blotting analysis revealed that Apaf-1 is located in both the mitochondron and cytosol fractions (Fig. 1A), and its expression level is not altered in CMS4.K79E cells. Caspase 9 is activated by FasL in both CMS4 and CMS4.Vector cells but not in CMS4.K79E cells. Consistent with caspase 9 activation, caspase 3 activation and PARP cleavage are also inhibited in CMS4.K79E cells (Fig. 1B). Taken together, our data suggest that IRF8 function is required for cytochrome C release and for cytochrome C-mediated caspase activation in the intrinsic apoptosis pathway.
Disruption of IRF8 function leads to inhibition of caspase 8 activation
Cytochrome C release is mediated by activated Bid in the intrinsic apoptosis pathway and activation of Bid by death receptor-initiated signals requires cleavage by activated caspase 8 (21). Our above observation that FasL-induced cytochrome C release is inhibited in CMS4.K79E cells suggests that caspase 8 activation might be blocked by loss of IRF8 function. To test this hypothesis, IFN-γ-treated tumor cells were incubated with oligomized Fas agonist mAb to induce death-inducing signaling complex (DISC) formation and to activate the Fas-mediated apoptosis signaling pathway. The immunoprecipitated Fas DISCs were then analyzed by Western blotting using caspase 8- and Fas-specific antibodies, respectively. It is clear that crosslinking the Fas receptor activated caspase 8 in the DISC in CMS4 and CMS4.Vector. The level of Fas DISC-associated procaspase 8 is much less in CMS4.K79E cells than in CMS4 and CMS4.Vector cells. Caspase 8 activation is not detected in CMS4.K79E cells (Fig. 2A). Initial attempts failed to detect FLIP in the Fas DISC in all three cell sublines (data not shown). To further determine the effect of loss of IRF8 function on caspase 8 activation, cultured tumor cells were sensitized with IFN-γ, followed by incubation with FasL. Cytosol and mitochondrion fractions were prepared and analyzed by Western blotting analysis using a caspase 8-specific antibody. A cleaved form of caspase 8 with a molecular weight of approximately 25 kDa was detected in CMS4 and CMS4.Vector cells (Fig. 2B). However, this cleaved form of caspase 8 was not detected in CMS4.K79E cells. Reproducible results were obtained with three different anti-active mouse caspase 8 antibodies (data not shown). No procaspase 8 protein was detected in the mitochondrion fraction (data not shown). Although IFN-γ can sensitize CMS4 cells to Fas-mediated apoptosis, IFN-γ treatment did not alter the procaspase 8 protein level (Fig. 2B). Therefore, we conclude that IRF8 mediates cytochrome C release via regulation of caspase 8 activation in sarcoma cells.
Figure 2. Disruption of IRF8 function inhibits caspase 8 and Bid activation.

A. IP-Western blotting analysis of caspase 8 activation in the Fas DISC. Tumor cells were treated with IFN-γ overnight and then incubated with oligomerized anti-Fas mAb (the antibody was attached to Dynad beads through biotin-streptavidin) for 10 min at room temperature with rotation. The antibody-bound cells were pulled down using a magnetic stand. Bead-bound cells were lysed and the protein complex associated with the anti-Fas antibody was resolved in 4-20% SDS-polyacrulamide gels and analyzed by Western blotting using Fas- (left panel) and caspase 8-specific antibodies (right panel). The protein molecular weight markers are indicated at the left. Fas, caspase 8 and the heavy chain of the antibody (IgGH) are indicated at the right. B. Western blotting analysis of caspase 8 activation. Tumor cells were cultured in the presence of IFN-γ overnight and followed by incubation with recombinant FasL for 4 h. Cytosol fractions were prepared and analyzed by Western blotting with caspase 8-specific antibody (upper panel). The cleaved caspase 8 is indicated by an arrow. The membrane was stripped and reprobed with anti-mouse β-actin. The protein molecular weight markers are indicated at the right and caspase 8 is indicated at the left (top panel). Bottom panel: Western blotting analysis of procaspase 8 in the indicated tumor cells. Tumor cells were treated as described above. To assess the procaspase 8 levels between the tumor cell sublines and effects of IFN-γ and FasL, less cell lysate (as compared to top panel) were loaded to the gels and analyzed by Western blotting analysis as in the top panel. C. Western blotting analysis of Bid activation. Tumor cells were treated as in B. The cytosol fractions were prepared and analyzed using tBid-specific antibody.
One of the direct downstream target of activated caspase 8 in the apoptosis pathway is Bid. Next, we examined the effect of loss of IRF8 function on Bid activation in the tumor cells. Tumor cells were sensitized with IFN-γ, followed by incubation with FasL. Cytosol and mitochondrion fractions were prepared and analyzed by Western blotting analysis using an antibody that is specific for mouse cleaved Bid (tBid). FasL treatment induced tBid formation in the cytosol fractions of CMS4 and CMS4.Vector cells but not in CMS4.K79E cells (Fig. 2C). Bid and tBid protein were not detected in the mitochondrion fractions of the 3 tumor cell sublines (data not shown).
IRF8 mediates caspase 8 activation through regulation of FLIP expression
Engagement of the death receptor Fas by its ligand recruits FADD and procaspase 8 to the death receptor cytoplasmic domain to form the DISC, leading to autocatalytic cleavage of procaspase 8 to activate caspase 8 (22, 23). It has also been shown that FLIP can bind to FADD in the DISC to inhibit caspase 8 activation (24). In light of the above observations, we next sought to determine whether caspase-8 activation is regulated by IRF8 through FLIP in sarcoma cells. We observed that the long form of FLIP (FLIPL) is expressed in all three cell lines (Fig. 3A). The short form of FLIP (FLIPs) is weakly detectable in the cytosol fractions of CMS4 and CMS4.Vector cells. In contrast, FLIPs protein level is much higher in the cytosol of CMS4.K79E cells (Fig. 3A). To determine whether IRF8 mediates the stability of FLIPs mRNA, CMS4.Vector and CMS4.K79E cells were incubated in the presence of actinomycin D and FLIPs mRNA levels were analyzed at different time points post actinomycin D treatment. The half-live of FLIPs mRNA is approximately 3 h in CMS4.Vector cells and approximately 12 h in CMS4.K79E cells, respectively (Fig. 3B), suggesting that IRF8 increases FLIPs protein level at least partly through stabilizing FLIPs mRNA in CMS4.K79E cells.
Figure 3. IRF8 functions as a repressor of FLIP.

A. Disruption of IRF8 function increased FLIP protein level in tumor cells. Cytosol and organelle-enriched mitochondrion fractions were prepared from the indicated tumor cell line/sublines and analyzed by Western blotting analysis using a FLIP-specific antibody. The blots were stripped and reprobed with anti-mouse β-actin antibody. The protein molecular weight markers are indicated at the right and the locations of FLIPL and FLIPs are indicated at the left. B. IRF8 mediates the stability of FLIPs mRNA. Tumor cells were cultured in the presence of actinomycin D (Act D). Total RNA was isolated from the tumor cells at different time points as indicated and analyzed by RT-PCR for FLIPs mRNA level (top panel). β-actin was used as reference. The FLIPs band intensity at each time point was quantified and normalized to the β-actin band intensity at the same time point. The FLIPs mRNA level in untreated cells was set at 100% and the FLIPs mRNA level at the various time points were expressed as percent of that of untreated cells (bottom panel). C. Effects of ectopic expression of vFLIP on apoptosis. Tumor cells were stably transfected with mammalian expression vector alone (CMS4.Vector) or vector containing the vFLIP coding sequence (CMS4.vFLIP), respectively. The transfected tumor cells were then treated with recombinant IFN-γ overnight, followed by incubation with recombinant FasL for 4 h for Western blotting analysis and 24 h for apoptosis analysis. The treated tumor cells were fractionated to prepare cytosolic fractions and analyzed by Western blotting analysis for active caspase 8 protein level (top panel). The blot was stripped and reprobed with anti-mouse β-actin antibody. The treated cells were also stained with PI and analyzed for by flow cytometry for apoptotic cell death (bottom panel). D. Western blotting analysis of IRF8 and FLIP in IRF8-deficient and wt myeloid cells isolated from IRF8 null and wt littermate control mice. Total cell lysate (for IRF8 analysis, top panel) and cytosol and organelle-enriched mitochondrion fractions (for FLIP analysis, bottom panel) were prepared and analyzed by Western blotting using IRF8- and FLIP-specific antibodies, respectively. The blots were stripped and reprobed with anti-mouse β-actin antibody.
To further validate the role of FLIP in regulation of caspase 8 activation and apoptosis, we carried out a proof of concept study. CMS4 cells were stably transfected with the viral form of FLIP (vFLIP) (25). Western blotting analysis revealed that ectopic expression of vFLIP blocked FasL-induced caspase 8 activation in CMS4 tumor cells (Fig. 3C). As expected, ectopic expression of vFLIP also significantly decreased the tumor cell sensitivity to Fas-mediated apoptosis (p<0.01; Fig. 3C).
To determine whether IRF8 is a general regulator of FLIP, we isolated primary myeloid cells from IRF8 null mice and wt control mice. Myeloid cells were chosen since they are a model primary cell for studying IRF8 function (26-28). Western blotting analysis revealed that FLIPs is undetectable in the cytosol fraction and FLIPL is weakly detectable in the mitochondrion fraction (Fig. 3D). Knocking out IRF8 expression dramatically increases FLIPL protein levels (Fig. 3D).
IRF8 protein level is inversely correlated with FLIP protein level in high grade human STS specimens
Our above observations suggest that IRF8 and FLIP are key determinants of the intrinsic apoptosis pathways in sarcoma cells. Consequently, we sought to examine IRF8 and FLIP protein levels in human STS specimens in a cohort of high grade pleiomorphic STS malignant fibrous histiocytoma specimens. A tissue microarray block consisting of tumor sections derived from 60 high grade human STS specimens was constructed. Immunohistochemical analysis revealed that only two (3%) of the STS specimens expressed nuclear IRF8, while 58 of the 60 (97%) exhibited no nuclear IRF8 protein but cytoplasmic IRF8 protein at varying levels of intensity: low=21, moderate-high=37; distribution: <50% of tumor cells in 4, 50-79% in 34, and ≥80 in 20; two specimens exhibited no IRF8 expression. As a positive control, normal human tonsil tissue was observed to express high levels of nuclear IRF8 protein (Fig 4A). These observations suggest that IRF8 protein is largely absent in the nuclei of human STS cells. Interestingly, a subset of cells in the STS microenvironment, defined by a STS specialist (A.L.), as lymphocytes, exhibited high nuclear IRF8 (Fig 4Ac). Studies to evaluate the role of nuclear IRF8 expression in STS-associated normal cells are currently ongoing.
Figure 4. Immunohistochemical analysis of IRF8 and FLIP protein levels in human STS.

Microsections of human STS specimens derived from 60 human STS patients were printed on glass slides as tissue microarray and immune-stained using IRF8-specifc antibody (A) or FLIP-specific antibody (B) as described in Material and Methods. The antibody-specific staining is shown as brown color. A. Four representative specimens with IRF8 staining: in the nuclei of tumor cells (a), cytosolic but no nuclear IRF8 staining of tumor cells (b), lymphoid aggregates in STS specimens showing strong nuclear staining (c), and IRF8 staining in the nuclei of tonsil lymphocytes (d). B. Three representative specimens with scattered low FLIP staining of tumor cells (a), strong FLIP staining of tumor cells (b), and FLIP staining is positive control tissue (c).
FLIP protein was detected in 59 of the 60 human STS specimens (98%) (Fig. 4B) (intensity: low=46, moderate to high=13; distribution: <50% of tumor cells=17, 50-79%=21, and ≥80%=21). Only one tumor (2%) exhibited no FLIP expression. These results suggest an inverse correlation between nuclear IRF8 protein level and FLIP protein level in high grade human STS specimens.
Silencing FLIP expression increased human sarcoma cells sensitivity to FasL and TRAIL-induced apoptosis
The presence of FLIP protein in the vast majority of human STS specimens suggests that FLIP might be responsible at least partly for the apoptosis-resistant phenotype in human STS (29). We next sought to silence FLIP expression and examined the effect of loss of FLIP expression on the tumor cells sensitivity to apoptosis. Two cell death detection methods were first compared for their sensitivity in detecting dead cells. In the first method, untreated and FasL-treated tumor cells were stained directly with PI and analyzed by flow cytometry. In the second method, tumor cells were analyzed for cell cycle status based on the DNA content of the cells. Apoptotic or dead cells are defined as cells in the subGo/G1 population. Because we measure the cell death at the late stage, the subG0/G1 cell population in the cell cycle analysis is actually a mixture of apoptotic and dead cells (Fig. 5A). Fig. 5A indicates that these two methods exhibit very similar sensitivity (Fig. 5A). Therefore, we used the economic PI direct staining method to detect FasL- and TRAIL-induced cell death. Next, four siRNA sequences were chosen from the conserved region of FLIPL and FLIPs. Human sarcoma HT1080 cells were transiently transfected with these 4 siRNAs and a scramble siRNA. Among the 4 siRNAs, only one siRNA effectively silenced FLIP expression (Fig. 5B). HT1080 cells were transiently transfected with scramble siRNA and the FLIP-specific siRNA, respectively, the tumor cells were then analyzed for apoptosis. It is clear that silencing FLIP expression significantly increased the tumor cell sensitivity to both FasL and TRAIL-induced apoptosis (p<0.01) (Fig. 5B).
Figure 5. Silencing FLIP expression enhances FasL and TRAIL-induced apoptosis in human sarcoma cells.
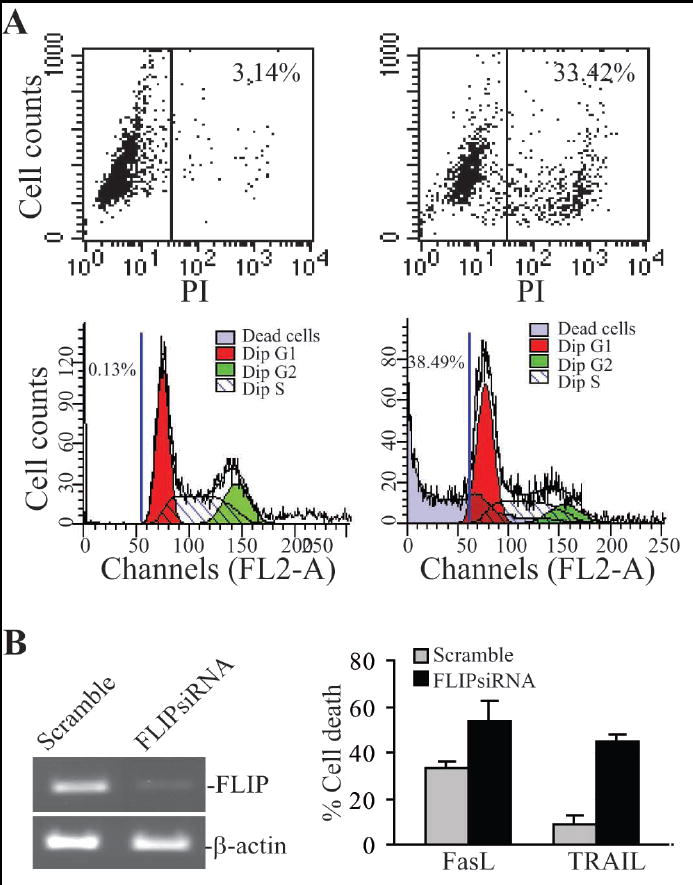
A. Comparison of cell death detection methods. HT1080 tumor cells were incubated in the absence (left 2 panels) or presence (right 2 panels) of FasL overnight and either directly stained with PI (top 2 panels), or fixed with 70% ethanol and then stained with PI as described in materials and methods (bottom 2 panel). The stained cells were analyzed by flow cytometry. B. Silencing FLIP expression increased the sensitivity of the tumor cells to FasL- and TRAIL-induced apoptosis. HT1080 cells were transiently transfected with scramble siRNA or FLIP-specific siRNA for approximately 24 h. The cells were analyzed for FLIP mRNA level using RT-PCR (left panel). The scramble- and FLIP siRNA-transfected cells were also treated with recombinant FasL or TRAIL protein overnight and analyzed for cell death by PI staining and flow cytometry analysis (right panel). Column, mean; bar, SD.
Ectopic expression of IRF8 increased the efficacy of TRAIL- and FasL-induced apoptosis in human STS cells
The observations that FLIP protein is present in the vast majority of human STS cells suggests that therapeutic agents that kill tumor cells by inducing cell death are possibly not going to be effective in the clinic. It has been demonstrated that suppression of FLIP expression by therapeutic agents can increase the sensitivity of tumor cells to death induction (30, 31). Because our data suggest that IRF8 is a key regulator of FLIP, we reasoned that restoring IRF8 expression could possibly sensitize the human sarcoma tumor cells to apoptosis, including FasL- and TRAIL-induced apoptosis. To test this hypothesis, we transfected human sarcoma cells to stably express IRF8 (Fig. 6A). Analysis of FLIP mRNA and protein levels in the transfected cells indicated that ectopic expression of IRF8 dramatically decreased FLIP transcript as well as protein levels in HT1080 cells (Fig. 6A). Next, HT1080, HT1080.Vector and HT1080.IRF8 cells were treated with various concentrations of recombinant FasL or TRAIL protein, and apoptosis was then analyzed. As shown in Fig 6B and C, FasL and TRAIL induced cell death in HT1080 cells in a dose-dependent manner. Ectopic expression of IRF8 significantly increased FasL-induced apoptotic cell death as compared to HT1080 and HT1080.Vector cells at all FasL concentrations tested (p<0.01) (Fig. 6B). Ectopic expression of IRF8 also significantly increased TRAIL-induced apoptotic cell death as compared to HT1080 and HT1080.Vector cells at all TRAIL concentrations (p<0.01) except at concentration of 5 ng/ml (Fig. 6C). Analysis of cell death kinetics revealed that FasL- and TRAIL-induced cell death reached highest rate at approximately 12 to18 hours, respectively (Fig. 6B & C). To determine whether IRF8-enhanced TRAIL-mediated apoptosis can possibly be attributed to an IRF8-regulated increase in the expression of TRAIL receptors DR4 and DR5, we analyzed cell surface DR4 and DR5 expression levels and observed no difference in DR4 and DR5 expression level between control and IRF8-expressing HT1080 cells (Fig. 6D). Taken together, our results suggest that targeting IRF8 expression in human sarcoma tumor is a potentially effective strategy to increase the efficacy of therapeutic agents (e.g. TRAIL) and perhaps thereby overcome human sarcoma chemoresistance.
Figure 6. Ectopic expression of IRF8 sensitized human sarcoma cells to FasL- and TRAIL-induced apoptosis.
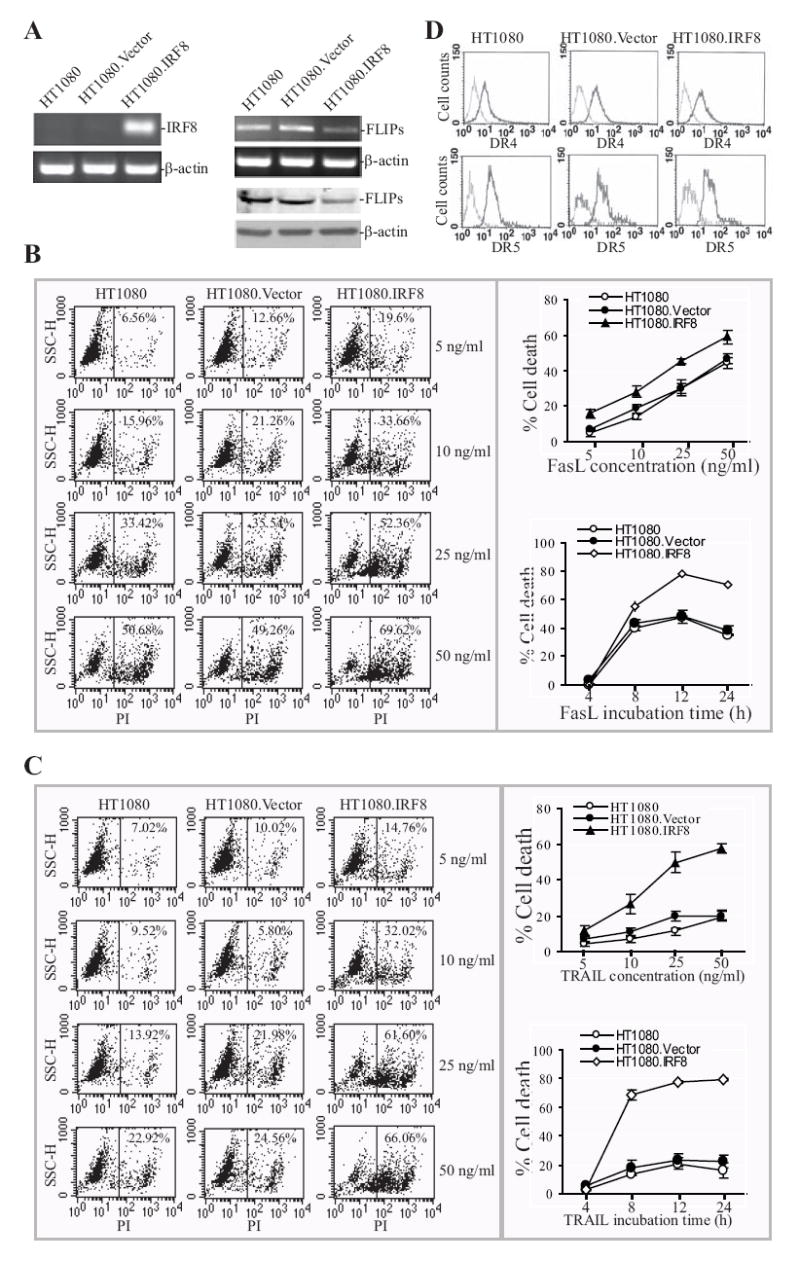
A. RT-PCR analysis of IRF8 expression in non-transfected (HT1080), vector-transfected (HT1080.Vector) and IRF8-transfected (HT1080.IRF8) human sarcoma cells. β-actin was used as normalization standard (left panel). RT-PCR and Western blotting analysis of FLIP expression in HT1080, HT1080.Vector and HT1080.IRF8 human sarcoma cells (right panel). Upper panel is FLIP mRNA level and lower panel shows FLIP protein level. B. Fas-mediated apoptosis in HT1080 sarcoma cell line/sublines. Tumor cells were cultured in the presence of different concentrations of recombinant human FasL for approximately 18 h. The cells were then stained with PI and analyzed by flow cytometry. Shown are histograms of one of three representative experiments (left panel) and plot of FasL concentration against percentage of cell death (top right panel). The tumor cells were also treated with FasL (25ng/ml) and analyzed for cell death at different time points (bottom right panel). C. TRAIL-induced apoptosis in HT1080 sarcoma cell line/sublines. Tumor cells were cultured in the presence of different concentrations of recombinant human TRAIL for approximately 18 h. The cells were then stained with PI and analyzed by flow cytometry. Shown are histograms of one of three representative experiments (left panel) and plot of FasL concentration against percentage of cell death (top right panel). The tumor cells were also treated with TRAIL protein (25ng/ml) and analyzed for cell death at different time points (bottom right panel). D. Cell surface TRAIL receptors DR4 and DR5 expression levels. Tumor cells were stained with anti-DR4- and DR5-specific antibodies, respectively, and analyzed with flow cytometry. Isotype-matched IgG control staining is depicted as gray areas, and DR4- or DR5-specific staining is depicted as solid lines.
Discussion
IRF8 is a transcription factor and current data suggest that IRF8 harbors dual functions as either a transcriptional activator or repressor. Genome-scale gene expression analysis has revealed that IRF8 regulates the expression of a large group of genes in hematopoietic cells (32, 33). Therefore, it is not surprising that IRF8 regulates a large set of gene expression in solid tumor cells as well. However, it is unexpected that IRF8 regulates the expression of several genes involved in apoptosis pathway, apparently at multiple levels. Because the expression of some apoptosis-related genes can be altered in the tumor microenvironment, defects in multiple mediators of the apoptosis pathway might thus confer the tumor cells with a stable apoptosis resistance phenotype. Therefore, tumor cells might use loss of IRF8 expression to acquire a stable apoptosis resistance phenotype.
Although IRF8 is constitutively expressed in macrophages, other myeloid cells, B cells, and T cells, expression of IRF8 can be dramatically up-regulated by IFN-γ (2). The relative roles of constitutively expressed IRF8 and IFN-γ-activated IRF8 are still not fully defined and remain an area of active research (2). IRF8 is also constitutively expressed in certain non-hematopoietic tumor cells, albeit at lower levels, but its expression can also be dramatically up-regulated by IFN-γ (8, 19) through the IFN-γR-mediated signaling pathway (34). IFN-γ has been shown to sensitize tumor cells to apoptosis through regulating caspase 8 or Bcl-2 expression (35, 36). In this study, IFN-γ treatment did not alter the expression levels of caspase 8 and Bcl-2 (Fig. 2B and data not shown) in mouse sarcoma cells. Because IFN-γ can regulate the expression of several IRF8-binding partner proteins in sarcoma cell lines (19), we suspect that IFN-γ treatment may induce IRF8-binding proteins that neutralize the effects of IFN-γ-regulated target genes. However, the exact molecular mechanisms under this phenomenon remain to be determined.
Although disrupting IRF8 expression significantly increased FLIPs expression in CMS4.K79E cells, disrupting IRF8 expression also diminished Fas receptor expression (16). Because Fas DISC formation requires Fas receptor engagement, diminished Fas receptor level will certainly lead to diminished Fas DISC level in CMS4.K79E cells, which may explain why less procaspase 8 is associated with the Fas DISC in CMS4.K79E cells (Fig. 2A). The diminished Fas expression level in CMS4.K79E cells also prevent us from determining whether increased FLIPs protein level inhibits procaspase 8 association with, and caspase activation in the Fas DISC.
Both Bcl-xL and Bcl-2 have been shown to be regulated by IRF8 in hematopoietic tumor cells (5, 12, 13). Although our initial attempts did not detect the effects of IRF8 on Bcl-xL and Bcl-2 in sarcoma cells (data not shown), other studies have shown important roles of Bcl-xL and Bcl-2 in apoptosis, including TRAIL-induced apoptosis, in both hematopoietic and non-hematopoietic tumor cells (36-38). Further studies are needed to examine the different regulatory mechanisms underlying IRF8 regulation of Bcl-xL and Bcl-2 between hematopoietic and non-hematopoietic tumor cells.
IRF8 protein is tyrosine-phosphorylated in certain myeloid lineages during monocytic leukemia development (13, 39-41). In myeloid leukemia, tyrosine phosphorylation increases IRF8 accumulation in the cytoplasm and inhibits IRF8 translocation to the nucleus in the blast crisis stage but not in the chronic stage (13). Phosphorylation-mediated inhibition of IRF8 nuclear localization in the blast crisis stage resulted in elevated Bcl-2 expression and apoptosis resistance (13), suggesting that IRF8 phosphorylation regulates IRF8 function in myeloid leukemia pathogenesis. In this study, we observed that IRF8 protein is largely absent in the nuclei of human STS tumor cells and weakly to moderately present in the cytoplasms (Fig. 4). Therefore, it is possible that the cytoplasmic IRF8 protein observed in the human STS patient specimens might be phosphorylated. Such phosphorylation could inhibit IRF8 nuclear translocation and function in sensitizing the tumor cells to apoptosis, which may particularly explain the chemoresistance of human STS. Further studies are needed to elucidate the phosphorylation status of IRF8 protein in human STS cells and to delineate its potential links to IRF8 subcellular localization and STS pathogenesis in human patients. IRF8 is also regulated by DNA methylation at the IRF8 promoter region in hematopoietic and non-hematopoietic tumor cells (8, 11, 42-44). However, the methylation status of the IRF8 promoter in human sarcoma cells has not been analyzed. Further studies are also needed to determine whether DNA methylation at the IRF8 promoter region also mediates IRF8 levels in human sarcoma.
A common feature of human sarcoma is resistance to conventional chemotherapy (1). Induction of apoptosis is the basis of many chemotherapeutic agents, including TRAIL, in cancer therapy (45, 46). Our data clearly suggests that FLIP is widely expressed in high grade human STS tumors in vivo, implying that cell death-inducing agents may not therefore be effective in sarcoma therapy. In support of our hypothesis that loss of IRF8 expression and/or function is responsible, at least in part, for the FLIP-mediated apoptosis resistance phenotype in human STS, restoration of IRF8 expression by transfection sensitized human sarcoma cells to apoptosis (Fig. 6). More importantly, forced expression of IRF8 also sensitized human sarcoma cells to TRAIL-induced apoptosis. TRAIL protein is considered a promising therapeutic agent for human sarcoma therapy (47) and is being tested in clinical trails to treat metastatic human cancers (48). Our findings collectively suggest that TRAIL therapy alone may not be effective, whereas combinational therapies targeting IRF8 expression in combination with conventional TRAIL therapy could potentially be an effective approach to overcoming chemoresistance in human STS therapy, a possibility that we are currently examining.
Acknowledgments
Grant Support: National Cancer Institute, NIH. CA 133085 (K. Liu)
We thank Dr. Scott I. Abrams for critical reading of the manuscript, Dr. Jeanene Pihkala for assistance in flow cytometry, and Ms. Kimberly K. Smith for technical assistance in Immunohistochemical staining.
References
- 1.Mocellin S, Rossi CR, Brandes A, Nitti D. Adult soft tissue sarcomas: conventional therapies and molecularly targeted approaches. Cancer Treat Rev. 2006;32:9–27. doi: 10.1016/j.ctrv.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Kanno Y, Levi BZ, Tamura T, Ozato K. Immune cell-specific amplification of interferon signaling by the IRF-4/8-PU.1 complex. J Interferon Cytokine Res. 2005;25:770–9. doi: 10.1089/jir.2005.25.770. [DOI] [PubMed] [Google Scholar]
- 3.Levi BZ, Hashmueli S, Gleit-Kielmanowicz M, Azriel A, Meraro D. ICSBP/IRF-8 transactivation: a tale of protein-protein interaction. J Interferon Cytokine Res. 2002;22:153–60. doi: 10.1089/107999002753452764. [DOI] [PubMed] [Google Scholar]
- 4.Holtschke T, Lohler J, Kanno Y, et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–17. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- 5.Gabriele L, Phung J, Fukumoto J, et al. Regulation of apoptosis in myeloid cells by interferon consensus sequence-binding protein. J Exp Med. 1999;190:411–21. doi: 10.1084/jem.190.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt M, Nagel S, Proba J, et al. Lack of interferon consensus sequence binding protein (ICSBP) transcripts in human myeloid leukemias. Blood. 1998;91:22–9. [PubMed] [Google Scholar]
- 7.Liu K, Abrams SI. Coordinate regulation of IFN consensus sequence-binding protein and caspase-1 in the sensitization of human colon carcinoma cells to Fas-mediated apoptosis by IFN-γ. J Immunol. 2003;170:6329–37. doi: 10.4049/jimmunol.170.12.6329. [DOI] [PubMed] [Google Scholar]
- 8.Yang D, Thangaraju M, Greeneltch K, et al. Repression of IFN regulatory factor 8 by DNA methylation is a molecular determinant of apoptotic resistance and metastatic phenotype in metastatic tumor cells. Cancer Res. 2007;67:3301–9. doi: 10.1158/0008-5472.CAN-06-4068. [DOI] [PubMed] [Google Scholar]
- 9.Greeneltch KM, Schneider M, Steinberg SM, et al. Host immunosurveillance controls tumor growth via IFN regulatory factor-8 dependent mechanisms. Cancer Res. 2007;67:10406–16. doi: 10.1158/0008-5472.CAN-07-1228. [DOI] [PubMed] [Google Scholar]
- 10.Egwuagu CE, Li W, Yu CR, et al. Interferon-γ induces regression of epithelial cell carcinoma: critical roles of IRF-1 and ICSBP transcription factors. Oncogene. 2006;25:3670–9. doi: 10.1038/sj.onc.1209402. [DOI] [PubMed] [Google Scholar]
- 11.Lee KY, Geng H, Ng KM, et al. Epigenetic disruption of interferon-γ response through silencing the tumor suppressor interferon regulatory factor 8 in nasopharyngeal, esophageal and multiple other carcinomas. Oncogene. 2008;27:5267–76. doi: 10.1038/onc.2008.147. [DOI] [PubMed] [Google Scholar]
- 12.Burchert A, Cai D, Hofbauer LC, et al. Interferon consensus sequence binding protein (ICSBP; IRF-8) antagonizes BCR/ABL and down-regulates bcl-2. Blood. 2004;103:3480–9. doi: 10.1182/blood-2003-08-2970. [DOI] [PubMed] [Google Scholar]
- 13.Middleton MK, Zukas AM, Rubinstein T, et al. Identification of 12/15-lipoxygenase as a suppressor of myeloproliferative disease. J Exp Med. 2006;203:2529–40. doi: 10.1084/jem.20061444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang W, Zhu C, Wang H, Horvath E, Eklund EA. The interferon consensus sequence-binding protein (ICSBP/IRF8) represses PTPN13 gene transcription in differentiating myeloid cells. J Biol Chem. 2008;283:7921–35. doi: 10.1074/jbc.M706710200. [DOI] [PubMed] [Google Scholar]
- 15.Thornton AM, Ogryzko VV, Dent A, et al. A dominant negative mutant of an IFN regulatory factor family protein inhibits both type I and type II IFN-stimulated gene expression and antiproliferative activity of IFNs. J Immunol. 1996;157:5145–54. [PubMed] [Google Scholar]
- 16.Yang D, Thangaraju M, Browning DD, et al. IFN Regulatory Factor 8 mediates apoptosis in nonhemopoietic tumor cells via regulation of Fas expression. J Immunol. 2007;179:4775–82. doi: 10.4049/jimmunol.179.7.4775. [DOI] [PubMed] [Google Scholar]
- 17.Yang D, Ud Din N, Browning DD, Abrams SI, Liu K. Targeting lymphotoxin β receptor with tumor-specific T lymphocytes for tumor regression. Clin Cancer Res. 2007;13:5202–10. doi: 10.1158/1078-0432.CCR-07-1161. [DOI] [PubMed] [Google Scholar]
- 18.Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang D, Stewart TJ, Smith KK, Georgi D, Abrams SI, Liu K. Downregulation of IFN-γR in association with loss of Fas function is linked to tumor progression. Int J Cancer. 2008;122:350–62. doi: 10.1002/ijc.23090. [DOI] [PubMed] [Google Scholar]
- 20.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–57. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 22.Medema JP, Scaffidi C, Kischkel FC, et al. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) EMBO J. 1997;16:2794–804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavrik IN, Mock T, Golks A, Hoffmann JC, Baumann S, Krammer PH. CD95 stimulation results in the formation of a novel death effector domain protein-containing complex. J Biol Chem. 2008;283:26401–8. doi: 10.1074/jbc.M800823200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ganten TM, Haas TL, Sykora J, et al. Enhanced caspase-8 recruitment to and activation at the DISC is critical for sensitisation of human hepatocellular carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic drugs. Cell Death Differ. 2004;11:S86–96. doi: 10.1038/sj.cdd.4401437. [DOI] [PubMed] [Google Scholar]
- 25.Wu Z, Roberts M, Porter M, et al. Viral FLIP impairs survival of activated T cells and generation of CD8+ T cell memory. J Immunol. 2004;172:6313–23. doi: 10.4049/jimmunol.172.10.6313. [DOI] [PubMed] [Google Scholar]
- 26.Konieczna I, Horvath E, Wang H, et al. Constitutive activation of SHP2 in mice cooperates with ICSBP deficiency to accelerate progression to acute myeloid leukemia. J Clin Invest. 2008;118:853–67. doi: 10.1172/JCI33742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tailor P, Tamura T, Kong HJ, et al. The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity. 2007;27:228–39. doi: 10.1016/j.immuni.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwieger M, Lohler J, Friel J, Scheller M, Horak I, Stocking C. AML1-ETO inhibits maturation of multiple lymphohematopoietic lineages and induces myeloblast transformation in synergy with ICSBP deficiency. J Exp Med. 2002;196:1227–40. doi: 10.1084/jem.20020824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siegmund D, Hadwiger P, Pfizenmaier K, Vornlocher HP, Wajant H. Selective inhibition of FLICE-like inhibitory protein expression with small interfering RNA oligonucleotides is sufficient to sensitize tumor cells for TRAIL-induced apoptosis. Mol Med. 2002;8:725–32. [PMC free article] [PubMed] [Google Scholar]
- 30.Park MA, Zhang G, Mitchell C, et al. Mitogen-activated protein kinase kinase 1/2 inhibitors and 17-allylamino-17-demethoxygeldanamycin synergize to kill human gastrointestinal tumor cells in vitro via suppression of c-FLIP-s levels and activation of CD95. Mol Cancer Ther. 2008;7:2633–48. doi: 10.1158/1535-7163.MCT-08-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang G, Park MA, Mitchell C, et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin Cancer Res. 2008;14:5385–99. doi: 10.1158/1078-0432.CCR-08-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamura T, Thotakura P, Tanaka TS, Ko MS, Ozato K. Identification of target genes and a unique cis element regulated by IRF-8 in developing macrophages. Blood. 2005;106:1938–47. doi: 10.1182/blood-2005-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee CH, Melchers M, Wang H, et al. Regulation of the germinal center gene program by interferon (IFN) regulatory factor 8/IFN consensus sequence-binding protein. J Exp Med. 2006;203:63–72. doi: 10.1084/jem.20051450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanno Y, Kozak CA, Schindler C, et al. The genomic structure of the murine ICSBP gene reveals the presence of the gamma interferon-responsive element, to which an ISGF3α subunit (or similar) molecule binds. Mol Cell Biol. 1993;13:3951–63. doi: 10.1128/mcb.13.7.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tekautz TM, Zhu K, Grenet J, Kaushal D, Kidd VJ, Lahti JM. Evaluation of IFN-gamma effects on apoptosis and gene expression in neuroblastoma--preclinical studies. Biochim Biophys Acta. 2006;1763:1000–10. doi: 10.1016/j.bbamcr.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 36.Zhou Y, Weyman CM, Liu H, Almasan A, Zhou A. IFN-γ induces apoptosis in HL-60 cells through decreased Bcl-2 and increased Bak expression. J Interferon Cytokine Res. 2008;28:65–72. doi: 10.1089/jir.2007.0025. [DOI] [PubMed] [Google Scholar]
- 37.Travert M, Ame-Thomas P, Pangault C, et al. CD40 ligand protects from TRAIL-induced apoptosis in follicular lymphomas through NF-κB activation and up-regulation of c-FLIP and Bcl-xL. J Immunol. 2008;181:1001–11. doi: 10.4049/jimmunol.181.2.1001. [DOI] [PubMed] [Google Scholar]
- 38.Petrella A, Ercolino SF, Festa M, et al. Dexamethasone inhibits TRAIL-induced apoptosis of thyroid cancer cells via Bcl-xL induction. Eur J Cancer. 2006;42:3287–93. doi: 10.1016/j.ejca.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 39.Sharf R, Meraro D, Azriel A, et al. Phosphorylation events modulate the ability of interferon consensus sequence binding protein to interact with interferon regulatory factors and to bind DNA. J Biol Chem. 1997;272:9785–92. doi: 10.1074/jbc.272.15.9785. [DOI] [PubMed] [Google Scholar]
- 40.Unlu S, Kumar A, Waterman WR, et al. Phosphorylation of IRF8 in a pre-associated complex with Spi-1/PU.1 and non-phosphorylated Stat1 is critical for LPS induction of the IL1B gene. Mol Immunol. 2007;44:3364–79. doi: 10.1016/j.molimm.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang W, Saberwal G, Horvath E, Zhu C, Lindsey S, Eklund EA. Leukemia-associated, constitutively active mutants of SHP2 protein tyrosine phosphatase inhibit NF1 transcriptional activation by the interferon consensus sequence binding protein. Mol Cell Biol. 2006;26:6311–32. doi: 10.1128/MCB.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tshuikina M, Nilsson K, Oberg F. Positive histone marks are associated with active transcription from a methylated ICSBP/IRF8 gene. Gene. 2008;410:259–67. doi: 10.1016/j.gene.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 43.Liu S, Ren S, Howell P, Fodstad O, Riker AI. Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment Cell Melanoma Res. 2008;21:545–58. doi: 10.1111/j.1755-148X.2008.00484.x. [DOI] [PubMed] [Google Scholar]
- 44.Tshuikina M, Jernberg-Wiklund H, Nilsson K, Oberg F. Epigenetic silencing of the interferon regulatory factor ICSBP/IRF8 in human multiple myeloma. Exp Hematol. 2008 Oct 13; doi: 10.1016/j.exphem.2008.08.001. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 45.Henson ES, Johnston JB, Gibson SB. The role of TRAIL death receptors in the treatment of hematological malignancies. Leuk Lymphoma. 2008;49:27–35. doi: 10.1080/10428190701713655. [DOI] [PubMed] [Google Scholar]
- 46.Almasan A, Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev. 2003;14:337–48. doi: 10.1016/s1359-6101(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 47.Kontny HU, Hammerle K, Klein R, Shayan P, Mackall CL, Niemeyer CM. Sensitivity of Ewing’s sarcoma to TRAIL-induced apoptosis. Cell Death Differ. 2001;8:506–14. doi: 10.1038/sj.cdd.4400836. [DOI] [PubMed] [Google Scholar]
- 48.Plummer R, Attard G, Pacey S, et al. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin Cancer Res. 2007;13:6187–94. doi: 10.1158/1078-0432.CCR-07-0950. [DOI] [PubMed] [Google Scholar]