

Abstract
Cigarette smoking impairs endothelial function. Hydroxymethylglutaryl (HMG) CoA reductase inhibitors (statins) may favorably affect endothelial function via nonlipid mechanisms. We tested the hypothesis that statins would improve endothelial function independent of changes in lipids in cigarette smokers. Twenty normocholesterolemic cigarette smokers and 20 matched healthy control subjects were randomized to atorvastatin 40 mg daily or placebo for 4 weeks, washed out for 4 weeks, and then crossed-over to the other treatment. Baseline low-density lipoprotein (LDL) levels were similar in smokers and healthy subjects, 103±22 versus 95±27 mg/dL, respectively (P=NS) and were reduced similarly in smokers and control subjects by atorvastatin, to 55±30 and 58±20 mg/dL, respectively (P=NS). Vascular ultrasonography was used to determine brachial artery, flow-mediated, endothelium-dependent, and nitroglycerin-mediated, endothelium-independent vasodilation. To elucidate potential molecular mechanisms that may account for changes in endothelial function, skin biopsy specimens were assayed for eNOS mRNA, eNOS activity, and nitrotyrosine. Endothelium-dependent vasodilation was less in smokers than nonsmoking control subjects during placebo treatment, 8.0±0.6% versus 12.1±1.1%, (P=0.003). Atorvastatin increased endothelium-dependent vasodilation in smokers to 10.5±1.3% (P=0.017 versus placebo) but did not change endothelium-dependent vasodilation in control subjects (to 11.0±0.8%, P=NS). Endothelium-independent vasodilation did not differ between groups during placebo treatment and was not significantly affected by atorvastatin. Multivariate analysis did not demonstrate any association between baseline lipid levels or the change in lipid levels and endothelium-dependent vasodilation. Cutaneous nitrotyrosine levels and skin microvessel eNOS mRNA, but not ENOS activity, were increased in smokers compared with controls but unaffected by atorvastatin treatment. Atorvastatin restores endothelium-dependent vasodilation in normocholesterolemic cigarette smokers independent of changes in lipids. These results are consistent with a lipid-independent vascular benefit of statins but could not be explained by changes in eNOS message and tissue oxidative stress. These findings implicate a potential role for statin therapy to restore endothelial function and thereby investigate vascular disease in cigarette smokers.
Keywords: cigarette smoking, endothelial nitric oxide synthase, hydroxymethylglutaryl CoA reductase inhibitor, endothelial function, oxidative stress
Cigarette smoking is a reversible risk factor for atherosclerosis. Smoking impairs endothelial function and creates a vascular environment that contributes to the development of atherosclerosis.1,2 Putative mechanisms through which cigarette smoking alters endothelial function and particularly the bioavailability of nitric oxide (NO) in humans include increased oxidative stress,3 alterations in endothelial nitric oxide synthase (eNOS),4 and activation of rho kinase.5
Hydroxymethylglutaryl CoA reductase inhibitors (statins) potently reduce total and low-density lipoprotein (LDL) cholesterol. The cholesterol-lowering effects are associated with improvements in endothelium-dependent vasodilation.6,7 Cellular and animal investigations have demonstrated non-lipid benefits of statins that include upregulation of eNOS, reduction of oxidative stress, and antagonism of isoprenoid-mediated activation of small GTP-binding proteins.8 Corroborating these experimental studies, statin-mediated improvements in endothelial function have been demonstrated within days in humans, even after a single dose and before any significant lipid affect.9,10 To investigate whether statins confer benefit on endothelial function independent of cholesterol reduction in humans, we sought to determine the effect of atorvastatin on endothelial function in normolipidemic cigarette smokers and healthy control subjects in a randomized, placebo-controlled, double-blind, crossover study. Moreover, we sought to determine whether the benefit was achieved by changes in oxidative stress or in eNOS mRNA levels and activity.
Methods
Subjects
Forty subjects, including 20 cigarette smokers and 20 age- and gender-matched healthy, nonsmoking controls were recruited through newspaper advertisement. All subjects underwent screening medical history, physical examination, and laboratory analysis including complete blood count, serum electrolytes, glucose, blood urea nitrogen, creatinine, transaminases, alkaline phosphatase, total cholesterol, and LDL. Subjects with hypertension, diabetes mellitus, LDL or total cholesterol greater than the 50th percentile for age and gender, cardiovascular disease, or other significant disease were excluded. All participants provided written informed consent. The protocol was approved by the Human Research Committees of the Brigham and Women’s Hospital.
Study Design
The effect of atorvastatin on endothelium-dependent and endothelium-independent vasodilation of the brachial artery was studied using a randomized, double-blind, placebo-controlled, crossover design. All subjects were studied in the morning in the postabsorptive state, fasting after the previous midnight. Cyclooxygenase inhibitors, alcohol, and caffeine were prohibited for 24 hours before the study. Cigarette smokers refrained from smoking for at least 2 hours before arrival in the vascular laboratory. Subjects were randomized to receive either atorvastatin 40 mg daily (Pfizer Inc) or matching placebo. After 30 days of medication, subjects underwent repeat vascular function testing. After testing, subjects had a 30-day washout period and then crossed over to the alternate treatment with placebo or atorvastatin. Vascular testing was performed again after 30 days of treatment.
Vascular Reactivity Studies
Subjects were studied in a quiet, temperature-controlled, dimly lit room after resting supine for a minimum of 5 minutes. High-resolution B-mode ultrasonography of the brachial artery was performed using a Toshiba Powervision 8000 (Toshiba America Medical Systems Inc) ultrasound machine and 7.5-MHz linear array probe. The brachial artery was imaged longitudinally, just proximal to the antecubital fossa. Transducer position was adjusted to obtain optimal images of the near and far walls of the intima. Images were simultaneously recorded on super VHS videotape. The video output and electrocardiographic signal of the ultrasound machine were connected to a computer equipped with a Data Translation frame-grabber videocard (Dataviz). The R wave on the electrocardiogram served as a trigger to acquire frames at end-diastole. After baseline image acquisition, a sphygmomanometric cuff was inflated to suprasystolic pressure (200 mm Hg) for 5 minutes. On cuff release, reactive hyperemia causes flow to increase through the brachial artery subserving the forearm. Flow-induced, endothelium-dependent vasodilation of the brachial artery was determined by acquiring images at 1 minute after cuff deflation. Ten minutes after cuff release, the brachial artery was imaged again to ensure a return to basal conditions. Then, to determine endothelium-independent vasodilation, subjects received 0.4 mg of nitroglycerin, sublingually. The brachial artery was imaged 3 minutes later. Brachial artery blood flow velocity was determined via time–velocity integral measurement. Nitroglycerin was not administered if the systolic blood pressure was <100 mm Hg. Thirty-six of the 40 subjects received nitroglycerin at all visits. The determination of endothelial function was performed in accordance with published guidelines.11
Laboratory Analyses
After each vascular function measurement, the subject underwent skin biopsy from the upper arm. Lidocaine 2% anesthesia was infused around the area of interest. A 4-mm bioptome was used to obtain a skin sample. The specimen was frozen immediately in liquid nitrogen and stored at −80°C until RNA preparation.
eNOS mRNA Expression
Samples were divided to enable assessment of both eNOS mRNA and eNOS activity. eNOS mRNA expression in skin was assessed by semiquantitative reverse-transcription polymerase chain reaction (RT-PCR). Total RNA isolation and RT-PCR were performed according to standard techniques.12 Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA was used as co-amplified internal control for sample normalization and relative quantitation of eNOS expression. Primer pairs spanning introns for eNOS and GAPDH were used as previously described and the conditions for each PCR reaction was chosen at linear phase of amplification. The PCR products containing eNOS and GAPDH were loaded on 1.5% agarose gels. Optical densities of ethidium bromide-stained bands were quantitated by densitometry and results were expressed as eNOS/GAPDH ratios.
eNOS Activity
Total protein was extracted from the skin and subcutaneous tissue samples. Protein concentration was determined with the Micro BCA Assay Kit (Pierce Chemical Co, Rockford, Ill). Ten μg of protein lysates from each sample were used for the eNOS activity assay. eNOS activity was measured as the conversion of [3H]L-arginine to [3H]L-citrulline at 37°C by 30 minutes using the eNOS assay kit (Calbiochem-Novabiochem Corp). Nonspecific activity was determined in the presence of excess unlabeled NG-monomethyl-L-arginine (LNMA, 1 mmol/L). eNOS activity values were normalized to reaction time and amount of protein and expressed as pmol L-citrulline/min per milligram of protein.
Tissue Nitrotyrosine Content
Nitrotyrosine was measured using a solid-phase enzyme-linked immunosorbent assay based on the sandwich principle according to the recommendations of the manufacturer (Nitrotyrosine ELISA Kit ab7371; Abcam). In brief, skin homogenates were lysed in cell lysis buffer (20 mmol/L Tris-HCl [pH 7.5], 150 mmol/L NaCl, 1 mmol/L Na2EDTA, 1 mmol/L EGTA, 1% Triton, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L beta-glycerophosphate, 1 mmol/L Na3VO4, 1 μg/mL leupeptin), rotated 1 hour at 4°C, and centrifuged at 14 000g for 30 minutes. Proteins of the supernatant were concentrated using Microcon centrifugal filter units with a cutoff of 10 000 Da (Millipore, Billerica, Mass) and protein concentration were measured using the BCA method (Pierce Biotechnology). Seventy-five μg of protein were incubated in microtiter wells coated with antibodies recognizing nitrotyrosine. A biotinylated second antibody to nitrotyrosine was added to the wells as a tracer to bind a streptavidin-peroxidase conjugate. Finally, tetramethylbenzidine was added as a substrate and the reaction was stopped by addition of citric acid. Absorbency was measured at 450 nm and compared with nitrotyrosine standards. Data are expressed as nitrotyrosine amounts in nmol per 1 mg of protein.
Statistical Methods
Descriptive measures are reported as means±SD. Experimental measures are reported as means±SE. Experimental measures, including flow-mediated and nitroglycerin-mediated vasodilation, eNOS mRNA, eNOS activity, and nitrotyrosine levels for each subject group were compared at the end of treatment using a paired Student t test. Comparisons made between groups were made using an independent t test. Multiple linear regression was performed to assess the effects of lipoprotein concentrations at baseline and the change with treatment on flow-mediated vasodilation. Statistical significance was accepted at the 95% confidence level (P<0.05). All statistics were run on SPSS Base 10.0 (SPSS Inc).
Results
Baseline characteristics are presented in Table 1. Cigarette smokers and nonsmokers were well matched for age, sex, total cholesterol, LDL cholesterol, and high-density lipoprotein (HDL) cholesterol levels, and body mass index. Both groups were normotensive; however, blood pressure was higher in cigarette smokers than in healthy subjects, 126/ 78 mm Hg versus 117/69 mm Hg, respectively (P=0.001). Blood pressure did not change significantly in either group with placebo or atorvastatin therapy and the difference between the groups remained the same.
TABLE 1.
Baseline Demographics
| Parameter | Smokers (n=20) | Controls (n=20) | P |
|---|---|---|---|
| Age (SD) | 42±11 | 38±13 | 0.26 |
| Gender (M) | 10 | 11 | 0.76 |
| BMI (SD) | 25.4±4.4 | 24.0±3.8 | 0.31 |
| T Chol (SD) | 188±23 | 176±23 | 0.11 |
| LDL (SD) | 103±22 | 95±26.7 | 0.33 |
| HDL (SD) | 54±22 | 54±13 | 0.98 |
| Trig (SD) | 184±145 | 131±81 | 0.18 |
| MAP (SD) | 94±10 | 85±5 | 0.001 |
| Glucose (SD) | 88±21 | 77±14 | 0.11 |
BMI indicates body mass index; SD, standard deviation; M, male; T Chol, total cholesterol mg/dL; LDL, low-density lipoprotein mg/dL; HDL, high-density lipoprotein mg/dL; MAP, mean arterial pressure mm Hg; Trig, triglycerides mg/dL.
Atorvastatin and Cholesterol Levels
After placebo treatment, total, LDL, and HDL cholesterol levels did not vary significantly between groups (Figure 1). Specifically, total cholesterol was 180±33 versus 166±40 mg/dL, LDL was 107±30 versus 87±36 mg/dL, and HDL was 56±15 versus 50±20 mg/dL in healthy subjects and cigarette smokers, respectively (all P=NS). Atorvastatin significantly reduced total and LDL cholesterol in both groups. In healthy controls, atorvastatin lowered total cholesterol to 123±30 mg/dL and LDL cholesterol to 58 mg/dL (both P<0.001). Similarly, in cigarette smokers, atorvastatin lowered total cholesterol to 137±42 mg/dL (P=0.023) and LDL to 55±30 mg/dL (P=0.003). Total, LDL, and HDL cholesterol levels did not differ between groups after atorvastatin treatment. Liver function tests and creatine kinase levels stayed within normal levels for all subjects at all times.
Figure 1.
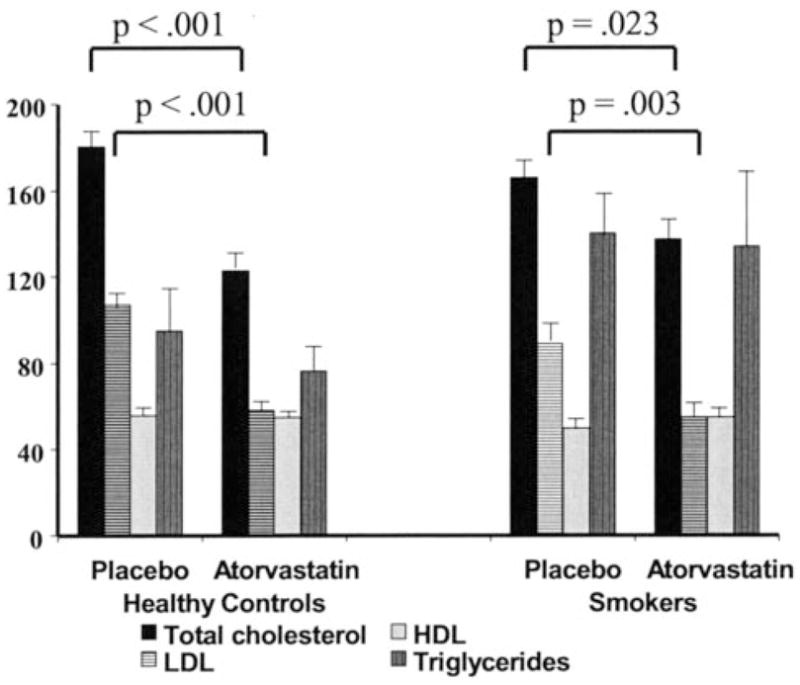
Effect of atorvastatin on lipid levels. The mean plasma concentrations (mg/dL) of total cholesterol, LDL cholesterol, HDL cholesterol, and triglycerides in cigarette smokers and healthy control subjects. During placebo treatment, there were no significant differences in lipid levels between smokers and healthy subjects. During atorvastatin treatment, total and LDL cholesterol levels decreased to similar levels in both groups.
Vascular Function Studies
Baseline arterial diameters after placebo and atorvastatin therapy did not differ within each group and between groups (Table 2). The increase in flow velocity with reactive hyperemia during placebo therapy was similar in healthy control subjects and cigarette smokers (P=NS). These values did not significantly differ during atorvastatin treatment in either group.
TABLE 2.
Brachial Artery Parameters
| Controls | Smokers | |||||
|---|---|---|---|---|---|---|
| Plac | Ator | P | Plac | Ator | P | |
| Baseline diameter (mm) | 3.6±0.7 | 3.6±0.6 | 0.89 | 3.8±0.6 | 3.8±0.6 | 0.34 |
| FMD (%) | 12.1±1.1* | 11.0±0.8 | 0.37 | 8.0±0.6* | 10.5±1.3 | 0.017 |
| Reactive hyperemia (% increase) | 656±256 | 676±205 | 0.78 | 520±223 | 517±186 | 0.94 |
| TNG hyperemia (% increase) | 100±39 | 109±40 | 0.51 | 104±51 | 96±51 | 0.63 |
P=0.003 for comparison between control and smoking subjects.
Data are mean±SD.
Plac indicates placebo; Ator, atorvastatin; FMD, flow-mediated vasodilation; TNG, nitroglycerin-mediated vasodilation.
Flow-mediated, endothelium-dependent vasodilation was less in cigarette smokers than healthy subjects during placebo treatment, 8.0±0.6% versus 12.1±1.1%, respectively (P=0.003) (Figure 2). Atorvastatin increased flow-mediated vasodilation in cigarette smokers from 8.0±0.6% to 10.5±1.3% (P=0.017) but had no significant effect on nonsmokers, 12.1±1.1% versus 11.0±0.8% (P=NS). During atorvastatin treatment, flow-mediated vasodilation did not differ significantly between cigarette smokers and healthy subjects, 11.0±0.8% versus 10.5±1.3%, respectively (P=NS). Multivariate analysis including all baseline variables revealed no significant relationship between change in total or LDL cholesterol or blood pressure and flow-mediated vasodilation, when the group was considered as a whole or cigarette smokers were considered separately.
Figure 2.
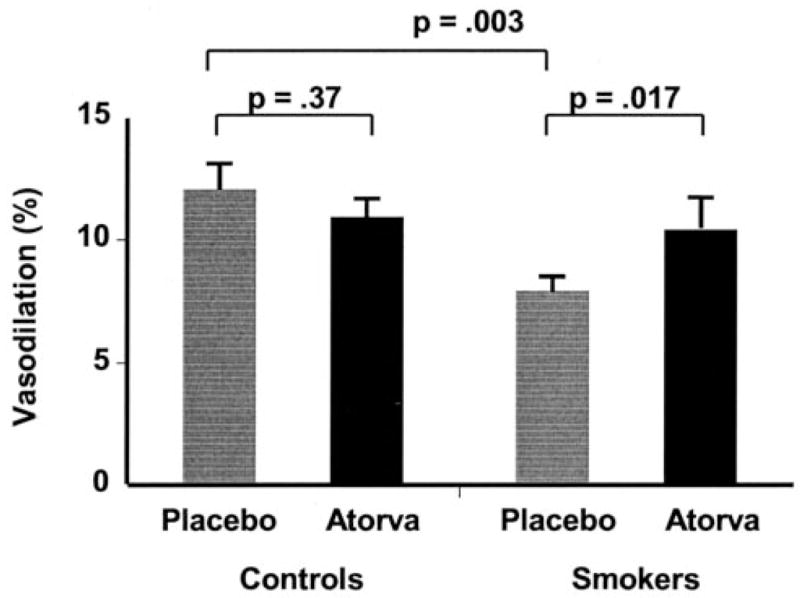
Effect of cigarette smoking and atorvastatin on flow-mediated vasodilation. The mean percent increase in brachial artery size 1 minute after cuff release compared with baseline is illustrated. Flow-mediated, endothelium-dependent vasodilation was significantly impaired in the brachial arteries of smoking subjects compared with control subjects. Atorvastatin increased flow-mediated, endothelium-dependent vasodilation in the smoking subjects but not in the healthy subjects.
Nitroglycerin-induced, endothelium-independent vasodilation did not differ between cigarette smokers and healthy subjects during placebo treatment, 21±1.8% versus 18.6±1.6%, respectively (P=0.34) (Figure 3). Atorvastatin did not significantly change nitroglycerin-mediated vasodilation in cigarette smokers or healthy subjects.
Figure 3.
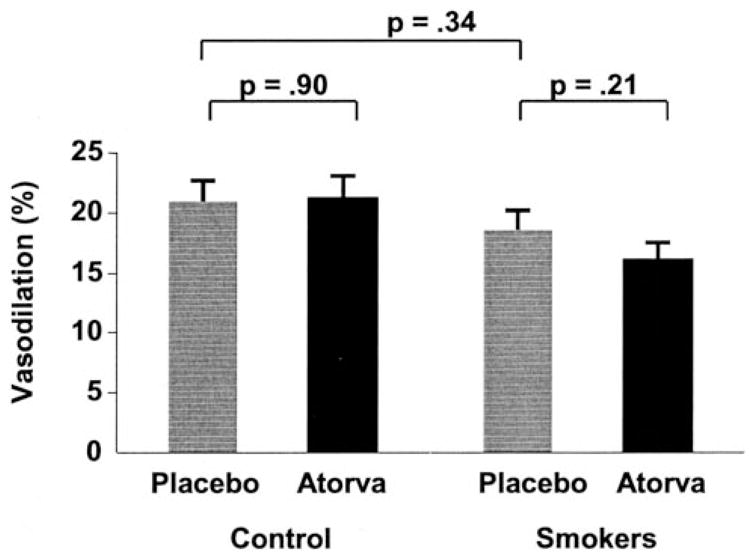
Effect of cigarette smoking and atorvastatin on nitroglycerin-mediated vasodilation. The mean percent increase in brachial artery size 3 minutes after sublingual nitroglycerin administration compared with baseline is illustrated. Nitroglycerin-mediated, endothelium-independent vasodilation in the brachial arteries was not significantly different between smoking and healthy controls subjects. Atorvastatin did not significantly affect nitroglycerin-mediated, endothelium-independent vasodilation in either group.
Tissue Studies
Subcutaneous vascular eNOS mRNA content was quantified as a ratio to GAPDH after placebo and atorvastatin treatment. eNOS mRNA content was higher in cigarette smokers than healthy control subjects, 0.78±0.08 versus 0.56±0.05 (P=0.04). Atorvastatin increased eNOS mRNA content in healthy subjects from 0.56±0.05 to 0.62±0.05 (P=−0.03) but had no effect on cigarette smokers, 0.78±0.08 to 0.77±0.08 (P=0.95). eNOS activity was similar in cigarette smokers and in healthy subjects during placebo treatment, 5.21±0.44 versus 5.08±0.35 pmol L-citrulline/min per milligram, respectively (P=0.72). Atorvastatin treatment did not significantly change eNOS activity in cigarette smokers or healthy subjects. Tissue nitrotyrosine content was higher in cigarette smokers than in healthy control subjects during placebo treatment (82.4±20.2 versus 38.1±7.9 nmol/mg, respectively, P=0.05). Atorvastatin treatment did not significantly change tissue nitrotyrosine content in cigarette smokers or in healthy subjects.
Discussion
This study has several salient findings. Flow-mediated, endothelium-dependent vasodilation is impaired in normocholesterolemic smokers. Statin therapy restores endothelium-dependent vasodilation in normolipidemic cigarette smokers independent of lipid levels, tissue oxidative stress, or eNOS mRNA levels. Microvessel eNOS mRNA content was higher in smokers than in healthy controls and increased only in healthy subjects with statin therapy. Finally, tissue nitrotyrosine content was higher in cigarette smokers than in healthy subjects but unaffected by statin therapy.
Cigarette Smoking and Endothelial Function
The first observation that endothelial function is impaired in cigarette smokers was made >10 years ago.1 Increasing cigarette number and duration worsen endothelium-dependent vasodilation, suggesting a dose-related phenomenon.1,13 Mechanisms whereby cigarette smoking may reduce the bioavailability of endothelium-derived NO include increased scavenging of NO by reactive oxygen species (ROS) and decreased content and functional activity of eNOS. In vitro14 and human investigation15 have demonstrated that cigarette smoking increases production of ROS, without a commensurate increase in antioxidant activity.16 Vitamin C plasma concentrations are reduced in cigarette smokers, suggesting an ongoing imbalance favoring oxidative stress.15 We measured tissue oxidative stress directly, demonstrating increased concentrations of nitrotyrosine, a specific marker of NO-derived oxidants,17 in smokers compared with healthy subjects. ROS, specifically superoxide anion, can scavenge NO and yield peroxynitrite,18 thereby diminishing the bio-availability of NO and endothelium-derived vasodilation. Scavenging superoxide anion acutely with vitamin C restores endothelium-dependent vasodilation in smokers but this effect cannot be maintained chronically.3,19 One important source of oxygen-derived free radicals may be increased angiotensin-converting enzyme activity and the resultant angiotensin II production.20 Angiotensin II increases the endothelial production of superoxide anion through activation of NADPH oxidases.21,22 In vitro and human investigation have demonstrated that angiotensin-converting enzyme inhibition restores endothelial function that had been impaired by cigarette smoking, suggesting an important role for vascular angiotensin in smoking.23,24 In addition to scavenging NO, generation of reactive oxygen species by cigarette smoking may also affect eNOS activity. The product of superoxide anion and NO, peroxynitrite, oxidizes tetrahydrobiopterin, the eNOS cofactor.25 Without tetrahydrobiopterin, eNOS becomes uncoupled and produces superoxide anion preferentially over NO.26,27 Supplementation of tetrahydrobiopterin restores endothelium-dependent vasodilation in cigarette smokers, lending support for a chronic deficiency of this eNOS cofactor.28,29
The effect of cigarette smoking on eNOS content and activity may depend on the origin of the endothelium. Studies in pulmonary arteries suggest that cigarette smoke, per se, decreases both endothelial eNOS content and activity.30,31 However, when serum from smokers was added to confluent human umbilical vein endothelial cells, eNOS NO production decreased, but eNOS protein expression increased.4 In our study, microvessel eNOS mRNA was increased in cigarette smokers compared with healthy control subjects. These findings suggest that in states of increased oxidative stress, eNOS mRNA content increases32 but effective NO production decreases.
Atorvastatin and Endothelial Function in Smokers
Atorvastatin administration decreased total and LDL cholesterol levels similarly in cigarette smokers and healthy controls. In cigarette smokers, atorvastatin treatment significantly increased endothelium-dependent vasodilation compared with placebo but did not affect endothelium-independent vasodilation. In nonsmoking, healthy subjects, atorvastatin treatment did not affect endothelium-dependent or endothelium-independent vasodilation. The improvement in endothelium-dependent vasodilation with atorvastatin in smokers was not associated with basal total or LDL cholesterol levels or the change in these lipid levels as a result of treatment.
Cigarette smokers had higher tissue eNOS mRNA concentrations than healthy controls, and these were unchanged by atorvastatin therapy. In vitro and ex vivo work demonstrate that atherosclerotic risk factors that increase oxidative stress, in general, and hydrogen peroxide, per se, increase eNOS concentration, as we found in this study.32,33 We expected that atorvastatin therapy would increase the eNOS mRNA concentration further on the basis of previous animal and in vitro experiments.34,35 Yet, eNOS mRNA concentrations increased only in the healthy subjects. These findings suggest that cigarette smoking, likely via ROS, maximally increases eNOS mRNA concentration, and further increases with statin therapy cannot occur as it does in healthy states. Statins have been demonstrated to attenuate hydrogen peroxide production.36 Therefore, the lack of change in eNOS mRNA levels in cigarette smokers may result from a balance between a statin-mediated decrease in ROS and a statin-mediated improved stabilization of eNOS mRNA. The control used in the study (GAPDH), unchanging with each treatment condition, is not specific to vascular cells. We cannot exclude the possibility that increases in oxidative stress or relative tissue hypoxia may have increased the skin microvessel concentration, thereby accounting for the increase in eNOS mRNA signal in smokers.
Finally, we tested the hypothesis that statins may improve endothelium-dependent vasodilation through decreases in oxidative stress. Animal and human investigation have demonstrated that statin therapy reduces oxidative stress.37,38 Statins decrease plasma NO-derived oxidants specifically.39 In our subjects, however, tissue nitrotyrosine content, which was higher in cigarette smokers, remained unaffected by atorvastatin treatment. Our observations may differ from previous work because of differences in the origin of ROS measurements: tissue and plasma. ROS in plasma may be produced by nonvascular cells (platelets and leukocytes) in addition to the vascular wall.40,41 Moreover, statins have been demonstrated to scavenge ROS directly.42,43 Thus, studies that recently demonstrated decreases in plasma nitrotyrosine may reflect a difference in statin-mediated reductions in ROS in tissue and plasma.39
We also investigated whether atorvastatin-mediated improvements in eNOS activity would improve the bioavailability of NO and, by extension, endothelium-dependent vasodilation. The addition of serum from cigarette smokers to HUVEC plates decreases eNOS activity,4 and statins may increase eNOS activity.44 In our population, there was no significant difference in eNOS activity between the smokers and healthy subjects during placebo treatment and no change with atorvastatin treatment. The difference in our findings from previous studies may result from a limitation in the experimental method used to measure eNOS activity. This method requires cofactor repletion, including tetrahydrobiopterin, in the medium and, as such, may have obscured an effect of increased plasma oxidative stress, which would deplete tetrahydrobiopterin. Thus, by repleting the cofactor typically depleted by ROS, we may not have detected statin-mediated changes in eNOS activity.
Additional mechanisms by which cigarette smoking chronically impairs endothelial function independent of oxidative stress include increases in inflammation45 and activation of rho kinase.5 Statins have been demonstrated to reduce activation of the GTPase rho and systemic inflammation.46,47 Components of inflammation decrease eNOS mRNA, protein expression, and activity in vitro.48,49 Rho kinase presents an appealing possibility as a statin-specific mechanism. Activation of rho has been demonstrated to decrease eNOS activation and increase oxidative stress,50 effects that are ameliorated by statins.
Conclusion
These data demonstrate that atorvastatin restores endothelial function in normocholesterolemic cigarette smokers independent of changes in lipids, tissue oxidative stress, and eNOS mRNA expression. The precise mechanism through which this occurs could not be determined in this investigation but does not appear to be related to changes in tissue oxidative stress or eNOS concentration or function. These findings implicate a potential role for statin therapy to restore endothelial function and thereby investigate vascular disease in cigarette smokers.
Acknowledgments
This study was supported by grants from the National Institutes of Health (HL 56607, HL 48743, and K23 HL-04169) and Pharmacia Upjohn. M.A.C. is the Simon C. Fireman Scholar in Cardiovascular Medicine at Brigham and Woman’s Hospital.
References
- 1.Celermajer DS, Sorensen KE, Georgakopoulos D, Bull C, Thomas O, Robinson J, Deanfield JE. Cigarette smoking is associated with dose-related and potentially reversible impairment of endothelium-dependent dilation in healthy young adults. Circulation. 1993;88:2149–2155. doi: 10.1161/01.cir.88.5.2149. [DOI] [PubMed] [Google Scholar]
- 2.Widlansky ME, Gokce N, Keaney JF, Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42:1149–1160. doi: 10.1016/s0735-1097(03)00994-x. [DOI] [PubMed] [Google Scholar]
- 3.Heitzer T, Just H, Munzel T. Antioxidant vitamin C improves endothelial dysfunction in chronic smokers [see comments] Circulation. 1996;94:6–9. doi: 10.1161/01.cir.94.1.6. [DOI] [PubMed] [Google Scholar]
- 4.Barua RS, Ambrose JA, Eales-Reynolds LJ, DeVoe MC, Zervas JG, Saha DC. Dysfunctional endothelial nitric oxide biosynthesis in healthy smokers with impaired endothelium-dependent vasodilatation. Circulation. 2001;104:1905–1910. doi: 10.1161/hc4101.097525. [DOI] [PubMed] [Google Scholar]
- 5.Noma K, Higashi Y, Jitsuiki D, Hara K, Kimura M, Nakagawa K, Goto C, Oshima T, Yoshizumi M, Chayama K. Smoking activates rho-kinase in smooth muscle cells of forearm vasculature in humans. Hypertension. 2003;41:1102–1105. doi: 10.1161/01.HYP.0000067062.92836.9E. [DOI] [PubMed] [Google Scholar]
- 6.Vogel RA, Corretti MC, Plotnick GD. Changes in flow-mediated brachial artery vasoactivity with lowering of desirable cholesterol levels in healthy middle-aged men. Am J Cardiol. 1996;77:37–40. doi: 10.1016/s0002-9149(97)89131-x. [DOI] [PubMed] [Google Scholar]
- 7.Perticone F, Ceravolo R, Maio R, Cloro C, Candigliota M, Scozzafava A, Mongiardo A, Mastroroberto P, Chello M, Mattioli PL. Effects of ator-vastatin and vitamin C on endothelial function of hypercholesterolemic patients. Atherosclerosis. 2000;152:511–518. doi: 10.1016/s0021-9150(00)00370-1. [DOI] [PubMed] [Google Scholar]
- 8.Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–1719. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto M, Akita H. Cerivastatin, a hydroxymethylglutaryl coenzyme a reductase inhibitor, improves endothelial function in elderly diabetic patients within 3 days. Circulation. 2002;105:E30–E31. [PubMed] [Google Scholar]
- 10.Omori H, Nagashima H, Tsurumi Y, Takagi A, Ishizuka N, Hagiwara N, Kawana M, Kasanuki H. Direct in vivo evidence of a vascular statin: a single dose of cerivastatin rapidly increases vascular endothelial responsiveness in healthy normocholesterolaemic subjects. Br J Clin Pharmacol. 2002;54:395–399. doi: 10.1046/j.1365-2125.2002.01677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard-Herman M, Herrington D, Vallance P, Vita J, Vogel R. Guidelines for the ultrasound assessment of endothelial-dependent flow- mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39:257–265. doi: 10.1016/s0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- 12.Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem. 1997;272:31725–31729. doi: 10.1074/jbc.272.50.31725. [DOI] [PubMed] [Google Scholar]
- 13.Nitenberg A, Antony I, Foult JM. Acetylcholine-induced coronary vaso-constriction in young, heavy smokers with normal coronary arterio-graphic findings. Am J Med. 1993;95:71–77. doi: 10.1016/0002-9343(93)90234-g. [DOI] [PubMed] [Google Scholar]
- 14.Nakayama T, Kodama M, Nagata C. Generation of hydrogen peroxide and superoxide anion radical from cigarette smoke. Gann. 1984;75:95–98. [PubMed] [Google Scholar]
- 15.Schectman G, Byrd JC, Gruchow HW. The influence of smoking on vitamin C status in adults. Am J Public Health. 1989;79:158–162. doi: 10.2105/ajph.79.2.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolzan AD, Bianchi MS, Bianchi NO. Superoxide dismutase, catalase and glutathione peroxidase activities in human blood: influence of sex, age and cigarette smoking. Clin Biochem. 1997;30:449–454. doi: 10.1016/s0009-9120(97)00047-7. [DOI] [PubMed] [Google Scholar]
- 17.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 18.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raitakari OT, Adams MR, McCredie RJ, Griffiths KA, Stocker R, Celermajer DS. Oral vitamin C and endothelial function in smokers: short-term improvement, but no sustained beneficial effect. J Am Coll Cardiol. 2000;35:1616–1621. doi: 10.1016/s0735-1097(00)00576-3. [DOI] [PubMed] [Google Scholar]
- 20.Sugiyama Y, Yotsumoto H, Okabe T, Takaku F. Measurement of angiotensin-converting enzyme activity in intact human alveolar macrophages and effect of smoking. Respiration. 1988;53:153–157. doi: 10.1159/000195408. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Schmeisser A, Garlichs CD, Plotze K, Damme U, Mugge A, Daniel WG. Angiotensin II-induced superoxide anion generation in human vascular endothelial cells: role of membrane-bound NADH-/NADPH-oxidases. Cardiovasc Res. 1999;44:215–222. doi: 10.1016/s0008-6363(99)00183-2. [DOI] [PubMed] [Google Scholar]
- 22.Rueckschloss U, Quinn MT, Holtz J, Morawietz H. Dose-dependent regulation of NAD(P)H oxidase expression by angiotensin II in human endothelial cells: protective effect of angiotensin II type 1 receptor blockade in patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2002;22:1845–1851. doi: 10.1161/01.atv.0000035392.38687.65. [DOI] [PubMed] [Google Scholar]
- 23.Ota Y, Kugiyama K, Sugiyama S, Ohgushi M, Matsumura T, Doi H, Ogata N, Oka H, Yasue H. Impairment of endothelium-dependent relaxation of rabbit aortas by cigarette smoke extract–role of free radicals and attenuation by captopril. Atherosclerosis. 1997;131:195–202. doi: 10.1016/s0021-9150(97)06106-6. [DOI] [PubMed] [Google Scholar]
- 24.Butler R, Morris AD, Struthers AD. Lisinopril improves endothelial function in chronic cigarette smokers. Clin Sci (Lond) 2001;101:53–58. [PubMed] [Google Scholar]
- 25.Milstien S, Katusic Z. Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem Biophys Res Commun. 1999;263:681–684. doi: 10.1006/bbrc.1999.1422. [DOI] [PubMed] [Google Scholar]
- 26.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahy-drobiopterin regulatory process. J Biol Chem. 1998;273:25804–25808. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 28.Ueda S, Matsuoka H, Miyazaki H, Usui M, Okuda S, Imaizumi T. Tetrahydrobiopterin restores endothelial function in long-term smokers. J Am Coll Cardiol. 2000;35:71–75. doi: 10.1016/s0735-1097(99)00523-9. [DOI] [PubMed] [Google Scholar]
- 29.Heitzer T, Brockhoff C, Mayer B, Warnholtz A, Mollnau H, Henne S, Meinertz T, Munzel T. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers : evidence for a dysfunctional nitric oxide synthase. Circ Res. 2000;86:E36–E41. doi: 10.1161/01.res.86.2.e36. [DOI] [PubMed] [Google Scholar]
- 30.Su Y, Han W, Giraldo C, De Li Y, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. 1998;19:819–825. doi: 10.1165/ajrcmb.19.5.3091. [DOI] [PubMed] [Google Scholar]
- 31.Barbera JA, Peinado VI, Santos S, Ramirez J, Roca J, Rodriguez-Roisin R. Reduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokers. Am J Respir Crit Care Med. 2001;164:709–713. doi: 10.1164/ajrccm.164.4.2101023. [DOI] [PubMed] [Google Scholar]
- 32.Cai H, Davis ME, Drummond GR, Harrison DG. Induction of endothelial NO synthase by hydrogen peroxide via a Ca(2+)/calmodulin-dependent protein kinase II/janus kinase 2-dependent pathway. Arterioscler Thromb Vasc Biol. 2001;21:1571–1576. doi: 10.1161/hq1001.097028. [DOI] [PubMed] [Google Scholar]
- 33.Barua RS, Ambrose JA, Srivastava S, DeVoe MC, Eales-Reynolds LJ. Reactive oxygen species are involved in smoking-induced dysfunction of nitric oxide biosynthesis and upregulation of endothelial nitric oxide synthase: an in vitro demonstration in human coronary artery endothelial cells. Circulation. 2003;107:2342–2347. doi: 10.1161/01.CIR.0000066691.52789.BE. [DOI] [PubMed] [Google Scholar]
- 34.Laufs U, Endres M, Stagliano N, Amin-Hanjani S, Chui DS, Yang SX, Simoncini T, Yamada M, Rabkin E, Allen PG, Huang PL, Bohm M, Schoen FJ, Moskowitz MA, Liao JK. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J Clin Invest. 2000;106:15–24. doi: 10.1172/JCI9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morikawa S, Takabe W, Mataki C, Kanke T, Itoh T, Wada Y, Izumi A, Saito Y, Hamakubo T, Kodama T. The effect of statins on mRNA levels of genes related to inflammation, coagulation, and vascular constriction in HUVEC. Human umbilical vein endothelial cells. J Atheroscler Thromb. 2002;9:178–183. doi: 10.5551/jat.9.178. [DOI] [PubMed] [Google Scholar]
- 36.Delbosc S, Cristol JP, Descomps B, Mimran A, Jover B. Simvastatin prevents angiotensin II-induced cardiac alteration and oxidative stress. Hypertension. 2002;40:142–147. doi: 10.1161/01.hyp.0000024348.87637.6f. [DOI] [PubMed] [Google Scholar]
- 37.Wassmann S, Laufs U, Baumer AT, Muller K, Ahlbory K, Linz W, Itter G, Rosen R, Bohm M, Nickenig G. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension. 2001;37:1450–1457. doi: 10.1161/01.hyp.37.6.1450. [DOI] [PubMed] [Google Scholar]
- 38.Wagner AH, Kohler T, Ruckschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol. 2000;20:61–69. doi: 10.1161/01.atv.20.1.61. [DOI] [PubMed] [Google Scholar]
- 39.Shishehbor MH, Aviles RJ, Brennan ML, Fu X, Goormastic M, Pearce GL, Gokce N, Keaney JF, Jr, Penn MS, Sprecher DL, Vita JA, Hazen SL. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA. 2003;289:1675–1680. doi: 10.1001/jama.289.13.1675. [DOI] [PubMed] [Google Scholar]
- 40.Takajo Y, Ikeda H, Haramaki N, Murohara T, Imaizumi T. Augmented oxidative stress of platelets in chronic smokers. Mechanisms of impaired platelet-derived nitric oxide bioactivity and augmented platelet aggregability. J Am Coll Cardiol. 2001;38:1320–1327. doi: 10.1016/s0735-1097(01)01583-2. [DOI] [PubMed] [Google Scholar]
- 41.Sharma RN, Deva C, Behera D, Khanduja KL. Reactive oxygen species formation in peripheral blood neutrophils in different types of smokers. Indian J Med Res. 1997;106:475–480. [PubMed] [Google Scholar]
- 42.Kalinowski L, Dobrucki LW, Brovkovych V, Malinski T. Increased nitric oxide bioavailability in endothelial cells contributes to the pleiotropic effect of cerivastatin. Circulation. 2002;105:933–938. doi: 10.1161/hc0802.104283. [DOI] [PubMed] [Google Scholar]
- 43.Suzumura K, Yasuhara M, Narita H. Superoxide anion scavenging properties of fluvastatin and its metabolites. Chem Pharm Bull (Tokyo) 1999;47:1477–1480. doi: 10.1248/cpb.47.1477. [DOI] [PubMed] [Google Scholar]
- 44.Feron O, Dessy C, Desager JP, Balligand JL. Hydroxymethylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation. 2001;103:113–118. doi: 10.1161/01.cir.103.1.113. [DOI] [PubMed] [Google Scholar]
- 45.Bermudez EA, Rifai N, Buring JE, Manson JE, Ridker PM. Relation between markers of systemic vascular inflammation and smoking in women. Am J Cardiol. 2002;89:1117–1119. doi: 10.1016/s0002-9149(02)02284-1. [DOI] [PubMed] [Google Scholar]
- 46.Laufs U, Marra D, Node K, Liao JK. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27(Kip1) J Biol Chem. 1999;274:21926–21931. doi: 10.1074/jbc.274.31.21926. [DOI] [PubMed] [Google Scholar]
- 47.Jialal I, Stein D, Balis D, Grundy SM, Adams-Huet B, Devaraj S. Effect of hydroxymethyl glutaryl coenzyme a reductase inhibitor therapy on high sensitive C-reactive protein levels. Circulation. 2001;103:1933–1935. doi: 10.1161/01.cir.103.15.1933. [DOI] [PubMed] [Google Scholar]
- 48.Verma S, Wang CH, Li SH, Dumont AS, Fedak PW, Badiwala MV, Dhillon B, Weisel RD, Li RK, Mickle DA, Stewart DJ. A self-fulfilling prophecy: C-reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation. 2002;106:913–919. doi: 10.1161/01.cir.0000029802.88087.5e. [DOI] [PubMed] [Google Scholar]
- 49.Venugopal SK, Devaraj S, Yuhanna I, Shaul P, Jialal I. Demonstration that C-reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation. 2002;106:1439–1441. doi: 10.1161/01.cir.0000033116.22237.f9. [DOI] [PubMed] [Google Scholar]
- 50.Laufs U. Beyond lipid-lowering: effects of statins on endothelial nitric oxide. Eur J Clin Pharmacol. 2003;58:719–731. doi: 10.1007/s00228-002-0556-0. [DOI] [PubMed] [Google Scholar]