

Abstract
Imbalanced protease expression and activities may contribute to the development of cancers including neuroblastoma. Neuroblastoma is a fatal childhood cancer of the sympathetic nervous system that frequently overexpresses mitogenic peptides, chemokines and their receptors. Dipeptidyl peptidase IV (DPPIV), a cell surface serine protease, inactivates or degrades some of these bioactive peptides and chemokines, thereby regulating cell proliferation and survival. Our studies show that DPPIV is expressed in normal neural crest-derived structures, including superior cervical and dorsal root ganglion cells, sciatic nerve, and in adrenal glands, but its expression is greatly decreased or lost in cells derived from neuroblastoma, their malignant counterpart. Restoration of DPPIV expression in neuroblastoma cells led to their differentiation in association with increased expression of the neural marker MAP2 and decreased expression of chemokines including stromal-derived factor 1 (SDF1) and its receptor CXCR4. Furthermore, DPPIV promoted apoptosis, and inhibited SDF1 mediated in vitro cell migration and angiogenic potential. These changes were accompanied by caspase activation, and decreased levels of phospho-AKT and MMP9 activity, down stream effectors of SDF1-CXCR4 signaling. Importantly, DPPIV suppressed the tumorigenic potential of neuroblastoma cells in a xenotransplantation mouse model. These data support a potential role for DPPIV in inhibiting neuroblastoma growth and progression.
Keywords: DPPIV, Neuroblastoma, Apoptosis, Chemokine, Migration and Invasion
Introduction
Proteases regulate autocrine and paracrine signaling of growth factors and chemokines, thereby promoting or inhibiting tumor growth and metastasis (Kenny et al., 1989; Ghersi et al., 2002; Nanus, 2003; Bauvois, 2004; Carl-McGrath et al., 2006; Bonfil et al., 2007; Golubkov and Strongin, 2007). However, the contribution of proteases and chemokines to neuroblastoma (NB) is not well understood. NB, the most common solid tumor of childhood, originates from the sympatho-adrenal lineage and remains the second cause of deaths among children (Matthay, 2008). NB consists of neural crest-derived, undifferentiated cells and is notable for its spontaneous regression to aggressive metastatic growth (Brodeur, 2003; Nakagawara, 2004; Tucker, 2004; Castel et al.,2007; Maris et al., 2007; Matthay, 2008). Therapy resistant aggressive NBs frequently overexpress and secrete high levels of growth factors and chemokines (Nakagawara et al.,1994; Eggert et al.,2000; Geminder et al., 2001; Vasudevan et al., 2005; Ren et al., 2006; Li and Thiele, 2007).
Chemokines and their receptors activate growth signaling pathways and matrix metalloproteinases, thereby providing a microenvironment suitable for tumor growth, migration, invasion, and angiogenesis (Gerard and Rollins, 2001; Barbero et al., 2003; Tanaka et al., 2005; Burger and Kipps, 2006; Chinni et al., 2006; Singh et al., 2007). Growing evidence implicates the chemokine stromal-derived factor 1 (SDF1/CXCL12) and its receptor CXCR4, which normally controls neural crest development, as having important roles in tumor growth, angiogenesis, and metastasis of various cancers including NB (Geminder et al., 2001; Payne and Cornelius, 2002; Tran et al., 2004; Belmadani et al., 2005; Airoldi et al., 2006; Guyon and Nahon, 2007). Elevated expression of SDF1 and CXCR4 in NB tumors are significantly correlated with tumor grade, poor clinical outcome, and metastasis to the bone marrow that expresses high levels of SDF1 (Russell et al., 2004).
Dipeptidyl peptidase IV (DPPIV), a serine protease is mainly expressed on the cell surface in neurons, epithelial cells, thymus, and melanocytes (Schrader et al., 1987; Dinjens et al., 1990; Abbott et al., 1997; Wesley et al., 1999; Gabrilovac et al., 2003; Wesley et al., 2004; Wesley et al., 2005). It is also present as a soluble form, circulating in the serum (Durinx et al., 2000; Christopherson et al., 2002; Havre et al., 2008). DPPIV cleaves N-terminal dipeptides from selected bioactive peptides, including some chemokines and neuropeptides, leading to their inactivation and/or degradation (Proost et al., 1998; Shioda et al., 1998; Mentlein, 1999; Durinx et al., 2000; Lambeir et al., 2001; Scharpe and De Meester, 2001). Indeed, loss or alteration of DPPIV expression is linked to the development of several tumors including prostate, lung, breast, hepatocellular carcinomas, ovarian, and melanomas (Morrison et al., 1993; Sakamoto et al., 1993; Bogenrieder et al., 1997; Tsuji et al., 2004; Wesley et al., 2005). Independent of its enzymatic activity, DPPIV interacts with extracellular matrix (ECM) components including collagen and fibronectin, thus regulating cell-cell and cell-ECM interactions (Piazza et al., 1989; Sedo and Kraml, 1994; Dobers et al., 2000; Havre et al., 2008). Through these functions, DPPIV regulates diverse biological processes including cell differentiation, adhesion, immune modulation, and apoptosis, functions that control neoplastic transformation. Previously, we and others have shown that DPPIV indeed suppresses the malignant phenotype of melanomas, prostate, ovarian, and lung cancers (Wesley et al., 1999; Pethiyagoda et al., 2000; Wesley et al., 2004; Wesley et al., 2005; Kajiyama et al., 2006). These studies support a role for DPPIV as a tumor suppressor gene.
In this study, we report that DPPIV is expressed in normal sympathetic nervous system-derived structures, but its expression and enzymatic activities are greatly decreased in cell lines derived from NB. We further demonstrate that DPPIV re-expression suppresses the malignant phenotype of NB cells, as indicated by inhibition of tumor growth, cell migration, and angiogenesis, in association with induction of apoptosis mediated by caspase activation. DPPIV restoration down-regulated several chemokines including SDF1 and its receptor CXCR4. These changes were accompanied by decreased levels of phospho-AKT, and gelatinase activity of MMP9, which are down stream effectors of SDF1-CXCR4 signaling. These data point to an important role for DPPIV in suppressing NB development and progression.
Materials and methods
Tissues and cell culture
Superior cervical and sensory dorsal root ganglia, and sciatic nerve were obtained from Sprague-Dawley rats (Charles River Laboratory, Wilmington, MA). Human neuroblastoma-derived cell lines, SK-N-AS, SK-N-SH, SK-N-DZ, SK-N-MC, SK-N-F1, and SH-SY5Y were from American Type Culture Collection (ATCC; Manassas, VA), and SMS-KCN and SMS-KCNR cells were kindly provided by Children’s Oncology Group. The cells were grown in RPMI with 10% fetal bovine serum. Normal human melanocytes were grown in media supplied by the provider (BioWhitaker, Walkersville, MD). To induce differentiation, SK-N-SH cells were treated with 1 mM dibutyryl adenosine 3’,5’-cyclic monophosphate (dbcAMP) (Sigma Aldrich, St. Louis, MO) for 8 days.
Immunohistochemical analysis
Human tissue microarrays were obtained from Cybrdi (Frederick, MD). After deparaffinization, slides were incubated with target retrieval solution (DAKO, Carpentaria, CA). Staining was performed using the rabbit ImmunoCruz staining system (Santa Cruz Biotechnology, Santa Cruz, CA) according to the manufacturer’s protocol. Anti-DPPIV antibody (Biomol, Plymouth Meeting, PA) was used at 1:400. Normal rabbit IgG was used as a negative control. Goat anti-rabbit Cy3-conjugated secondary antibody (Jackson Laboratories, Bar Harbor, ME) was used at 1:500. DAPI was used as a nuclear stain (Molecular Probes, Eugene, OR). Tumor specimens were processed for staining as described above. Anti-CD31 (Fisher Scientific, Fremont, CA) was used at 1:200. Apoptosis was detected by TUNEL assay using an APOPTAG kit (Chemicon International Inc, CA) according to manufacturer’s protocol. Slides were viewed using a Nikon ECLIPSE TE2000-U inverted microscope connected to a RT Slider Spot digital camera (Diagnostic Instruments, Sterling Heights, MI). Images were acquired using SPOT software version 3.2.
Semiquantitative RT-PCR
cDNAs synthesized from total RNA (2 μg) using superscript-II reverse transcriptase (Invitrogen, Carlsbad, CA) were used as templates for RT-PCR. Primer sets used: DPPIV (5’-tcatatgacatttatgattta-3’ and 5’-caaaatgaggaggcaagatca tc-3’.), MAP2 (5’ tgtcacagtggaggaagcag-3’ and 5’-cctgggatagctaggggttc-3’), and GAPDH as a control (5’-atcttccaggagcgagatcc-3’ and 5’-accactgacacgttggcagt-3’). PCR amplification was carried out as previously described (Wesley et al., 2005).
Flow cytometry
Cell surface expression of DPPIV was determined using an EPICS XL / XL-MCL Flow Cytometry System (Beckman Coulter, Fullerton, CA). Cells were stained with DPPIV primary antibody (S27 mAb, ATCC). Fluorescein-conjugated rabbit anti-mouse IgG (DAKO) was used as the secondary antibody. Cells stained with only IgG were analyzed in parallel as negative controls.
Dipeptidyl peptidase enzymatic activity
DPPIV peptidase activity was measured by colorimetric assay using Gly-Pro p-nitroanilide substrate, as previously described (Wesley et al., 2005).
Establishment of in vitro model system
SK-N-AS and SK-N-SH cells were co-transfected with the tetracycline-inducible pTRE vector containing DPPIV cDNA and the selectable plasmid pTET-on (Clontech, Mountain View, CA), using lipofectamine reagent (Invitrogen). Stably transfected cells were selected in presence of G418 (400 μg/ml) and screened for DPPIV expression. Cells transfected with the vector alone were used as controls.
Immunofluorescence microscopy
Cells grown on coverslips were stained with the respective mAbs (S27, SDF1, CXCR4 and MAP2, Santa Cruz Biotechnology) and incubated with either AlexaFluor 488 (Invitrogen) or Cy3-conjugated secondary antibodies at 1:500 dilution. Cells were counter stained with DAPI. Stained cells were viewed and imaged with an inverted microscope.
Measurements of neurite-like out growth
Cells possessing neurite-like processes longer than the diameter of the cell body were considered to be positive for counting. Images of the cells were captured with an inverted microscope. All neurites in each region of interest (ROI) were manually traced and the length was measured using SPOT software. The average neurite length was calculated by dividing the sum of neurite length by the number of cells in each ROI. For each group, at least 100 cells in 10 randomly selected fields were measured. The data are presented as mean values +/- SD.
MTT Cell Proliferation Assay
Control and DPPIV-transfected cells were plated in triplicate on 96-well plates and cultured in the presence of doxycycline (1 μg/ml). Colorimetric MTT assays were performed on day 3 following manufacturer’s instructions (ATCC). The absorbance of released purple formazan was measured at 570 nm. The experiment was repeated twice and the data are presented as mean values +/- SD.
Detection of apoptotic cells by TUNEL assay
Parental, vector control, and DPPIV-transfected SK-N-AS cells were cultured in media with or without serum for 48 hours in presence of doxycycline (1 μg/ml). DPPIV-transfected SK-N-AS cells were cultured in presence or absence of 5 mM diprotin A (Sigma Aldrich). Cells were fixed in 1% parafarmaldehyde and ice cold 70% ethanol. TUNEL assay was performed following manufacturer’s instructions and percent apoptosis was determined using flow cytometry.
Wound induced cell migration assay and matrigel cell invasion assay
Cell monolayers were wounded with a sterile micropipette tip and fed with medium containing SDF1 (100 ng/ml). Cells were photographed at 2 and 24 hours after wounding using a phase contrast microscope. Invasion was assessed using Matrigel-coated Biocoat cell culture inserts (BD Biosciences, Bedford, MA) with 8 μm pores in 24 wells. A total of 3 x 104 cells were placed in the upper compartment and the lower compartment was filled with 500 μl growth medium containing SDF1 (100 ng/ml). DPPIV-transfected SK-N-AS cells were cultured in presence or absence of 5 mM diprotin A for 15 min at 37°C. After 24 hours, cells on the lower surface were stained with crystal violet and solubilized in extraction buffer. Optical densities (OD) values at 540 nm correlating with cell migration were plotted. Results are presented as mean values +/- SD of triplicates.
In vitro angiogenesis assay
Fifteen μl of Matrigel matrix (BD Biosciences) was distributed per well in a 96-well plate. Human Umbilical cord Vein Endothelial Cell (HUVEC; ATCC) (7×103 cells/well) suspended in 100 μl of RPMI supplemented with 0.2% FBS were added to each well and co-cultured in triplicate with 7×103 of either SK-NAS, SK-NAS+Vector, or SK-NAS+DPPIV cells at 37°C. DPPIV activity was inhibited by pretreatment of cells with 5 mM diprotin A for 15 min at 37°C. After 20 hours, images were captured and the total length of tube-like angiogenic structures of five randomly chosen microscopic fields were measured by Image J software (NIH, Bethesda, MD).
MMP9 gelatinase activity
For gelatin zymography, ten μg protein from each cell supernatant was applied to 10% SDS-PAGE gels containing gelatin-A (1 mg/ml; Sigma Aldrich). After electrophoresis, gels were washed in 2.5% Triton X-100, incubated for 18 hours at 37°C and stained in 0.1% Coomassie brilliant blue. The gelatinolytic regions were observed as white bands against a blue background. In situ detection of gelatinolytic activity was carried out by overlaying cells grown in an 8-well chamber slide with 100 μg/ml quenched fluorescein-labeled gelatinase substrate, DQ-gelatin (Molecular Probes) for 2 hours at 37°C. Slides were fixed with 4% paraformaldehyde, and imaged using an inverted microscope.
Western blot analysis
30 μg of total protein was separated by SDS-PAGE and probed with respective antibodies (1:1000 for phospho-Akt (Ser-473), total AKT, and caspases 3, 8 and 9, Cell Signaling, Danvers, MA; 1:500 for actin, Sigma Aldrich), followed by incubation with secondary antibody conjugated to horseradish peroxidase at room temperature for 1 hour. Signals were developed with chemiluminescence using a PerkinElmer ECL kit (Waltham, MA). Total AKT and actin were used as loading controls.
Tumorigenicity in nude mice
Six-week old BALB/c (nu/nu) nude mice (Taconic, Hudson, NY) were injected subcutaneously into the flanks with 5X106 control and DPPIV re-expressing SK-N-AS cells suspension in matrigel (BD Biosciences). Five animals were used in each group. All mice were fed doxycycline containing food (Bio-Serve, Frenchtown, NJ). Tumor volume was estimated by using the formula width2 x length x 0.52 in cm3. At day 29, mice were euthanized, and the tumors were removed for further experiments. Animal experiments were approved by the Institutional Animal Care and Use Committee. Apoptosis and tumor vascularity were assessed in each tumor specimen as described above. For quantification of apoptosis, the number of TUNEL positive cells was counted in a total of 6 high power fields and expressed as mean percentage of total cells in these fields of the tumor.
Chemokine Pathway focused gene expression profiling by quantitative real-time RT-PCR (qRT-PCR)
A PCR array (PAHS-022A, SuperArray, Frederick, MD) that profiles the expression of 84 genes encoding chemokines and their receptors was used according to manufacturer’s protocol. Briefly, the cDNA generated from 2 μg of total RNA was combined with SYBR green qPCR master mix. Equal aliquots of this mixture were added to each well of the PCR array plates containing pre-dispensed gene specific primer sets. qRT-PCR analysis was performed in an Applied Biosystems Prism 5700 Sequence Detection system and analyzed using GeneAmp 5700 SDS software. Relative quantification was performed using standard curves generated for each gene-specific primer pair. The values obtained from each set of gene-specific primers were normalized to endogenous control genes and used to determine relative expression levels. Levels of SDF1 and CXCR4 mRNA were further validated using assay on demand primer sets (Applied Biosystems, Foster City, CA).
Statistical Analysis
All in vitro studies were performed in triplicate and results are expressed as mean ± standard deviation (SD). Statistical significances were determined by means of Student’s t test or by ANOVA and multiple comparisons test. A probability p <0.05 was considered statistically significant in all calculations.
Results
DPPIV is expressed in normal neural crest-derived structures and decreased in NB-derived cell lines
DPPIV expression was readily observed by RT-PCR in rat neural crest-derived structures including dorsal root ganglia, superior cervical ganglia, and sciatic nerve (Figure 1A). Immunohistochemical analysis of a normal human tissue microarray revealed DPPIV expression in the adrenal glands and peripheral nerves, and DPPIV expression was comparable to that of thymus tissue that is known to express DPPIV. However, DPPIV expression was not detectable in thyroid gland tissues in the same array demonstrating the tissue specific expression of DPPIV. (Figure 1B). We then examined the cell surface expression of DPPIV in a panel of NB-derived cell lines, including SK-N-SH, SH-SY5Y, SMS-KCN, SMS-KCNR, SK-N-MC, SK-N-DZ, SK-N-AS, and SK-N-F1. As compared with neural crest-derived melanocytes, cell surface expression of DPPIV was greatly decreased or almost undetectable in all cell lines tested except SMS-KCN, which showed moderate levels of DPPIV (Figure 1C). The DPPIV enzyme activities in these cell lines correlated with the levels of cell surface protein expression. Melanocyte DPPIV enzyme activities ranged from 180-220 pmoles/μg protein/min, while in SMS-KCN, enzyme activities ranged from 80-90 pmoles/μg protein/min. In all the other cell lines tested, DPPIV enzyme activities were reduced to 30-50 pmoles/μg protein/min (Figure 1D). Furthermore, differentiation of SK-N-SH cells with dbcAMP (Figure 1Ea) resulted in up regulation of DPPIV mRNA levels in association with increased expression of a known differentiated neural marker, microtubule-associated protein 2 (MAP2) (Figure 1Eb). Thus, the presence of DPPIV in differentiated neuronal cells and its absence in NB-derived cells suggests that DPPIV loss correlates with NB development and/or progression.
Figure 1.
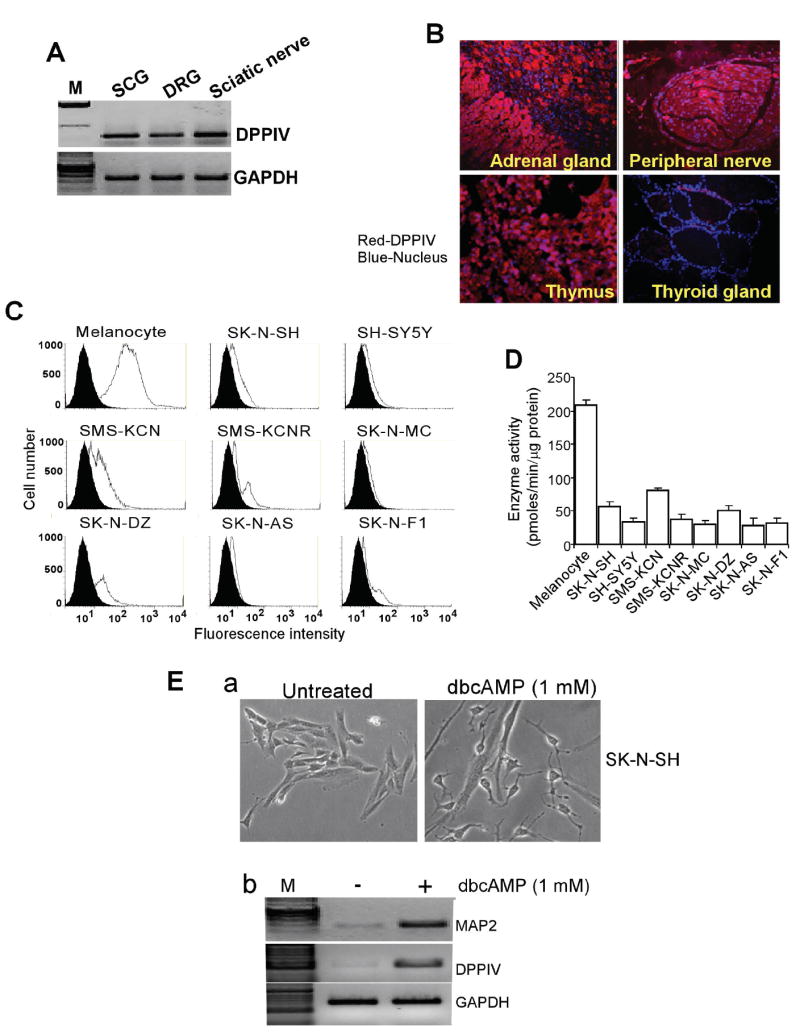
Expression of DPPIV in neural crest-derived structures and neuroblastoma-derived cell lines. A. Semiquantitative RT-PCR indicating the expression of DPPIV in superior cervical ganglia (SCG), dorsal root ganglia (DRG), and sciatic nerve. B. Representative photomicrographs of immunohistochemical analysis showing DPPIV expression in adrenal glands, peripheral nerve, and thymus, but not in thyroid gland present on the same tissue array. Red fluorescence staining represents DPPIV expression and blue staining represents nuclei. C. Flow cytometric analysis showing decreased cell surface expression of DPPIV in neuroblastoma-derived cell lines (SK-N-SH, SH-SY5Y, SMS-KCN, SMS-KCNR, SK-N-MC, SK-N-DZ, SK-N-AS, and SK-N-F1) as compared to neural crest-derived melanocytes. Cells were stained with the DPPIV-specific mAb S27 and fluorescein-conjugated rabbit anti-mouse secondary antibody. The Y-axis shows relative cell number and the X-axis shows the log of relative fluorescence intensity. The solid black line depicts DPPIV expression and filled histogram is control IgG1 antibody. D. DPPIV enzymatic activity in total cell lysates obtained from the same set of neuroblastoma cell lines and melanocytes as measured by colorimetric assay. Results are mean values of DPPIV activity ± SD of triplicates. E. DPPIV expression is associated with dbcAMP-induced differentiation of neuroblastoma (SK-N-SH) cells. a. Bright field images of undifferentiated and differentiated SK-N-SH cells treated with 1 mM dbcAMP for 8 days; b. RT-PCR showing mRNA levels of neuronal marker MAP2 and DPPIV.
DPPIV induces differentiated phenotype in NB cells (SK-N-SH and SK-N-AS)
To further understand the functional role of DPPIV in NB, we established SK-N-SH and SK-N-AS cell lines stably expressing DPPIV under the control of a tetracycline responsive promoter. Stable restoration of DPPIV cell surface expression was confirmed by immunofluorescence staining (Figure 2A). These stable cell lines retained the catalytic activity as demonstrated by peptidase enzyme activity ranging from 160-200 pmoles/μg/min protein and was comparable to the normal melanocytes (180-220 pmoles/μg protein/min) (Figure 2Ba). Furthermore, RT-PCR clearly indicated that the levels of DPPIV mRNA in transfected cells were similar to the physiological levels seen in normal human adrenal glands (Figure 2Bb). Restoration of DPPIV expression resulted in morphological changes in these cells. Parental and vector transfected SK-N-SH and SK-N-AS cells grew in a disorganized array of focal clumps. In contrast, SK-N-SH and SK-N-AS cells expressing DPPIV for 10-14 days acquired either neuronal-like or larger flat-epithelial-like phenotypes (Figure 2Ca). Neurite length was significantly increased in SK-N-SH cells re-expressing DPPIV, indicating neuronal differentiation (Figure 2Cb). Changes in phenotype of these cells was associated with increased expression of the neural marker MAP2 (Figure 2D). Not surprisingly, the proliferation rate was greatly decreased in both cell types as detected by decreased number of viable cells (Figure 2E). These data show that DPPIV inhibits NB proliferation in vitro by promoting differentiation.
Figure 2.
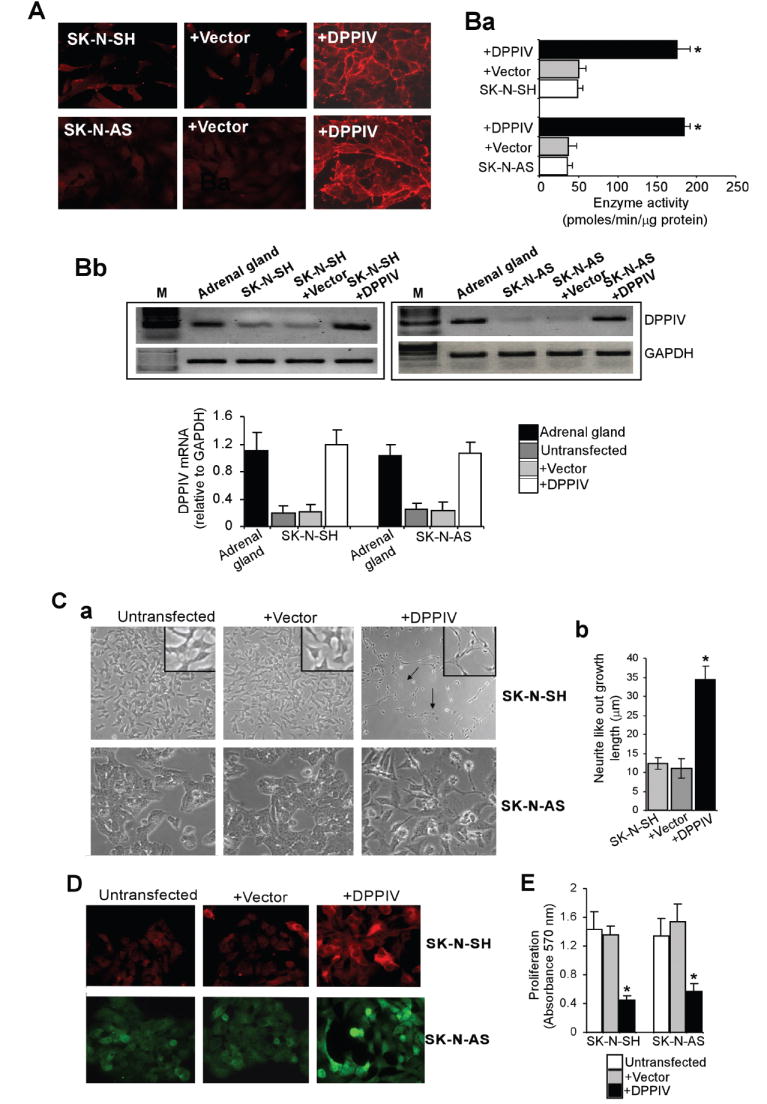
DPPIV induces differentiated phenotype in neuroblastoma cells (SK-N-SH and SK-N-AS). A. Immunofluorescence staining showing cell surface expression of DPPIV. Parental, vector control, and DPPIV transfected SK-N-SH and SK-N-AS cells were cultured in the presence of doxycycline (1 μg/ml) for 48 h and stained with mAb S27 against DPPIV. Original magnification 200X. Ba. DPPIV enzyme activities in parental and DPPIV or vector transfected SK-N-SH and SK-N-AS cells. Results shown are mean values ± SD of triplicates. Bb. Top panel: RT-PCR showing DPPIV mRNA levels in normal human adrenal glands and parental, vector, and DPPIV transfected SK-N-AS cells. Bottom panel: Densitometric analysis showing DPPIV mRNA levels relative to GAPDH mRNA levels C. Phenotypic changes associated with DPPIV expression. a. Morphology of untransfected parental cells showed short spindle-shaped and polygonal morphology and grew in unorganized clusters. Vector transfected cells were morphologically similar to the parental cell line. The SK-N-SH and SK-N-AS cells re-expressing DPPIV acquired morphological changes including either neuronal like or larger flat-epithelial dendritic phenotypes. Magnification, 200X. b. Neurite length was significantly increased in SK-N-SH cells re-expressing DPPIV. Images of the SK-N-SH, SK-N-SH+Vector and SK-N-SH+DPPIV cells were captured with an inverted microscope. All neurites in each region of interest (ROI) were manually traced and the length was measured using the software SPOT version 3.2. The data are presented as mean values +/- SD. D. Immunofluorescence staining showing the expression of the neuron-specific marker MAP2. Increased MAP2 reactivity was localized in representative images of DPPIV expressing SK-N-SH and SK-N-AS as compared to parental and vector transfected cells. E. MTT assay showing decreased proliferation in DPPIV re-expressing SK-N-SH and SK-N-AS cells as compared to parental and vector transfected cells (mean ± S.D.; n = 4; *p < 0.05).
DPPIV induces apoptosis in NB cells (SK-N-AS) through caspase activation and decreased Akt phosphorylation
Acquiring resistance to apoptosis is a critical step in NB growth and progression. Our data show that DPPIV increases sensitivity of SK-N-AS cells to apoptosis in serum withdrawal conditions. Cells undergoing apoptosis exhibited decreased cell size, round morphology, and cellular fragmentation. The proportion of apoptotic cells 3 days following serum withdrawal ranged from 25% to 30% in DPPIV-expressing cells, while the control cells exhibited significantly lower percent (5-7%) of apoptosis (Figure 3A). Induction of apoptosis by DPPIV was mediated through activation of both intrinsic and extrinsic apoptotic pathways as shown by generation of cleaved caspase 8 as well as caspase 9, leading to activation of caspase 3, the downstream effector (Figure 3B). Furthermore, Akt showed a marked decrease in phosphorylated levels at site Ser 473, in SK-N-AS cells that re-expressed DPPIV compared with control SK-N-AS cells (Figure 3C). DPPIV inhibitor diprotin A greatly decreased the DPPIV induced caspase activation and restored the Akt phosphorylation. These data demonstrate a role for DPPIV in interrupting the survival signaling pathways that may contribute to NB growth and progression.
Figure 3.
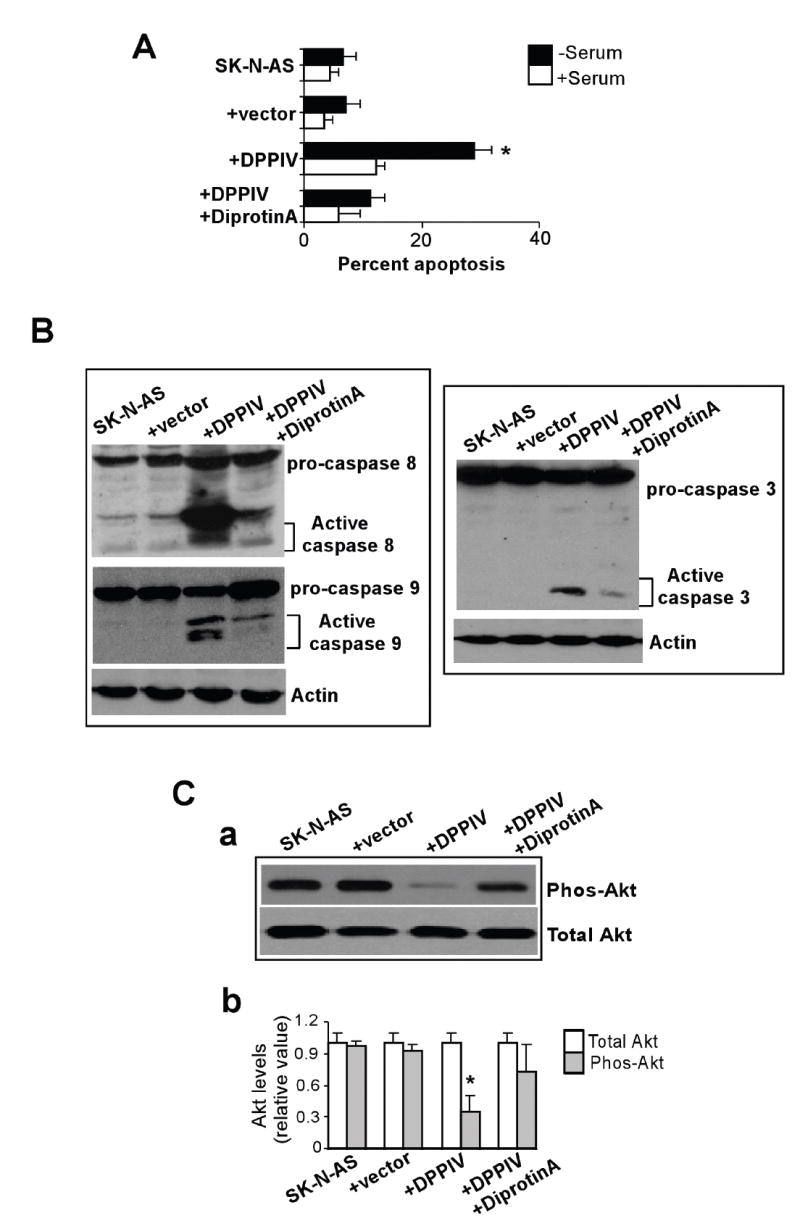
DPPIV induces apoptosis in SK-N-AS cells through caspase activation. A. Control (parental and vector-transfected) SK-N-AS cells or SK-N-AS cells transfected with DPPIV constructs were assessed for apoptosis in presence or absence of serum by TUNEL assay as described in Materials and Methods. The percent of apoptotic cells was measured by flow cytometry. (mean ± S.D.; n = 4; *p < 0.05). B. Apoptosis induced by DPPIV involves activation of caspase 8 and caspase 9 leading to activation of caspase 3 as shown by western blot analysis. Ca. Western blot analysis showing decreased phosphorylated Akt levels in DPPIV expressing SK-N-AS cells. Cb. Densitometric analysis showing the levels of total and phosphorylated Akt levels (mean ± S.D.; n = 3; *, p < 0.05).
DPPIV inhibits SDF1-induced NB cell migration and invasion, and angiogenesis in vitro
Cell migration and invasion are known to be facilitated by chemokines including SDF1 and its receptor CXCR4. In a wound-induced migration assay, control SK-N-AS cells treated with SDF1 migrated to fill the wounded area after 24 hours of wounding, indicating their strong migratory potential. However, restoration of DPPIV greatly reduced this SDF1 mediated migratory potential as indicated by an unfilled wound area after 24 hours (Figure 4A, bottom panel). Furthermore, the ability of DPPIV expressing SK-N-AS cells to invade through matrigel-coated filters was significantly reduced when compared with control SK-N-AS cells. This effect of DPPIV was blocked by diprotin A pretreatment indicating the specific effects of DPPIV enzyme activity (Figure 4B). Angiogenesis is necessary for tumor cell growth and metastasis. Therefore, we determined the effect of DPPIV on the formation of pro-angiogenic structures arising from HUVEC. Control SK-N-AS cells stimulated the angiogenenic process as indicated by the formation of closed intercellular compartments, while DPPIV re-expressing SK-N-AS cells showed a greatly decreased ability to form closed intercellular compartments and this effect was blocked by DPPIV inhibitor (Figure 4Ca). The length of tubular structures in SK-N-AS+DPPIV cells was significantly shorter than those from control SK-N-AS cells (52% reduction, p<0.05) (Figure 4Cb). These effects were accompanied by decreased activities of matrix metalloproteinase MMP9, a pro-angiogenic and invasive factor, as shown by in situ gelatinase activity (Figure 4Da) and gel zymography (Figure 4Db). Interestingly, diprotin A treatment resulted in restoration of MMP9 activity indicating the specific effects of DPPIV on decreasing MMP9 levels. Together, these results demonstrate that restoration of DPPIV expression attenuates the malignant phenotype of NB cells in vitro.
Figure 4.

DPPIV expression inhibits in vitro cell migration, invasion, and angiogenesis of neuroblastoma cells (SK-N-AS). A. Wound healing assay showing SDF1 (100 ng/ml) mediated migration of SK-N-AS, vector or DPPIV transfected SK-N-AS cells with or without DPPIV inhibitor diprotin A, after 2 and 24 hours of wounding (Magnification 100X). B. Quantitative cell invasion assay as done using Boyden chamber cell invasion assay kit. Absorbance correlating with the number of cells invaded were read at 540 nm (mean ± S.D; n = 3; *, p < 0.05). C. DPPIV inhibits formation of closed rings arising from HUVEC sprouting (pro-angiogenic structure) in vitro. HUVECs were co-cultured with control or DPPIV expressing SK-N-AS cells for 18 h on matrigel basement. (a) Representative photomicrographs of HUVEC pro-angiogenic structure formation in co-culutre experiments. (b) Tubular length was quantified in five randomly selected fields (mean ± S.D.; n = 5; *, p < 0.05). D. DPPIV re-expression leads to decreased activation of MMP9 in SK-N-AS cells. In situ (a) and gel zymography analysis (b) showing MMP9 gelatinase activity in SK-N-AS, SK-N-AS+Vector and SK-N-AS+DPPIV cells.
DPPIV suppresses the tumorigenic potential of NB cells (SK-N-AS) in athymic nude mice
We examined the effects of DPPIV on SK-N-AS xenografts in nude mice. Tumors grew in all five mice injected with vector control SK-N-AS cells. However, mice injected with SK-N-AS cells re-expressing DPPIV showed no tumor growth except one animal that developed a tumor at a greatly reduced rate (Figure 5A). This tumor was available for immunohistochemistry. TUNEL assay clearly indicated an increased number of apoptotic cells in the DPPIV-expressing SK-N-AS tumor (Figure 5Ba). Quantification of TUNEL-positive cells revealed significant increase in apoptosis (25-30%) in DPPIV expressing SK-N-AS tumors when compared with control tumor (4-6%) (Figure 5Bb). Furthermore, tumors in control mice were highly angiogenic and showed increased CD31-positivity compared to the tumor developed from SK-N-AS+DPPIV cells (Figure 5C). These data suggest that DPPIV suppresses NB tumor growth in vivo through induction of apoptosis and probably through inhibition of angiogenesis.
Figure 5.
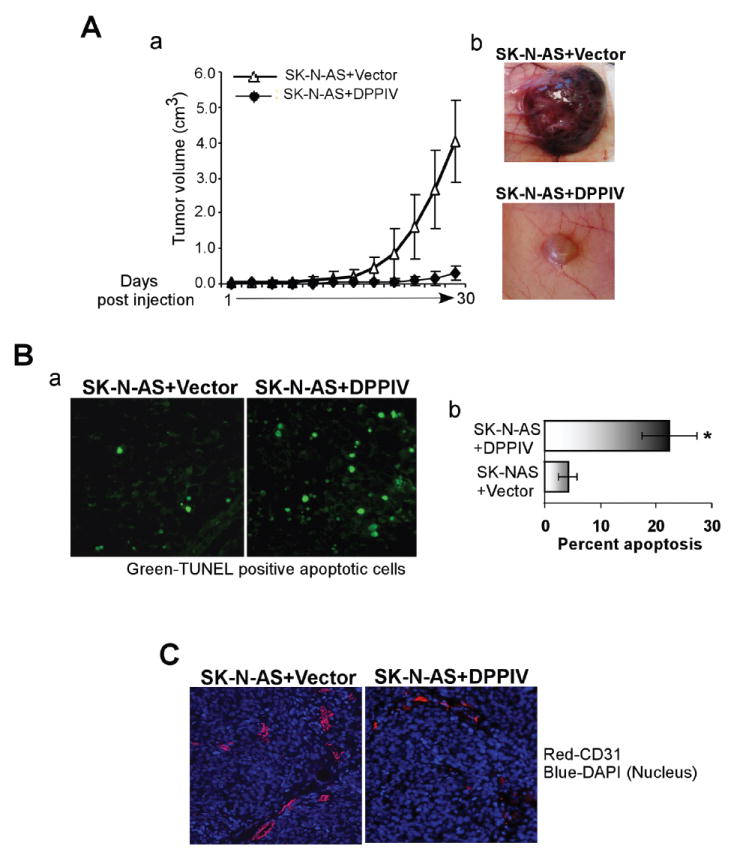
DPPIV re-expression suppresses the tumorigenic potential of SK-N-AS cells in a xenotransplantation mouse model. Two different sets of nude mice (BALB/C nu/nu, n = 5 for each group, SK-N-AS+Vector or SK-N-AS+DPPIV) were injected subcutaneously with 5X106 cells as a 50% suspension in matrigel. Tumors were measured every 3 days. A. a. Effects of DPPIV on tumor growth. Results are presented as average tumor volume +/-SD. b. Photographs of tumors excised from SK-N-AS+Vector and SK-N-AS+ DPPIV mice. B. a. Representative photomicrographs of TUNEL assay performed on excised tumors showing increased number of apoptotic cells (green) in tumors developed from SK-N-AS+DPPIV cells as compared to tumors developed from SK-N-AS+Vector control cells. Magnification 200X. b. Quantification of DPPIV induced apoptosis in tumors. The number of TUNEL positive cells was counted in a total of 6 high power fields and expressed as mean percentage of total cells in these fields of the tumor C. Immunohistochemical analysis showing CD31 staining as a measure of vascularity in tumors developed from SK-N-AS+Vector or SK-N-AS+DPPIV cells. Magnification 200X.
DPPIV down-regulates chemokine SDF1 and its receptor CXCR4 in NB (SK-N-AS) cells
Chemokines and their receptors play critical roles in tumor growth and metastasis. In order to examine if DPPIV’s effects on NB cells (SK-N-AS) are mediated through regulation of chemokines and their receptors, we screened a chemokine pathway-specific PCR array. RNAs from control SK-N-AS and DPPIV re-expressing SK-N-AS cells were analyzed in parallel. Interestingly, DPPIV re-expression resulted in down-regulation of 13 genes, 7 of which were down-regulated more than 5 fold. In contrast, 9 genes that showed up-regulation were changed by only 2-3 fold (Figure 6A). Because SDF1 and CXCR4 showed significant down regulation (24 and 7 fold, respectively), their mRNA and protein levels were further validated by quantitative real-time RT-PCR (Figure 6B) and immunofluorescence staining (Figure 6C). These results suggest that DPPIV negatively regulates NB cell growth via interruption of chemokine signaling.
Figure 6.

DPPIV regulates expression of chemokines and chemokine receptors in NB cells (SK-N-AS). A. Restoration of DPPIV alters chemokine and chemokine receptor mRNA levels in SK-N-AS cells as indicated by chemokine pathway-specific PCR array screening. B. Quantitative real-time PCR confirming array data for SDF1 and CXCR4 expression. C. Immunofluorescence staining indicating the decreased levels of SDF1 and CXCR4 in DPPIV re-expressing SK-N-AS cells as compared to vector transfected SK-N-AS cells. Magnification 200X.
Discussion
Although dysregulated protease expression has been recognized as a hallmark of cancer, our understanding of proteases that function as tumor and/or metastasis suppressors is limited. Recently, cell surface proteases including DPPIV and neutral endopeptidase (NEP) have emerged as important tumor suppressor genes for prostate, ovarian, and lung cancers, as well as hepatocarcinoma and melanoma (Wesley et al., 1999; Dai et al., 2001; Wesley et al., 2004; Terauchi et al., 2005; Wesley et al., 2005; Kajiyama et al., 2006). However, the role of cell surface proteases in suppressing NB is virtually unknown. Our studies have shown that DPPIV is expressed in neural crest-derived structures, and in agreement with previous observations in other human neoplasias, DPPIV expression is greatly decreased in NB-derived cells suggesting that DPPIV is a potential tumor suppressor gene for NB. In support of this idea, we have demonstrated that DPPIV indeed suppresses the malignant phenotype of NB cells, characterized by inhibition of survival, migration/invasion, angiogenesis, and tumor growth in nude mice. It is intriguing that a cell surface peptidase can have such pleiotropic effects on the malignant phenotype of NB cells. One possible explanation is that DPPIV degrades or inactivates growth factors and chemokines required for growth and survival of neuroblastic cells. Also, DPPIV may modulate the extracellular microenvironment through its interaction with cell surface receptors and ECM components, thereby providing inhibitory effects on tumor progression.
Children with aggressive and metastatic NB are highly refractory to current treatments and have poor survival rates (Maris and Woods, 2008; Matthay, 2008). Thus, further understanding the mechanisms that facilitate aggressive tumor growth and identifying the molecules that inhibit this process is of great importance. Recent studies provide compelling evidence that dysregulation of chemokine-receptor signaling, including SDF1-CXCR4, is associated with growth and metastasis of NB and many other types of cancers (Geminder et al., 2001; Payne and Cornelius, 2002; Barbero et al., 2003; Balkwill, 2004; Fernandis et al., 2004; Tran et al., 2004; Belmadani et al., 2005; Burger and Kipps, 2006; Scala et al., 2006; Guyon and Nahon, 2007; Singh et al., 2007; Airoldi et al., 2008). Although, no CXCL12-driven chemotaxis of NB cells was observed by Airoldi et al., 2006, other studies have shown that SDF1-CXCR4 expression correlates with aggressive NB with increased migratory potential and higher vascularity in tumors. Furthermore, autocrine stimulation of CXCR4 by its ligand SDF1 was shown to be necessary for the survival of some NB cells in vitro (Geminder et al., 2001; Nevo et al., 2004; Russell et al., 2004; Vasudevan et al., 2005). Despite these important observations, the mechanisms responsible for activation of the SDF1-CXCR4 pathway remain elusive.
DPPIV is known to regulate the function of certain chemokines. Thus, loss of DPPIV allows accumulation of these chemokines leading to increased interaction with their receptors, and activation of downstream signaling molecules. In support of this view, our studies demonstrate that DPPIV decreases the expression of chemokines in NB cells. Of note, SDF1 and its receptor CXCR4 were down-regulated by more than 5 fold. SDF1 and CXCR4 produced by tumors and stromal cells not only promote migration, but also can stimulate growth, survival, and angiogenesis through activation of survival signaling pathways, and MMP9 required for degradation of nearby matrix (Rundhaug, 2005; Zigrino et al., 2005; Raman et al., 2007). In fact, DPPIV re-expressing SK-N-AS cells showed decreased MMP9 activity and exhibited reduced in vitro angiogenic and migratory potential. These effects were overcome by pre-treatment with diprotin A that resulted in restoration of MMP9. Diprotin A is a competitive inhibitor of DPPIV enzyme activity and thus it appears that DPPIV enzyme activity is required for suppressing the angiogenic and migratory potential of NB cells. In addition, DPPIV also stimulated an apoptotic cascade by activating caspase 8 and 9 as well as their downstream effector caspase 3. These effects of DPPIV were linked to decreased levels of phospho-AKT, the down stream effectors of SDF1-CXCR4 signaling. Interestingly, DPPIV inhibitor diprotin A greatly blocked the DPPIV induced caspase activation with the increased levels of phosphorylated Akt. Furthermore, evaluation of tumor growth in vivo revealed that inhibited tumor growth of SK-N-AS cells by DPPIV was associated with increased apoptosis and decreased angiogenesis.
These functions of DPPIV may be mediated through regulating SDF1-CXCR4 signaling pathway. DPPIV is shown to inactivate SDF1 by proteolytic cleavage. Interestingly, DPPIV also interacts with CXCR4 in hematopoetic cells. However, such interactions have not yet been identified in neuronal cells. Our studies have shown that DPPIV decreases the levels of both SDF1 and its receptor CXCR4. In fact it has been reported that SDF1 not only stimulates CXCR4 signaling but also regulates its expression by positive feed back mechanism. It is possible that in neural crest-derived cells, DPPIV disrupts this positive autocrine signaling loop not only by direct cleavage of SDF1 but also by associating with receptor CXCR4 eventually leading to decreased levels of CXCR4. Independent of its protease activity, DPPIV has other functions, including binding to ECM and other proteins such as CD45, and adenosine deaminase as shown in hematopoetic cells (Havre et al., 2008). It will be of great importance to elucidate the specific mechanisms by which DPPIV regulates SDF1-CXCR4 in NB-derived cells. This interruption of the SDF1-CXCR4 axis is probably of significance for inhibition of growth and metastasis of NB (Meitar et al., 1996; Eggert et al., 2000; Geminder et al., 2001; Russell et al., 2004; Tran et al., 2004; Tucker, 2004).
In summary, our data strongly support a role for DPPIV as a potential tumor suppressor gene for NB. Given its ability to regulate differentiation, apoptosis, angiogenesis, and cell motility through its modulation of chemokines and the ECM, DPPIV provides a new and potentially significant approach for development of treatment strategies aimed at blocking mitogenic and angiogenic signaling. Thus, an emerging appreciation for the role of proteases in suppressing the malignant phenotype could prove especially beneficial in understanding and treating tumors such as NB.
Acknowledgments
This work was supported by the Grant, Neuroscience COBRE 2 P20 RR016435-P4Y6 from the NCRR. We thank the Department of Microbiology and Molecular Genetics and the Vermont Cancer Center (VCC) for their support. We thank Drs. Cynthia Forehand, Diane Jaworski, Rodney Parsons, Marcus Bosenberg, Rae Nishi, and Cedric Wesley for helpful discussions and critical review of the manuscript. We thank the staff at the VCC and COBRE-Neuroscience core facilities for their technical help.
References
- Abbott CA, Gorrell MD, Levy MT, McCaughan GW. Molecular analyses of human and rat dipeptidyl peptidase IV. Adv Exp Med Biol. 1997;421:161–9. doi: 10.1007/978-1-4757-9613-1_21. [DOI] [PubMed] [Google Scholar]
- Airoldi I, Cocco C, Morandi F, Prigione I, Pistoia V. CXCR5 may be involved in the attraction of human metastatic neuroblastoma cells to the bone marrow. Cancer Immunol Immunother. 2008;57:541–8. doi: 10.1007/s00262-007-0392-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Airoldi I, Raffaghello L, Piovan E, Cocco C, Carlini B, Amadori A, et al. CXCL12 does not attract CXCR4+ human metastatic neuroblastoma cells: clinical implications. Clin Cancer Res. 2006;12:77–82. doi: 10.1158/1078-0432.CCR-05-1376. [DOI] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–50. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Barbero S, Bonavia R, Bajetto A, Porcile C, Pirani P, Ravetti JL, et al. Stromal cell-derived factor 1alpha stimulates human glioblastoma cell growth through the activation of both extracellular signal-regulated kinases 1/2 and Akt. Cancer Res. 2003;63:1969–74. [PubMed] [Google Scholar]
- Bauvois B. Transmembrane proteases in cell growth and invasion: new contributors to angiogenesis? Oncogene. 2004;23:317–29. doi: 10.1038/sj.onc.1207124. [DOI] [PubMed] [Google Scholar]
- Belmadani A, Tran PB, Ren D, Assimacopoulos S, Grove EA, Miller RJ. The chemokine stromal cell-derived factor-1 regulates the migration of sensory neuron progenitors. J Neurosci. 2005;25:3995–4003. doi: 10.1523/JNEUROSCI.4631-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenrieder T, Finstad CL, Freeman RH, Papandreou CN, Scher HI, Albino AP, et al. Expression and localization of aminopeptidase A, aminopeptidase N, and dipeptidyl peptidase IV in benign and malignant human prostate tissue. Prostate. 1997;33:225–32. doi: 10.1002/(sici)1097-0045(19971201)33:4<225::aid-pros1>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Bonfil RD, Chinni S, Fridman R, Kim HR, Cher ML. Proteases, growth factors, chemokines, and the microenvironment in prostate cancer bone metastasis. Urol Oncol. 2007;25:407–11. doi: 10.1016/j.urolonc.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–16. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–7. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- Carl-McGrath S, Lendeckel U, Ebert M, Rocken C. Ectopeptidases in tumour biology: a review. Histol Histopathol. 2006;21:1339–53. doi: 10.14670/HH-21.1339. [DOI] [PubMed] [Google Scholar]
- Castel V, Grau E, Noguera R, Martinez F. Molecular biology of neuroblastoma. Clin Transl Oncol. 2007;9:478–83. doi: 10.1007/s12094-007-0091-7. [DOI] [PubMed] [Google Scholar]
- Chinni SR, Sivalogan S, Dong Z, Filho JC, Deng X, Bonfil RD, et al. CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: the role of bone microenvironment-associated CXCL12. Prostate. 2006;66:32–48. doi: 10.1002/pros.20318. [DOI] [PubMed] [Google Scholar]
- Christopherson KW, 2nd, Hangoc G, Broxmeyer HE. Cell surface peptidase CD26/dipeptidylpeptidase IV regulates CXCL12/stromal cell-derived factor-1 alpha-mediated chemotaxis of human cord blood CD34+ progenitor cells. J Immunol. 2002;169:7000–8. doi: 10.4049/jimmunol.169.12.7000. [DOI] [PubMed] [Google Scholar]
- Dai J, Shen R, Sumitomo M, Goldberg JS, Geng Y, Navarro D, et al. Tumor-suppressive effects of neutral endopeptidase in androgen-independent prostate cancer cells. Clin Cancer Res. 2001;7:1370–7. [PubMed] [Google Scholar]
- Dinjens WN, Ten Kate J, Kirch JA, Tanke HJ, Van der Linden EP, Van den Ingh HF, et al. Adenosine deaminase complexing protein (ADCP) expression and metastatic potential in prostatic adenocarcinomas. J Pathol. 1990;160:195–201. doi: 10.1002/path.1711600303. [DOI] [PubMed] [Google Scholar]
- Dobers J, Grams S, Reutter W, Fan H. Roles of cysteines in rat dipeptidyl peptidase IV/CD26 in processing and proteolytic activity. Eur J Biochem. 2000;267:5093–100. doi: 10.1046/j.1432-1327.2000.01571.x. [DOI] [PubMed] [Google Scholar]
- Durinx C, Lambeir AM, Bosmans E, Falmagne JB, Berghmans R, Haemers A, et al. Molecular characterization of dipeptidyl peptidase activity in serum: soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-Pro dipeptides. Eur J Biochem. 2000;267:5608–13. doi: 10.1046/j.1432-1327.2000.01634.x. [DOI] [PubMed] [Google Scholar]
- Eggert A, Ikegaki N, Kwiatkowski J, Zhao H, Brodeur GM, Himelstein BP. High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin Cancer Res. 2000;6:1900–8. [PubMed] [Google Scholar]
- Fernandis AZ, Prasad A, Band H, Klosel R, Ganju RK. Regulation of CXCR4-mediated chemotaxis and chemoinvasion of breast cancer cells. Oncogene. 2004;23:157–67. doi: 10.1038/sj.onc.1206910. [DOI] [PubMed] [Google Scholar]
- Gabrilovac J, Abramić M, Uzarević B, Andreis A, Poljak L. Dipeptidyl peptidase IV (DPPIV) enzyme activity on immature T-cell line R1.1 is down-regulated by dynorphin-A (1-17) as a non-substrate inhibitor. Life sci. 2003;73(2):151–66. doi: 10.1016/s0024-3205(03)00257-1. [DOI] [PubMed] [Google Scholar]
- Geminder H, Sagi-Assif O, Goldberg L, Meshel T, Rechavi G, Witz IP, et al. A possible role for CXCR4 and its ligand, the CXC chemokine stromal cell-derived factor-1, in the development of bone marrow metastases in neuroblastoma. J Immunol. 2001;167:4747–57. doi: 10.4049/jimmunol.167.8.4747. [DOI] [PubMed] [Google Scholar]
- Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–15. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- Ghersi G, Dong H, Goldstein LA, Yeh Y, Hakkinen L, Larjava HS, et al. Regulation of fibroblast migration on collagenous matrix by a cell surface peptidase complex. J Biol Chem. 2002;277:29231–41. doi: 10.1074/jbc.M202770200. [DOI] [PubMed] [Google Scholar]
- Golubkov VS, Strongin AY. Proteolysis-driven oncogenesis. Cell Cycle. 2007;6:147–50. doi: 10.4161/cc.6.2.3706. [DOI] [PubMed] [Google Scholar]
- Guyon A, Nahon JL. Multiple actions of the chemokine stromal cell-derived factor-1alpha on neuronal activity. J Mol Endocrinol. 2007;38:365–76. doi: 10.1677/JME-06-0013. [DOI] [PubMed] [Google Scholar]
- Havre PA, Abe M, Urasaki Y, Ohnuma K, Morimoto C, Dang NH. The role of CD26/dipeptidyl peptidase IV in cancer. Front Biosci. 2008;13:1634–45. doi: 10.2741/2787. [DOI] [PubMed] [Google Scholar]
- Kajiyama H, Shibata K, Terauchi M, Ino K, Nawa A, Kikkawa F. Involvement of DPPIV/CD26 in epithelial morphology and suppressed invasive ability in ovarian carcinoma cells. Ann N Y Acad Sci. 2006;1086:233–40. doi: 10.1196/annals.1377.007. [DOI] [PubMed] [Google Scholar]
- Kenny AJ, O’Hare MJ, Gusterson BA. Cell-surface peptidases as modulators of growth and differentiation. Lancet. 1989;2:785–7. doi: 10.1016/s0140-6736(89)90841-6. [DOI] [PubMed] [Google Scholar]
- Lambeir AM, Proost P, Durinx C, Bal G, Senten K, Augustyns K, et al. Kinetic investigation of chemokine truncation by CD26/dipeptidyl peptidase IV reveals a striking selectivity within the chemokine family. J Biol Chem. 2001;276:29839–45. doi: 10.1074/jbc.M103106200. [DOI] [PubMed] [Google Scholar]
- Li Z, Thiele CJ. Targeting Akt to increase the sensitivity of neuroblastoma to chemotherapy: lessons learned from the brain-derived neurotrophic factor/TrkB signal transduction pathway. Expert Opin Ther Targets. 2007;11:1611–21. doi: 10.1517/14728222.11.12.1611. [DOI] [PubMed] [Google Scholar]
- Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- Maris JM, Woods WG. Screening for neuroblastoma: a resurrected idea? Lancet. 2008;371:1142–3. doi: 10.1016/S0140-6736(08)60500-0. [DOI] [PubMed] [Google Scholar]
- Matthay KK. Chemotherapy for neuroblastoma: does it hit the target? Lancet Oncol. 2008;9:195–6. doi: 10.1016/S1470-2045(08)70046-9. [DOI] [PubMed] [Google Scholar]
- Meitar D, Crawford SE, Rademaker AW, Cohn SL. Tumor angiogenesis correlates with metastatic disease, N-myc amplification, and poor outcome in human neuroblastoma. J Clin Oncol. 1996;14:405–14. doi: 10.1200/JCO.1996.14.2.405. [DOI] [PubMed] [Google Scholar]
- Mentlein R. Dipeptidyl-peptidase IV (CD26)--role in the inactivation of regulatory peptides. Regul Pept. 1999;85:9–24. doi: 10.1016/s0167-0115(99)00089-0. [DOI] [PubMed] [Google Scholar]
- Morrison ME, Vijayasaradhi S, Engelstein D, Albino AP, Houghton AN. A marker for neoplastic progression of human melanocytes is a cell surface ectopeptidase. J Exp Med. 1993;177:1135–43. doi: 10.1084/jem.177.4.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawara A. Neural crest development and neuroblastoma: the genetic and biological link. Prog Brain Res. 2004;146:233–42. doi: 10.1016/s0079-6123(03)46015-9. [DOI] [PubMed] [Google Scholar]
- Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14:759–67. doi: 10.1128/mcb.14.1.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanus DM. Of peptides and peptidases: the role of cell surface peptidases in cancer. Clin Cancer Res. 2003;9:6307–9. [PubMed] [Google Scholar]
- Nevo I, Sagi-Assif O, Meshel T, Geminder H, Goldberg-Bittman L, Ben-Menachem S, et al. The tumor microenvironment: CXCR4 is associated with distinct protein expression patterns in neuroblastoma cells. Immunol Lett. 2004;92:163–9. doi: 10.1016/j.imlet.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Payne AS, Cornelius LA. The role of chemokines in melanoma tumor growth and metastasis. J Invest Dermatol. 2002;118:915–22. doi: 10.1046/j.1523-1747.2002.01725.x. [DOI] [PubMed] [Google Scholar]
- Pethiyagoda CL, Welch DR, Fleming TP. Dipeptidyl peptidase IV (DPPIV) inhibits cellular invasion of melanoma cells. Clin Exp Metastasis. 2000;18:391–400. doi: 10.1023/a:1010930918055. [DOI] [PubMed] [Google Scholar]
- Piazza GA, Callanan HM, Mowery J, Hixson DC. Evidence for a role of dipeptidyl peptidase IV in fibronectin-mediated interactions of hepatocytes with extracellular matrix. Biochem J. 1989;262:327–34. doi: 10.1042/bj2620327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proost P, Struyf S, Schols D, Durinx C, Wuyts A, Lenaerts JP, et al. Processing by CD26/dipeptidyl-peptidase IV reduces the chemotactic and anti-HIV-1 activity of stromal-cell-derived factor-1alpha. FEBS Lett. 1998;432:73–6. doi: 10.1016/s0014-5793(98)00830-8. [DOI] [PubMed] [Google Scholar]
- Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Lett. 2007;256:137–65. doi: 10.1016/j.canlet.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Chan HM, Fan J, Xie Y, Chen YX, Li W, et al. Inhibition of tumor growth and metastasis in vitro and in vivo by targeting macrophage migration inhibitory factor in human neuroblastoma. Oncogene. 2006;25:3501–8. doi: 10.1038/sj.onc.1209395. [DOI] [PubMed] [Google Scholar]
- Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. 2005;9:267–85. doi: 10.1111/j.1582-4934.2005.tb00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell HV, Hicks J, Okcu MF, Nuchtern JG. CXCR4 expression in neuroblastoma primary tumors is associated with clinical presentation of bone and bone marrow metastases. J Pediatr Surg. 2004;39:1506–11. doi: 10.1016/j.jpedsurg.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Sakamoto J, Watanabe T, Teramukai S, Akiyama S, Morimoto T, Takagi H, et al. Distribution of adenosine deaminase binding protein in normal and malignant tissues of the gastrointestinal tract studied by monoclonal antibodies. J Surg Oncol. 1993;52:124–34. doi: 10.1002/jso.2930520214. [DOI] [PubMed] [Google Scholar]
- Scala S, Giuliano P, Ascierto PA, Ierano C, Franco R, Napolitano M, et al. Human melanoma metastases express functional CXCR4. Clin Cancer Res. 2006;12:2427–33. doi: 10.1158/1078-0432.CCR-05-1940. [DOI] [PubMed] [Google Scholar]
- Scharpe S, De Meester I. Peptide truncation by dipeptidyl peptidase IV: a new pathway for drug discovery? Verh K Acad Geneeskd Belg. 2001;63:5–32. discussion 32-3. [PubMed] [Google Scholar]
- Schrader WP, West CA, Strominger NL. Localization of adenosine deaminase and adenosine deaminase complexing protein in rabbit brain. J Histochem Cytochem. 1987;35:443–51. doi: 10.1177/35.4.3546489. [DOI] [PubMed] [Google Scholar]
- Sedo A, Kraml J. Dipeptidyl peptidase IV in cell proliferation and differentiation. Sb Lek. 1994;95:285–88. [PubMed] [Google Scholar]
- Shioda T, Kato H, Ohnishi Y, Tashiro K, Ikegawa M, Nakayama EE, et al. Anti-HIV-1 and chemotactic activities of human stromal cell-derived factor 1alpha (SDF-1alpha) and SDF-1beta are abolished by CD26/dipeptidyl peptidase IV-mediated cleavage. Proc Natl Acad Sci U S A. 1998;95:6331–6. doi: 10.1073/pnas.95.11.6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Sadanandam A, Singh RK. Chemokines in tumor angiogenesis and metastasis. Cancer Metastasis Rev. 2007;26:453–67. doi: 10.1007/s10555-007-9068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Bai Z, Srinoulprasert Y, Yang BG, Hayasaka H, Miyasaka M. Chemokines in tumor progression and metastasis. Cancer Sci. 2005;96:317–22. doi: 10.1111/j.1349-7006.2005.00059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi M, Kajiyama H, Shibata K, Ino K, Mizutani S, Kikkawa F. Anti-progressive effect of neutral endopeptidase 24.11 (NEP/CD10) on cervical carcinoma in vitro and in vivo. Oncology. 2005;69:52–62. doi: 10.1159/000087476. [DOI] [PubMed] [Google Scholar]
- Tran PB, Ren D, Veldhouse TJ, Miller RJ. Chemokine receptors are expressed widely by embryonic and adult neural progenitor cells. J Neurosci Res. 2004;76:20–34. doi: 10.1002/jnr.20001. [DOI] [PubMed] [Google Scholar]
- Tsuji T, Sugahara K, Tsuruda K, Uemura A, Harasawa H, Hasegawa H, et al. Clinical and oncologic implications in epigenetic down-regulation of CD26/dipeptidyl peptidase IV in adult T-cell leukemia cells. Int J Hematol. 2004;80:254–60. doi: 10.1532/ijh97.04066. [DOI] [PubMed] [Google Scholar]
- Tucker RP. Neural crest cells: a model for invasive behavior. Int J Biochem Cell Biol. 2004;36:173–7. doi: 10.1016/s1357-2725(03)00243-7. [DOI] [PubMed] [Google Scholar]
- Vasudevan SA, Nuchtern JG, Shohet JM. Gene profiling of high risk neuroblastoma. World J Surg. 2005;29:317–24. doi: 10.1007/s00268-004-7820-7. [DOI] [PubMed] [Google Scholar]
- Wesley UV, Albino AP, Tiwari S, Houghton AN. A role for dipeptidyl peptidase IV in suppressing the malignant phenotype of melanocytic cells. J Exp Med. 1999;190:311–22. doi: 10.1084/jem.190.3.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley UV, McGroarty M, Homoyouni A. Dipeptidyl peptidase inhibits malignant phenotype of prostate cancer cells by blocking basic fibroblast growth factor signaling pathway. Cancer Res. 2005;65:1325–34. doi: 10.1158/0008-5472.CAN-04-1852. [DOI] [PubMed] [Google Scholar]
- Wesley UV, Tiwari S, Houghton AN. Role for dipeptidyl peptidase IV in tumor suppression of human non small cell lung carcinoma cells. Int J Cancer. 2004;109:855–66. doi: 10.1002/ijc.20091. [DOI] [PubMed] [Google Scholar]
- Zigrino P, Loffek S, Mauch C. Tumor-stroma interactions: their role in the control of tumor cell invasion. Biochimie. 2005;87:321–8. doi: 10.1016/j.biochi.2004.10.025. [DOI] [PubMed] [Google Scholar]