

Abstract
Summary
The molecular chaperone Heat Shock Protein 90 (Hsp90) is required for the folding and activation of numerous essential signaling proteins. Hsp90 is generally thought to transition between an open, apo and a closed, ‘ATP’ conformation in response to nucleotide. Here, 3D single particle reconstructions of E. coli and yeast Hsp90 homologs establish the existence of two distinct nucleotide-stabilized conformations (ATP, ADP) in addition to an apo extended state, supporting previous structural work. However, single particle matching methods reveal that, rather than being irreversibly determined by nucleotide, a species-dependent dynamic conformational equilibrium exists between states. Using crosslinking methods we trap transient nucleotide-specific states of yeast and human Hsp90 and establish that the apo, ATP and ADP states are universal. These data support a conserved three-state chaperone cycle where the conformational equilibrium varies between species, implicating evolutionary tuning to meet the particular client protein and metabolic environment of an organism.
Keywords: Hsp90, molecular chaperone, protein folding, electron microscopy
Introduction
The diverse class of proteins known as molecular chaperones plays essential roles in maintaining the population of correctly folded proteins in the cell. This generally occurs by recognition of exposed hydrophobic regions on the unfolded substrates and repeated rounds of ATP-dependent binding and release (Hartl and Hayer-Hartl, 2002; Young et al., 2004). Well-described chaperones include the heat shock proteins 70 (Hsp70) (Zhu et al., 1996) and 60 (GroEL) (Martin et al., 1991) which act early in the folding process – stabilizing nascent polypeptide chains and facilitating the folding of relatively unstructured intermediates (Bukau and Horwich, 1998). By contrast, the abundant heat shock protein 90 (Hsp90) family preferentially interacts with its substrate (client) proteins late in the folding pathway, facilitating essential protein-protein and protein-ligand interactions (Nathan et al., 1997; Picard, 2002; Zhao et al., 2005). Despite characterization of numerous client proteins that require Hsp90-mediated activation, much of the conformational rearrangements, protein interaction sites and nucleotide-dependence of the chaperone process remain unclear.
Hsp90 is highly conserved from E. coli (HtpG) to yeast (Hsc82 and Hsp82 isoforms) to humans (Hsp90α and Hsp90β isoforms). While client proteins have not been definitively identified for HtpG, in higher organisms Hsp90 activity is essential for the activation of many proteins including steroid hormone nuclear receptors (Picard et al., 1990; Pratt et al., 2004), protein kinases (Richter and Buchner, 2001; Sato et al., 2000), nitric oxide synthase (Garcia-Cardena et al., 1998), and telomerase (Holt et al., 1999). For eukaryotes, both client-specific and general co-chaperones are required during the chaperone process (Felts and Toft, 2003; Johnson et al., 1998). Hsp90 specific inhibitors, including geldanamycin and its derivatives, promote the inactivation and degradation of numerous client oncoproteins and cell cycle kinases (Neckers, 2002; Roe et al., 1999).
Hsp90 contains three distinct domains – a C-terminal, high-affinity dimerization domain (CTD), a middle domain (MD) important for substrate maturation, and an N-terminal ATPase domain (NTD). Crystal structures of the ATPase domain identified it as a member of the GHKL family that includes DNA gyrase and topoisomerase II (Dutta and Inouye, 2000; Prodromou et al., 1997). From comparisons of these family members and biochemical studies, it has been proposed that the energy from nucleotide binding triggers a conformational switch where the NTDs dimerize to form a closed state that is required both for hydrolysis and client protein activation (Ban et al., 1999; Prodromou et al., 2000). While distinct client binding sites have yet to be identified, regions of significant exposed hydrophobic surface area within each domain indicate potential interaction sites (Shiau et al., 2006; Harris et al., 2004; Meyer et al., 2003). Notably, a negative-stain electron microscopy (EM) reconstruction of a yeast Hsp90:Cdc37:Cdk4 complex shows the Cdk4 kinase client interacting with a single monomer near the NTD, suggesting a more asymmetric mode of client interaction (Vaughan et al., 2006).
Crystal structures of intact Hsp90 homologs from yeast (Ali et al., 2006), E. coli (Shiau et al., 2006) and mammalian endoplasmic reticulum (ER) (Dollins et al., 2007) reveal dramatically different conformations that result from large rigid-body movements of the domains. The structure of apo HtpG identified an open conformation with a large CTD-MD angle and an NTD arrangement that results in an 80 Å separation of N-terminal residues. The Grp94 structures have essentially the same NTD conformation, but a different open, CTD-MD angle. The crystal structure of yeast Hsp82 bound to AMPPNP (nonydrolyzable ATP analog) and in a complex with the p23 co-chaperone identified a closed, ‘ATP’ conformation, supporting models from previous biochemical work (Prodromou et al., 2000). This closed conformation has a reduced CTD-MD angle compared to either apo HtpG or Grp94 and a 90° rotation of the NTD resulting in dimerization of the NTDs. For HtpG, ADP binding also resulted in a significant conformational change, yielding a unique tetramer in the crystallographic asymmetric unit. The packing arrangement along with 2D EM data led to a model for a compact, ‘ADP’ dimer that retained the large buried surface area (3700Å2 per monomer) observed in the crystal form.
Recent solution small angle x-ray scattering (SAXS) experiments with HtpG have identified additional complexity by modeling an apo conformation that is more extended than the crystal form and a mixture of open and closed states in the presence of AMPPNP, but no changes were seen with ADP compared to apo (Krukenberg et al., 2008). Cryo-EM and SAXS studies of human Hsp90 showed two different apo conformations (Bron et al., 2008), while no significant conformational changes have been observed in the presence of nucleotide (Zhang et al., 2004). Thus, while previous structural work illustrates substantial domain rearrangements of Hsp90, critical questions remain about the relevant conformational states, including the nucleotide dependence, the extent to which they are populated during the chaperone cycle and the degree of conservation across species.
Although challenging because of the small size of Hsp90, single particle electron microscopy (EM) is particularly well suited to address these questions because it provides direct visualization of structural states unconstrained by crystal lattice contacts better reflecting the solution state of Hsp90. In the following work we investigate the nucleotide-dependent conformational states of E. coli, yeast and human Hsp90s. We provide 3D reconstructions of HtpG:AMPPNP and HtpG:ADP that support a model for the hydrolysis cycle involving three distinct states: an extended apo state, a closed ATP state and a compact ADP state. Our reconstruction of yeast Hsp90 (Hsc82):AMPPNP establishes that nucleotide binding alone drives NTD dimerization. Detailed comparison with the crystal structure suggests an altered NTD-MD conformation in the absence of p23 that reflects an active state on-pathway to hydrolysis. Through crosslinking, we were able to trap transiently populated states and identify for the first time the closed ATP conformation in human Hsp90 and the compact ADP conformation in both human and yeast Hsp90s.
Finally, our work reveals that the Hsp90 nucleotide cycle operates quite differently than related GHKL family members where nucleotide binding triggers a discrete conformational change. Analysis of single particle images reveals that the Hsp90 conformational states are not deterministic; rather, the different states coexist in a dynamic equilibrium. Nucleotide binding is found to provide only modest stabilizing energy, biasing the equilibrium towards, but not dictating the conformational state. This work supports a model involving a universal three-state Hsp90 conformational cycle, where the equilibrium between states is species-dependent and thus evolutionarily optimized to meet the demands and influences of different client proteins and co-chaperones found in each organism.
Results
Species-dependent occupancy of open (apo), closed (ATP), and compact (ADP) conformations of Hsp90
Previous structural studies identified dramatically different conformational states of Hsp90 but raised critical questions about the nucleotide-dependence and the conservation of theses states between species. Therefore we sought to determine how nucleotide binding alone affects the conformational state of E. coli, yeast and human Hsp90s using negative stain EM. Recombinantly expressed and purified HtpG (E. coli), Hsc82 (yeast), and Hsp90α (human) were incubated in the presence or absence of saturating concentrations of AMPPNP and ADP. From the example particles (Figure 1) and micrographs (Figure S1, S2, S3) it is clear that HtpG adopts three distinct nucleotide dependent conformations, Hsc82 adopts two conformations, while for Hsp90α no significant changes are observed. In the absence of nucleotide HtpG, Hsc82 and Hsp90α adopt a heterogeneous ensemble of open and extended conformations with an overall shape that is consistent with previous studies (Krukenberg et al., 2008; Bron et al., 2008). Interestingly, in micrographs of apo HtpG other low-population conformations that appear more compact in shape can be identified. These are reminiscent of the proposed nucleotide-bound conformations (see below).
Figure 1.
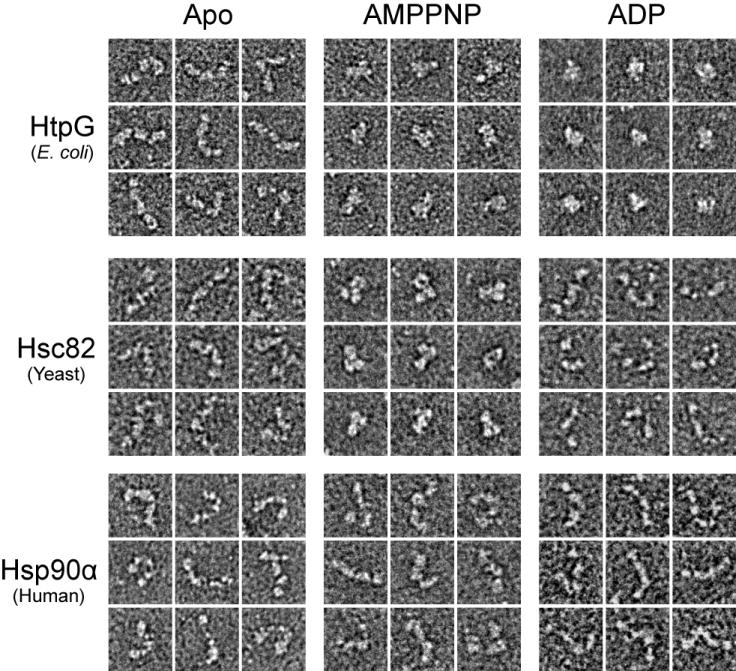
Three Nucleotide-Stabilized Conformational States of Hsp90 Vary Between Species. Typical 2D negative stain single particle views of E. coli HtpG, yeast Hsc82 and human Hsp90α are shown following incubations in the absence (apo) or presence of AMPPNP and ADP. Box sizes equal 330 Å.
In the presence of AMPPNP, both HtpG and Hsc82 adopt a significantly different, closed conformation that appears to match the yeast Hsp82:AMPPNP:p23 crystal structure (Ali et al., 2006). From the micrographs (Figure S1, S2), a mixture of open and closed particles is observed for HtpG but for Hsc82 a significantly greater fraction appear closed. Conversely, when Hsp90α is incubated with AMPPNP no conformational changes are observed (Figure 1, S3). Incubations with ATP and ATPγS also resulted in no apparent changes in Hsp90α conformation (data not shown). Experiments performed with the Hsp90β isoform gave identical results to Hsp90α (data not shown). Finally, in the presence of saturating amounts of ADP, HtpG adopts a third, highly compact conformation that differs significantly from apo HtgG and HtpG:AMPPNP. These HtpG:ADP particles look more globular and similar to the compact dimer conformation modeled from the HtpG:ADP crystal structure. By contrast, no significant conformational changes are observed when either Hsc82 or Hsp90α are incubated with ADP.
These results establish that the extended, apo conformation is conserved, that AMPPNP binding alone stabilizes a closed conformation in HtpG and Hsc82, and that ADP binding results in an alternate, highly compact conformation in HtpG. What is unclear from these 2D views is how similar the closed, AMPPNP-bound conformations are to the Hsp82:AMPPNP:p23 crystal structure and whether the HtpG:ADP particles closely match the proposed ADP model. Finally, the observation that no conformational changes were seen with Hsp90α:AMPPNP, Hsp90α:ADP and Hsc82:ADP indicates that there are fundamental species-dependent differences and suggests the possibility that either these states are not universal or that they are only transiently sampled.
3D EM reconstructions of HtpG:AMPPNP and HtpG:ADP identify distinct closed and compact conformations
Our 2D images of HtpG:AMPPNP and HtpG:ADP are strikingly different than apo HtpG, revealing significant nucleotide-dependent conformational changes. However, detailed 3D analysis was required in order to properly distinguish these conformations and make comparisons to the observed Hsp82:AMPPNP:p23 and modeled HtpG:ADP structures. Therefore, we performed 3D single particle reconstructions. An initial set of HtpG:AMPPNP particles were collected for reference-free class averaging (Figure 2A, S4A). Given the two-fold symmetry of the crystal structures of Hsp90, the same symmetry was imposed for the initial model (Figure S4B) and during the refinement. The resolution of the final model was estimated to be 22 Å using the Fourier Shell Correlation method (FSC = 0.5 for all reconstructions) (Figure S6A).
Figure 2. Three-Dimensional Reconstruction of HtpG:AMPPNP.
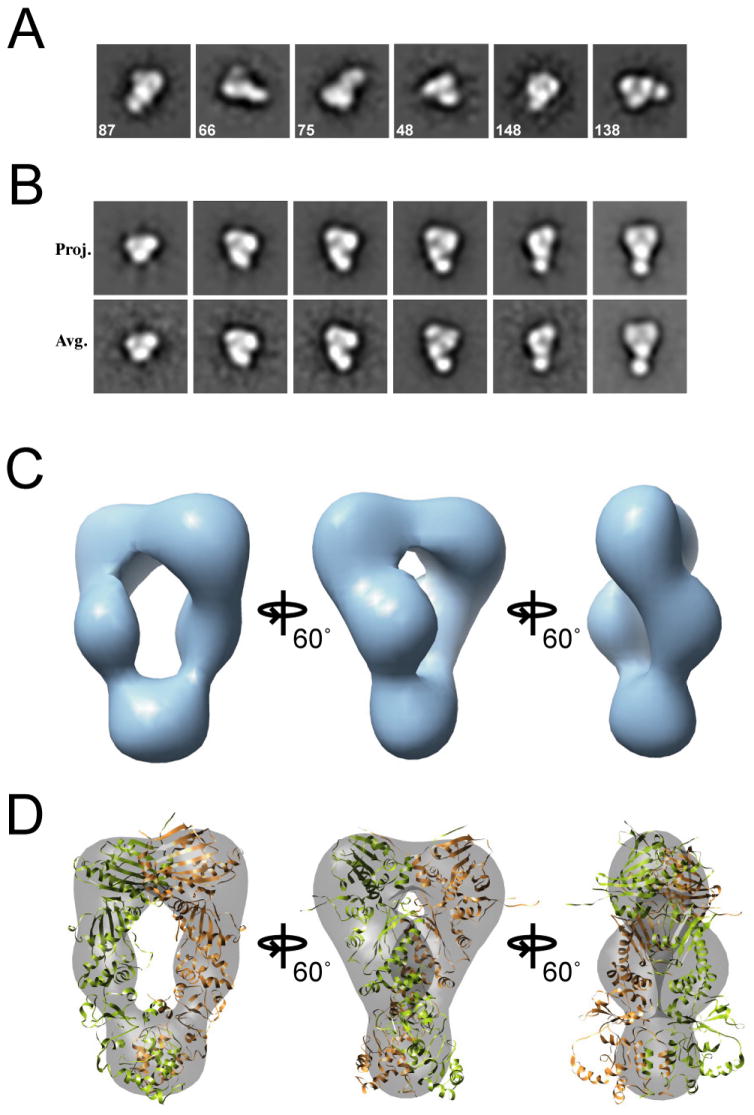
Reference-free class averages are shown (A), with the number of single particles used to generate the averages listed on each image. A set of projections and class averages for the final model (B) are shown. Box sizes equal to 250 Å. In (C) the final HtpG:AMPPNP 3D map is shown and in (D) the yeast Hsp90 structure (Ali et al., 2006) was fit into the EM map. The monomers of the homodimer are colored green and brown.
In the HtpG:AMPPNP reconstruction, two large globular domains, predicted to be the NTDs, appear dimerized along the inside surface and separate into extended middle domains, then re-connect in a smaller globular domain (Figure 2B,C). A surprisingly large cavity (roughly 55 by 15 Å) that was not apparent from the reference-free averages separates the two middle domains. The yeast Hsp82:AMPPNP:p23 crystal structure (Figure 2D) and a model based on the HtpG sequence (data not shown) both fit well into the 3D reconstruction. The fit of the structures confirms the proposed architecture for the HtpG:AMPPNP reconstruction and identifies a closed conformation indistinguishable from the yeast Hsp82:AMPPNP:p23 structure. Regions that are outside the density (when contoured to the molecular weight) include flexible loops at the CTD-MD junction and the flexible amphipathic helices in the CTD. Importantly, this 3D reconstruction of HtpG:AMPPNP establishes that the closed ATP conformation is conserved in E. coli and that nucleotide binding alone is sufficient to drive a large-scale rearrangement resulting in NTD dimerization.
Initial 2D views of HtpG:ADP reveal a distinct compact conformation supporting our previous work. Reference free class averages confirmed the globular shape (typical dimensions of 60 by 95 Å) (Figure 3A). From the averages an initial model was obtained using the cross common lines method with two-fold symmetry imposed (Figure S4C,D). The 2D projections of the final model and the corresponding class averages match well and present an overall compact trapezoid shape (Figure 3B). The resolution of the final model was estimated to be 23 Å (Figure S6B).
Figure 3.
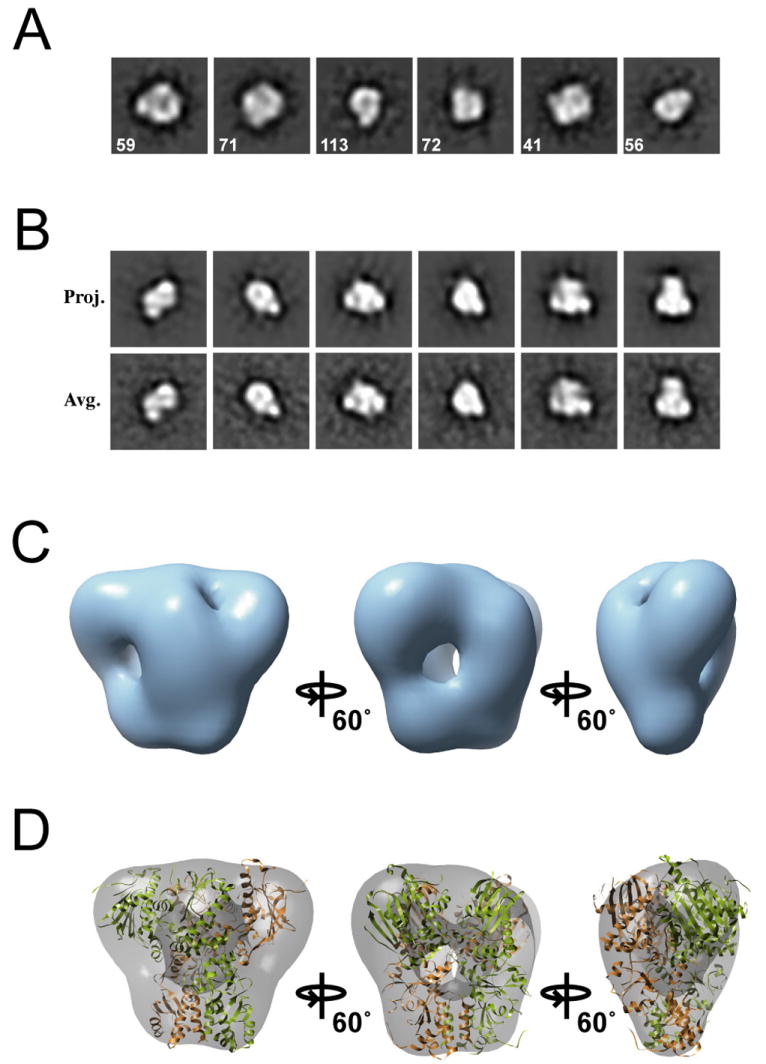
Three-Dimensional Reconstruction of HtpG:ADP. Reference-free class averages (A) and example projections with corresponding averages for the final model (B) are shown. The box sizes are equal to 210 Å. The final HtpG:ADP 3D map (C) and the compact HtpG structure model (Shiau et al., 2006) fit into the EM map (D) are shown, identifying the compact conformation.
Our reconstruction illustrates that HtpG:ADP has adopted a conformation that is much more compact than HtpG:AMPPNP (Figure 3C). Higher density regions appear to be the NTDs that have rotated down, making alternate inter-monomer contacts. The HtpG:ADP compact model predicted from our tetramer Htpg:ADP crystal structure (Shiau et al., 2006) fits with significant accuracy into the EM volume (Figure 3D). The overall shape of the EM map and the docked model indicate that these structures are essentially identical, establishing the existence of a compact ADP-dependent conformation that is dramatically different than the ATP-bound state for Hsp90.
3D EM reconstruction of yeast Hsc82:AMPPNP establishes nucleotide-driven NTD dimerization
2D images of Hsc82:AMPPNP established that nucleotide-binding alone promotes a closed conformation in yeast Hsp90 (Figure 1). To ascertain the role of p23 in determining the closed conformation, it was necessary to obtain a complete single particle 3D reconstruction and compare it to the Hsp82:AMPPNP:p23 crystal structure. Reference-free class averages were generated from an initial data set and clearly resemble the HtpG:AMPPNP averages (compare Figure 4A and Figure 2A). Therefore, the HtpG:AMPPNP initial model (Figure S4A,B) was used for the Hsc82:AMPPNP reconstruction. A set of projections and corresponding class averages of the final round of refinement are shown (Figure S5A) and the resolution was estimated to be 24 Å (Figure S6C). The reconstruction is quite similar to the Hsp82:AMPPNP:p23 structure, clearly demonstrating the dimerized, globular NTDs, an extended MD, and a CTD that is better resolved than for the HtpG reconstruction (Figure 4B). As expected, the Hsp82:AMPPNP:p23 crystal structure fits extremely well in the density. This fit is an improvement over that found for the HtpG:AMPPNP reconstruction, with a larger fraction of atoms fitting within the EM map: 0.72 compared to 0.66 for HtpG:AMPPNP. Interestingly, the span of density between the NTDs is reduced compared to the crystal structure, indicated by portions of the NTD and middle domains that are outside the EM volume at the appropriate contour.
Figure 4. Three-Dimensional Reconstruction of Hsc82:AMPPNP.
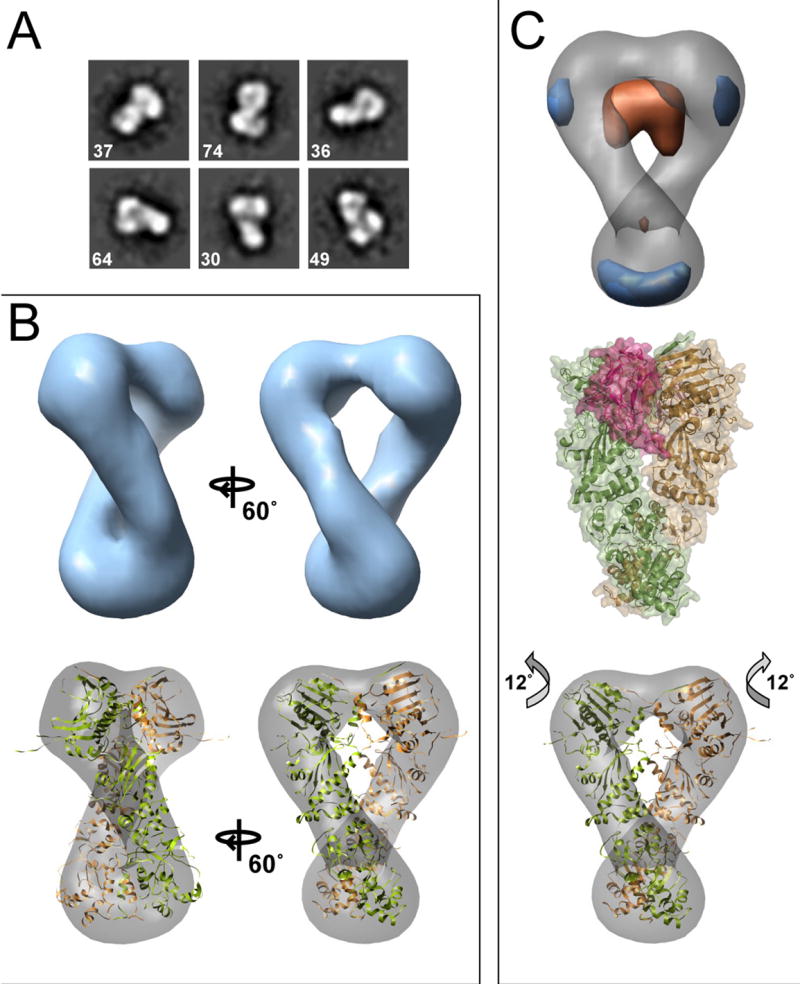
Reference-free class averages are shown in (A). In (B) the final Hsc82:AMPPNP reconstruction is shown along with the Hsp82:AMPPNP:p23 crystal structure docked into the map. In (C) a difference map (experimental - calculated) is shown aligned to the yeast Hsc82:AMPPNP reconstruction. In orange is density present in the crystal structure but missing in the EM volume, and blue is additional EM map density. For comparison, the yeast Hsp82:AMPPNP:p23 complex is shown (C, middle panel). Two p23 molecules (raspberry) flank the NTD dimerization interface. An improved, rigid-body docking of the domains into the EM map (C, lower panel) highlights an alternate closed conformation with a 12° rotation of the NTD.
To better address the potential for an altered NTD-middle domain arrangement, we calculated a difference map of the aligned Hsp82:AMPPNP crystal structure and our 3D reconstruction (Figure 4C). This facilitates the identification of non-overlapping regions, indicating a different arrangement between the crystal structure and our reconstruction. In the difference map (experimental – calculated), positive density (blue, Figure 4C, upper panel) is present in several small regions near the CTD. Interestingly, significant negative density (orange), representing regions of the crystal structure that are not present in the reconstruction, spans area between the monomers at the base of the NTDs. From the structure of the Hsp82:AMPPNP:p23 complex it is clear that p23 interacts directly in this region (Figure 4C, middle panel), likely providing additional stabilizing interactions along the NTD dimerization interface. As a control we performed a reconstruction with the total data set using the crystal structure volume as the initial model (low-pass filtered to 30 Å). After 8 rounds of refinement the data converged to a model identical to when the common-lines generated model was used, eliminating model bias in our analysis and further substantiating the differences between our final model and crystal structure (data not shown).
We performed rigid body fitting of flexible domains to determine if an altered NTD-MD arrangement could better describe the Hsc82:AMPPNP reconstruction. This method significantly improved the fit; the fraction of atoms within the EM density increased from 0.72 to 0.74 and the cross-correlation value improved from 0.60 to 0.63 (Figure 4C lower panel). The primary structural change was a rotation of 12 degrees about the NTD-MD interface, extending the NTDs slightly up and outward with an overall untwisting of the dimer. Some MD density still remains outside the map, but fits appropriately at a lower contour threshold (Figure S5B). Thus, our improved fit presents a plausible alternate arrangement of the closed yeast Hsc82:AMPPNP dimer, one where p23 is not providing additional stabilizing interactions.
EM analysis of the equilibrium of Hsp90 conformational states
During ATP binding, Hsp90 is generally thought to convert from an open to closed conformation through a two-state cycle. However, our recent SAXS studies demonstrated more nuanced behavior in solution: while apo HtpG primarily exists in an open and extended state, in the presence of AMPPNP only a portion of the population shifts to a closed conformation (Krukenberg et al., 2008). By providing direct views of individual single particles, EM images allow us to better determine relative populations of the conformations in different nucleotide states. This in turn yields insight into how nucleotide binding energy is utilized to drive a population of molecules to an alternate conformation.
As discussed above, HtpG primarily adopts an open, extended arrangement in the absence of nucleotide (Figure 1). However, additional closed conformations were also observed at a low frequency. Further analysis reveals that these closed particles match quite well with the closed ATP and compact ADP forms of HtpG (Figure 5A,B). This is striking because it indicates that in the absence of the binding energy from AMPPNP or ADP HtpG is able to sample these alternate conformations. Thus, these data suggest that in the absence of nucleotide, HtpG adopts an ensemble of states that reflect the complete nucleotide cycle.
Figure 5. Single Particle Analysis Identifies and Equilibrium Between the Different Conformational States.
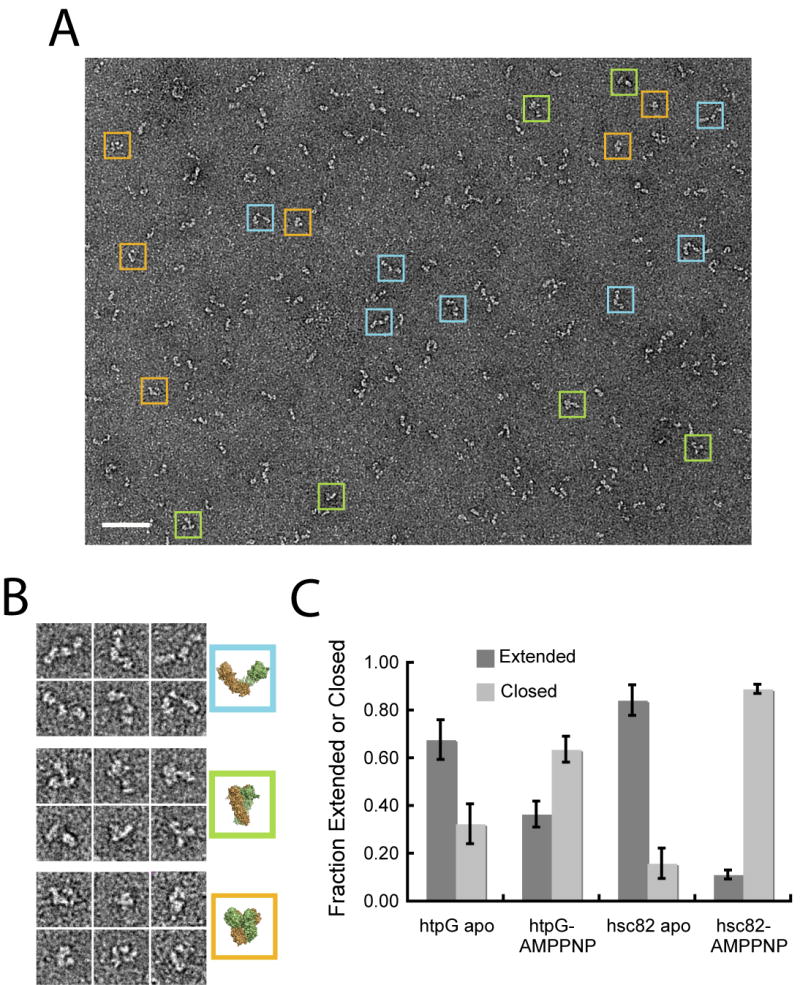
In (A) is a typical micrograph showing multiple conformations of apo HtpG. Extended (blue), closed ATP-like (green) and compact ADP-like (orange) particles are boxed and enlarged in (B) along with comparable views of the corresponding structures. Scale bar equals 500 Å and the box sizes are 330 Å. For (C) a set of single particles of HtpG or Hsc82 in the absence or presence of AMPPNP were collected and matched to a set of unique 2D projections of extended apo and closed ATP structures in order to determine the fraction of extended (dark gray) and closed (light gray) conformations. Error bars shown represent the standard deviation of three independent experiments.
Ideally we would like to quantitatively explore species-dependent differences in the Hsp90 conformational equilibrium. However, because there were no visually discernable differences between Hsc82:ADP, Hsp90α:AMPPNP and Hsp90a:ADP and their respective apo states, we chose to focus on HtpG and Hsc82 and determine the conformational equilibrium of their extended apo and closed ATP states. A multireference-based approach was used in which single particles of HtpG and Hsc82 were matched to either the open or closed conformation. 2D projections of the apo HtpG and closed Hsp82:AMPPNP models were generated and first aligned to each other in order to determine, based on a correlation value, similar and distinct views. Single particle data sets of apo and AMPPNP-bound HtpG and Hsc82 were then collected and scored against the total set of references. To avoid ambiguous views, only particle images that showed a distinct preference for either the open or the closed model were used in calculating the fraction of the open and closed states.
Overall, the data reveal two main points (Figure 5C): 1) both HtpG and Hsc82 exhibit an incomplete shift to the closed conformation in the presence of nucleotide; and 2) the conformational equilibrium is significantly different between the two Hsp90s, with a more significant population shift from the open to closed state for Hsc82. For HtpG, in the absence of nucleotide, 68% of the particles match the extended conformation, while 32% match the closed ATP conformation. This essentially reverses to a 33% open and 67% closed when AMPPNP is present (Figure 8A). The data for Hsc82 show that vast majority of the particles (82% and 84% respectively) match either the open conformation in the absence of nucleotide or the closed conformation when AMPPNP is present, indicating a more substantial equilibrium shift to the closed state.
Crosslinking establishes that the ATP and ADP conformational states are universal to Hsp90
Despite significant sequence and structural conservation, the closed conformation has not been observed with human Hsp90. In fact, previous studies have suggested that for human Hsp90 the NTDs act independently, possibly not dimerizing during the nucleotide cycle (McLaughlin et al., 2004). Furthermore, our results here failed to reveal any significant conformational changes in human Hsp90α in the presence of saturating amounts of AMPPNP (Figure 1). However, recent kinetic studies support a conserved ATPase cycle and show evidence for NTD dimerization in human Hsp90 (Frey et al., 2007; Richter et al., 2008). Given these conflicting studies and our observation that HtpG and Hsc82 have dramatically different conformational equilibria, we hypothesized that while a closed conformation can exist for human Hsp90:ATP, the conformational equilibrium must be strongly shifted towards the open state. Thus, in the presence of nucleotide only a small fraction of human Hsp90 would exist in the closed conformation.
In order to trap rarely populated conformational states we crosslinked Hsp90α by treating samples with a small amount of glutaraldehyde (0.005%) in the presence or absence of nucleotide before visualization. Remarkably, when Hsp90α is crosslinked following incubation with AMPPNP, numerous closed particles predominate and the class averages appear to be identical to the HtpG and Hsc82 closed conformation (Figure 6A and S7C). Incubation with glutaraldehyde in the absence of nucleotide does not produce significant numbers of closed particles, indicating that this closed state is nucleotide-dependent (Figure S7A,B). Interestingly, the apo crosslinked particles appear circular in shape, with a small NM angle and an open MC angle, resulting in a conformation that is distinct from the ATP and ADP states. This conformation is comparable to the ‘semi-open’ arrangement of mammalian apo Hsp90 identified in recent cryo-EM studies (Bron et al., 2008). A low population (< 5%) of closed Hsp90α ATP-like conformations was observed in the apo crosslinked data, suggesting that the closed state is also sampled at a very low level in the absence of nucleotide (data not shown). Importantly, these data reveal that although human Hsp90α exists predominantly in the open conformation in the presence of AMPPNP, the closed ATP state is clearly sampled in a nucleotide-dependent manner. This is the first structural evidence that human Hsp90α forms the closed state and supports a conserved conformational change during the nucleotide cycle.
Figure 6. Crosslinking Reveals the Closed ATP and compact ADP Conformations in Human and Yeast Hsp90.
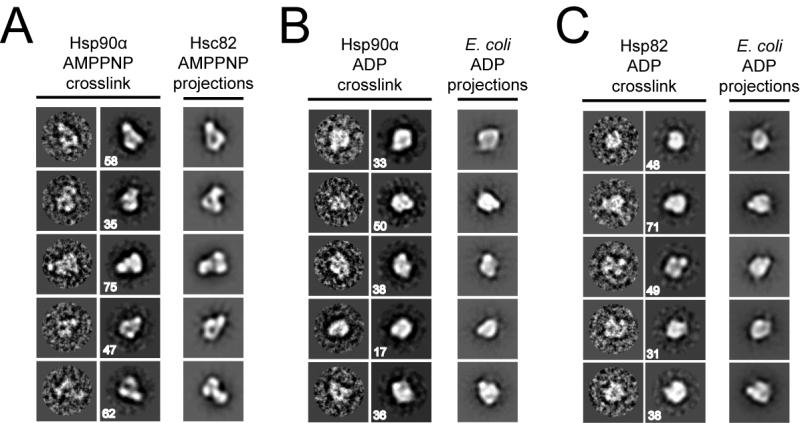
In (A) Hsp90α was incubated with AMPPNP and 0.005% glutaraldehyde crosslinker prior to negative staining. Aligned single images, reference free class averages and aligned 2D projections of the Hsc82:AMPPNP reconstruction are shown (right to left). Box sizes are 250 Å. Hsp90α (B) and yeast Hsc82 (C) incubated with ADP and crosslinker are shown as in (A) and compared to the HtpG:ADP reconstruction.
Given the success of this method in revealing the closed conformation in Hsp90α, crosslinking was attempted in the presence of ADP. Hsp90α was incubated with saturating amounts of ADP along with low concentrations of glutaraldehyde as before, followed by negative staining. From the micrographs and class averages Hsp90α:ADP appears highly compact and identical to projections of the HtpG:ADP reconstruction (Figure 6B, S7D). Next, we crosslinked yeast Hsc82 in the presence of ADP and strikingly, Hsc82 also appears highly compact and identical to HtpG:ADP (Figure 6C, S8B). As a control, Hsc82 alone was also incubated in the presence of glutaraldehyde and no significant differences were observed compared to the absence of crosslinker (Figure S8A). These results identify that, as with HtpG, both human Hsp90α and yeast Hsc82 transiently form a compact conformation in an ADP dependent manner. The nucleotide dependence clearly establishes that the compact ADP conformation is a distinct and well-conserved conformational state that occurs during the hydrolysis cycle of Hsp90 and, as with the ATP state, the conformational equilibrium is species-dependent.
Discussion
Recent full-length crystal structures of different Hsp90 homologs have revealed identical domain architecture but dramatically different conformational states. Coupled to these conformations, the nucleotide cycle of Hsp90 has been thought to be analogous to other related GHKL family members, where ATP binding drives a discreet conformational switch from an open to closed state. The differences in the structures, the speculative nature of the HtpG ADP state, and the failure to observe some conformational states have raised significant questions including the conservation between species and the degree to which nucleotide stabilizes the different states. Here we have investigated the apo and nucleotide-bound conformations of the E. coli, yeast and human Hsp90 homologs using single particle EM methods. Our results reveal a unique conformational cycle for Hsp90, involving at least three dramatically different states that exist in equilibrium. Unexpectedly, we find that nucleotide binding only provides modest stabilization energy and that the occupancy of the different states is highly species dependent. Our results with human Hsp90 illustrate the most striking difference; nucleotide binding (ATP or ADP) does not substantially shift the conformational equilibrium away from the apo state, unlike E. coli and yeast Hsp90. However, identified for the first time by our crosslinking results, the ATP closed and ADP compact nucleotide-stabilized states indeed exist for human Hsp90, establishing a universal three-state conformational cycle.
The closed, NTD dimerized conformation of Hsp90 is conserved from E. coli to humans and stabilized by ATP binding
Our 3D reconstructions of HtpG:AMPPNP and Hsc82:AMPPNP identify that AMPPNP binding alone stabilizes a closed conformation that closely matches the yeast Hsp82:AMPPNP:p23 complex. Furthermore, crosslinking analysis of Hsp90α reveals for the first time that this conformation is conserved in human Hsp90s as well, validating previous experiments identifying that human Hsp90 can compliment yeast Hsp90 function in vivo (Picard et al., 1990). From our reconstructions and the accurate fit of the crystal structure it is clear that ATP binding induces a large-scale rotation of the NTDs from the orientation observed in either the apo-HtgG or Grp94 structures. This closed conformation has been identified as catalytically significant and involves a direct interaction between a middle domain arginine residue and the γ-phosphate of ATP (Ali et al., 2006; Meyer et al., 2003). The reconstructions further support the importance of the closed conformation in the hydrolysis cycle by identifying that the energy from nucleotide binding alone is sufficient to drive the large conformational rearrangements in Hsp90, establishing an essential role for nucleotide in the chaperone mechanism.
In the yeast Hsp82:AMPPNP:p23 crystal structure the closed conformation is clearly stabilized by two p23 co-chaperones interacting adjacent to the NTD dimerization interface. This interaction is known to significantly slow the hydrolysis rate and stabilize client-bound Hsp90 complexes (McLaughlin et al., 2006). In our 3D reconstruction of Hsc82:AMPPNP we observe differences compared to the crystal structure of the p23-bound complex that reflect a different arrangement, where the NTDs have rotated up and away slightly. This is the first structural evidence for an alternate, closed conformation and suggests a model where the p23-bound state is off-pathway for ATP hydrolysis. In support of this idea, biochemical work from our lab has identified a T22F mutation in Hsc82 that results in a significant increase in the hydrolysis rate but is sterically incompatible with the Hsp82:AMPPNP:p23 structure (Cunningham et al., 2008). In the crystal structure this mutation is located in a helix that is sandwiched between p23 and the opposing NTD and is predicted to destabilize NTD dimerization. Therefore, a conformational change where the NTDs rotate away slightly following p23 release may be required for maximum hydrolysis activity. This potential mechanism would addressed from high resolution structures of Hsc82:AMPPNP alone or possibly with Aha1, a co-chaperone that interacts nearby and stimulates ATP hydrolysis (Meyer et al., 2004).
The compact ADP conformation represents a third state in the hydrolysis cycle
Our 3D reconstruction of HtpG:ADP clearly establishes the existence of a more compact conformation that likely results following ATP hydrolysis. Remarkably our newly determined 3D volume quantitatively matches the compact dimer model proposed based on domain-swapping of the observed crystallographic tetramer. From comparisons of this crystal structure model and other Hsp90 structures it is clear that the ADP conformation involves a significant rotation of the NTD down towards the MD while the MD-CTD angle closely matches the closed ATP conformation. In support of this arrangement, previous biochemical studies on Grp94 identified inter-domain crosslinks that are only consistent with the domain geometry of the ADP compact state (Chu et al., 2006).
Since the vast majority of the HtpG:ADP molecules are in this conformation in our EM micrographs this appears to be an authentic stable state in the E. coli Hsp90 ATPase cycle. By contrast, Hsc82 and Hsp90α do not form the compact state to an appreciable degree under similar conditions. Furthermore, this conformation has not been observed by solution SAXS analysis from our lab (HtpG) and others (human) (Zhang et al., 2004), leading to questions about its existence. Given our EM data, we argue that experimental differences including the 1,000-fold higher protein concentrations necessary in SAXS experiments and possible transient higher order interactions (Chadli et al., 1999) may deter formation of the dimeric ADP state. More importantly, just as crosslinking established the existence of a transient closed Hsp90α:AMPPNP state, it also confirms that the dimeric ADP conformation is indeed populated in an ADP-dependent manner for both human and yeast Hsp90s. These data establish that the ADP state is in fact conserved among different organisms, supporting a functional role in the chaperone cycle.
Model for the Hsp90 hydrolysis cycle
From the work presented here we propose a model for the Hsp90 chaperone cycle that involves three specific conformational states that are in a dynamic and species-dependent equilibrium (Figure 7). ATP binding and hydrolysis provide modest amounts of stabilization energy and help move Hsp90 through the cycle. In the absence of nucleotide, Hsp90 predominantly exists in an open conformation (1) that is significantly more extended than the apo-HtgG crystal structure and variable in the open angle, supporting recent Cryo-EM data (Bron et al., 2008). Importantly, apo Hsp90 also samples the closed and compact states (Figure 5; Figure7-1b, 3) indicating the conformational equilibrium exists in the absence of nucleotide. ATP binding biases this equilibrium to the closed state (2), however the shift is drastically different between the three species of Hsp90s studied here. In E. coli the conversion to the closed state is modest, in yeast there is a dramatic shift, while in human Hsp90 very little change occurs. These results highlight potential differences in the utilization of ATP binding energy in stabilizing the closed conformation. Binding of p23 further stabilizes a closed Hsp90 but restricts NTD-MD rotations necessary for hydrolysis (2b). Following hydrolysis Hsp90 transiently adopts the compact conformation (3) and then cycles back to the open, extended state.
Figure 7.
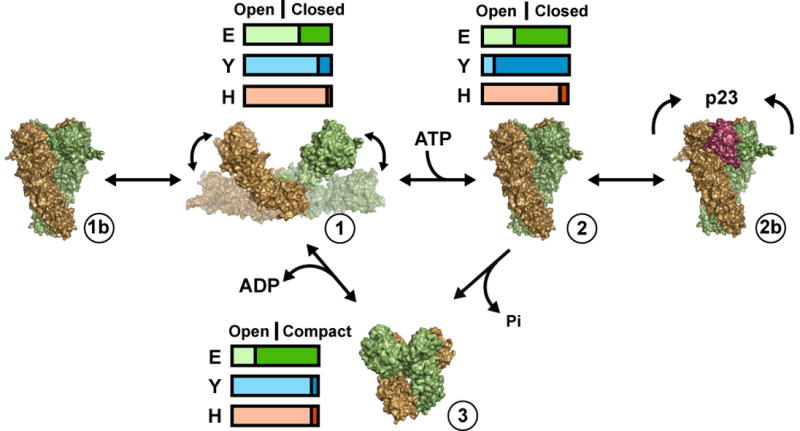
Species-Dependent Conformational Equilibrium Model for the Hsp90 Chaperone Cycle. The Hsp90 chaperone cycle is represented as a dynamic conformational equilibrium where ATP stabilizes, but does not completely determine the closed conformation. The horizontal bars represent the different fractions of open (light color) and closed (dark color) conformations for E. coli (green), yeast (blue) and human (orange) Hsp90 in the apo form (1) or bound to ATP (2) or ADP (3). The fraction of open and closed conformations for E. coli and yeast were quantitatively determined (Figure 5) while human Hsp90 data and the fraction of compact ADP conformations were visually estimated.
Finally, the different conformational equilibria we have identified point to optimization of the kinetic cycle within different organisms while the overall chaperone mechanism remains conserved. These observed equilibrium differences parallel the known differences in hydrolysis rates as well, with the fastest turnover observed for yeast Hsp90 (1.3 × 10-2 sec-1)(Prodromou et al., 2000), a moderate rate for HtpG (approx. 5×10-3 sec-1) (Cunningham, unpublished data) and a very low rate for human Hsp90 (1.5 ×10-3 sec-1)(McLaughlin et al., 2002), suggesting that NTD rearrangement and dimerization is rate limiting for the overall hydrolysis cycle. Interestingly, our data indicate that the energetics of NTD dimerization are quite different between organisms. For HtpG, dimerization appears to be relatively isoenergetic with nucleotide binding producing only a slight increase in the fraction of closed complexes and thus a very small ΔG for the closed conformation. For yeast, the equilibrium data suggest that the ΔG is much larger, with nucleotide binding resulting in a significant shift to the closed state, but for human Hsp90 the conformational change remains unfavorable. In vitro experiments have identified a higher human Hsp90 ATPase rate in the presence of a known client protein (McLaughlin et al., 2002). Therefore, one possible reason for these differences is that for human Hsp90, productive client protein interactions along with nucleotide binding stabilize the closed state. Additional structural and biochemical experiments will be critical in determining whether the interactions made by client proteins and co-chaperones contribute to the conformational equilibrium and how these dramatic conformational changes promote stabilization and activation of specific substrates.
Experimental Procedures
Protein Expression and Purification
Full-length HtpG (E. coli), cloned into a pET29b vector (Novagen) and under the control of an IPTG-inducible T7 promoter was expressed in BL21 cells (Invitrogen). Full-length HtpG and Hsc82 protein were purified as described (Cunningham et al., 2008; Krukenberg et al., 2008). Human Hsp90α DNA, provided by the David Toft lab, was cloned into the pET151 vector containing a 6x His-tag and TEV cleavage site (Invitrogen). Hsp90α protein was purified by ion exchange (DE52 resin, Whatman), Ni-NTA affinity, and size exclusion (Sephacryl S-200 column, GE Healthcare), followed by TEV cleavage of the His-tag. We assessed the quality of each purification by measuring the ATP hydrolysis rates (Cunningham et al., 2008) and comparing to known or published rates.
Electron Microscopy
HtpG, Hsc82 and Hsp90α proteins were negatively stained with uranyl formate (pH 5.5-6.0) on thin carbon-layered (40-50 Å thick) 400 mesh copper grids (Pelco) essentially as described (Ohi et al., 2004). Initial experiments were performed with other stains including ammonium molybdate, methylamine vanadate and uranyl acetate. The particles appeared similar, however the low contrast and granularity prevented further analysis. Prior to staining, protein samples were typically incubated for 20 minutes at 150nM with or without 5mM AMPPNP or 2mM ADP (Sigma-Aldrich) at 37° for HtpG and Hsp90α and 30° for Hsc82 in an incubation buffer containing 20mM Tris pH 8.0, 20mM KCl, 5mM MgCl2 and 1mM DTT. KCl and MgCl2 concentrations were varied (0.5-10 and 10-100mM, respectively) in order to maximize the formation of closed or compact nucleotide-bound protein complexes.
Negative-stained samples were imaged using a Tecnai T20 and G2 Spirit TEMs (FEI) operated at 120 keV. Data used in the final 3D reconstructions was collected from a single source. Micrograph images were recorded using a 4k × 4k CCD camera (Gatan) at 67,000x and 68,000x magnification with 2.25 and 2.21 Å pixel size for the T20 and T12 microscopes, respectively.
Three-Dimensional Reconstructions and Analysis
3D reconstructions were performed using EMAN (Ludtke et al., 1999). For the HtpG:AMPPNP and HtpG:ADP reconstructions a subset of class averages were chosen and used to generate an initial model by the cross common lines method (Figure S4). For HtpG:AMPPNP some preferential orientation of the side views was observed, therefore a 60° tilted data set was also collected and added to the 0° data in the refinement. For the Hsc82:AMPPNP reconstruction, the HtpG:AMPPNP initial model was used. Defocus values ranged between 1.2 and 1.5 μM defocus, calculated by ctfit. The reconstructions involved 10 (HtpG:ADP) and 12 (HtpG:AMPPNP and Hsc82:AMPPNP) rounds of refinement and the final models were low-pass filtered to 20 Å. A total of 5,778 (4,421 for the final model), 5,035 (3,812 for the final model) and 7,549 (6,197 for the final model) particles were used in the HtpG:AMPPNP, HtpG:ADP, and Hsc82:AMPPNP reconstructions, respectively. During early rounds of refinement, large ‘classkeep’ (1.0) and ‘classiter’ (8) values were used to reduce initial model bias. These values were made more stringent in later rounds to improve resolution. For visualization, the maps were contoured to the approximate molecular weight of HtpG (142 kDa) or Hsc82 (162 kDa). The resolution of the final maps was measured by the even-odd test in EMAN where the Fourier shell correlation method is used to compare two models generated from a random split of the data. The resolution was set to the spatial frequency at an FSC value equal to 0.5 (Figure S2).
Reconstructions were compared to crystal structure data by rigid body docking of the structure maps using the Chimera ‘fit model in map’ command (Pettersen et al., 2004). The fit of the yeast Hsp90:AMPPNP structure and the model docked by rigid body domain fitting were compared by calculating a cross-correlation value using the Laplacian filter in the Situs software package (Chacon and Wriggers, 2002). Difference maps (Figure 6A) were calculated using SPIDER (Frank et al., 1996) by generating a 3D volume (low-pass filtered to 20 Å) of the Hsp82:AMPPNP crystal structure model aligned to the Hsc82:AMPPNP map. The aligned volumes were subtracted and the resulting difference maps were contoured to equivalent molecular weight values for visualization. Rigid body domain fitting of the Hsp82:AMPPNP structure was achieved by a combination of a three-dimensional grid search in angle space and simultaneous least-squares optimization of rotations and translations using custom-developed software.
Conformational Matching by 2D Multireference Alignment
We used a multireference method to identify the fraction of open and closed conformations under different conditions. Three independent data sets (500-800 single particles for each) of apo HtgG, HtpG:AMPPNP, apo Hsc82 and Hsc82:AMPPNP were first collected. For the references, 2D projections were generated from 3D volumes of the closed yeast Hsp90:AMPPNP structure and an extended apo-HtgG model derived from solution SAXS studies (Krukenberg et al., 2008). The reference projections of the closed and extended conformations were first aligned to each other to determine the top distinct views by correlation value (0.7 cutoff). The single particle data sets were then aligned to the combined set references and the fraction that matched the distinct closed or extended references was determined under the given conditions. Particles that aligned to top-down views of the closed state were excluded due to similarities with the compact conformation.
Crosslinking and 2D analysis of human Hsp90α and yeast Hsc82 proteins
Hsp90 proteins were incubated at 150 nM as discussed above except samples were diluted into a buffer containing 20mM Hepes, pH 7.5. Following a 20-minute equilibration step, glutaraldehyde was added to 0.005% and samples were then incubated for 10 minutes. The crosslinking reaction was quenched with 20mM Tris pH 7.5. Samples were then negatively stained as discussed above. Single particle data sets of approximately 1,000 were collected for each and reference-free class averages were then generated (EMAN), and aligned to 2D projections of the appropriate reconstructions for comparison.
Supplementary Material
Acknowledgments
Funding for this project was provided by the Howard Hughes Medical Institute, University of California Discovery Grant bio03-10401, and an American Cancer Society postdoctoral fellowship. Special thanks to U. Boettcher, Y. Chang, C. Cunningham, J. Kollman, K. Krukenberg, T. Street, L. Lavery, and L. Rice for many helpful discussions and comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban C, Junop M, Yang W. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell. 1999;97:85–97. doi: 10.1016/s0092-8674(00)80717-5. [DOI] [PubMed] [Google Scholar]
- Bron P, Giudice E, Rolland JP, Buey RM, Barbier P, Diaz JF, Peyrot V, Thomas D, Garnier C. Apo-Hsp90 coexists in two open conformational states in solution. Biol Cell. 2008;100:413–425. doi: 10.1042/BC20070149. [DOI] [PubMed] [Google Scholar]
- Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- Chacon P, Wriggers W. Multi-resolution contour-based fitting of macromolecular structures. J Mol Biol. 2002;317:375–384. doi: 10.1006/jmbi.2002.5438. [DOI] [PubMed] [Google Scholar]
- Chadli A, Ladjimi MM, Baulieu EE, Catelli MG. Heat-induced oligomerization of the molecular chaperone Hsp90. Inhibition by ATP and geldanamycin and activation by transition metal oxyanions. J Biol Chem. 1999;274:4133–4139. doi: 10.1074/jbc.274.7.4133. [DOI] [PubMed] [Google Scholar]
- Chu F, Maynard JC, Chiosis G, Nicchitta CV, Burlingame AL. Identification of novel quaternary domain interactions in the Hsp90 chaperone, GRP94. Protein Sci. 2006;15:1260–1269. doi: 10.1110/ps.052065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CN, Krukenberg KA, Agard DA. Intra-and inter-monomer interactions are required to synergistically facilitate ATP hydrolysis in Hsp90. J Biol Chem. 2008 doi: 10.1074/jbc.M800046200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci. 2000;25:24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- Felts SJ, Toft DO. p23, a simple protein with complex activities. Cell Stress Chaperones. 2003;8:108–113. doi: 10.1379/1466-1268(2003)008<0108:paspwc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Frey S, Leskovar A, Reinstein J, Buchner J. The ATPase cycle of the endoplasmic chaperone Grp94. J Biol Chem. 2007;282:35612–35620. doi: 10.1074/jbc.M704647200. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- Harris SF, Shiau AK, Agard DA. The crystal structure of the carboxy-terminal dimerization domain of htpG, the Escherichia coli Hsp90, reveals a potential substrate binding site. Structure (Camb) 2004;12:1087–1097. doi: 10.1016/j.str.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Holt SE, Aisner DL, Baur J, Tesmer VM, Dy M, Ouellette M, Trager JB, Morin GB, Toft DO, Shay JW, et al. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 1999;13:817–826. doi: 10.1101/gad.13.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BD, Schumacher RJ, Ross ED, Toft DO. Hop modulates Hsp70/Hsp90 interactions in protein folding. J Biol Chem. 1998;273:3679–3686. doi: 10.1074/jbc.273.6.3679. [DOI] [PubMed] [Google Scholar]
- Krukenberg KA, Forster F, Rice LM, Sali A, Agard DA. Multiple Conformations of E. coli Hsp90 in Solution: Insights into the Conformational Dynamics of Hsp90. Structure. 2008;16:755–765. doi: 10.1016/j.str.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Martin J, Langer T, Boteva R, Schramel A, Horwich AL, Hartl FU. Chaperonin-mediated protein folding at the surface of groEL through a ’molten globule’-like intermediate. Nature. 1991;352:36–42. doi: 10.1038/352036a0. [DOI] [PubMed] [Google Scholar]
- McLaughlin SH, Smith HW, Jackson SE. Stimulation of the weak ATPase activity of human hsp90 by a client protein. J Mol Biol. 2002;315:787–798. doi: 10.1006/jmbi.2001.5245. [DOI] [PubMed] [Google Scholar]
- McLaughlin SH, Sobott F, Yao ZP, Zhang W, Nielsen PR, Grossmann JG, Laue ED, Robinson CV, Jackson SE. The co-chaperone p23 arrests the Hsp90 ATPase cycle to trap client proteins. J Mol Biol. 2006;356:746–758. doi: 10.1016/j.jmb.2005.11.085. [DOI] [PubMed] [Google Scholar]
- McLaughlin SH, Ventouras LA, Lobbezoo B, Jackson SE. Independent ATPase activity of Hsp90 subunits creates a flexible assembly platform. J Mol Biol. 2004;344:813–826. doi: 10.1016/j.jmb.2004.09.055. [DOI] [PubMed] [Google Scholar]
- Meyer P, Prodromou C, Hu B, Vaughan C, Roe SM, Panaretou B, Piper PW, Pearl LH. Structural and functional analysis of the middle segment of hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol Cell. 2003;11:647–658. doi: 10.1016/s1097-2765(03)00065-0. [DOI] [PubMed] [Google Scholar]
- Meyer P, Prodromou C, Liao C, Hu B, Mark Roe S, Vaughan CK, Vlasic I, Panaretou B, Piper PW, Pearl LH. Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. Embo J. 2004;23:511–519. doi: 10.1038/sj.emboj.7600060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan DF, Vos MH, Lindquist S. In vivo functions of the Saccharomyces cerevisiae Hsp90 chaperone. Proc Natl Acad Sci U S A. 1997;94:12949–12956. doi: 10.1073/pnas.94.24.12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8:S55–61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- Ohi M, Li Y, Cheng Y, Walz T. Negative Staining and Image Classification - Powerful Tools in Modern Electron Microscopy. Biol Proced Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Picard D. Heat-shock protein 90, a chaperone for folding and regulation. Cell Mol Life Sci. 2002;59:1640–1648. doi: 10.1007/PL00012491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature. 1990;348:166–168. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- Pratt WB, Galigniana MD, Morishima Y, Murphy PJ. Role of molecular chaperones in steroid receptor action. Essays Biochem. 2004;40:41–58. doi: 10.1042/bse0400041. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Panaretou B, Chohan S, Siligardi G, O’Brien R, Ladbury JE, Roe SM, Piper PW, Pearl LH. The ATPase cycle of Hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. Embo J. 2000;19:4383–4392. doi: 10.1093/emboj/19.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Richter K, Buchner J. Hsp90: chaperoning signal transduction. J Cell Physiol. 2001;188:281–290. doi: 10.1002/jcp.1131. [DOI] [PubMed] [Google Scholar]
- Richter K, Soroka J, Skalniak L, Leskovar A, Hessling M, Reinstein J, Buchner J. Conserved conformational changes in the ATPase cycle of human Hsp90. J Biol Chem. 2008 doi: 10.1074/jbc.M800540200. [DOI] [PubMed] [Google Scholar]
- Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau AK, Harris SF, Southworth DR, Agard DA. Structural Analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell. 2006;127:329–340. doi: 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- Vaughan CK, Gohlke U, Sobott F, Good VM, Ali MM, Prodromou C, Robinson CV, Saibil HR, Pearl LH. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol Cell. 2006;23:697–707. doi: 10.1016/j.molcel.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- Zhang W, Hirshberg M, McLaughlin SH, Lazar GA, Grossmann JG, Nielsen PR, Sobott F, Robinson CV, Jackson SE, Laue ED. Biochemical and structural studies of the interaction of Cdc37 with Hsp90. J Mol Biol. 2004;340:891–907. doi: 10.1016/j.jmb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Zhao R, Davey M, Hsu YC, Kaplanek P, Tong A, Parsons AB, Krogan N, Cagney G, Mai D, Greenblatt J, et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell. 2005;120:715–727. doi: 10.1016/j.cell.2004.12.024. [DOI] [PubMed] [Google Scholar]
- Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.