

Abstract
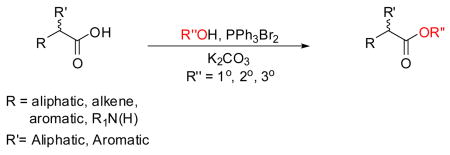
We report a one-pot, expedient protocol for the conversion of carboxylic acids to their esters using excess triphenylphosphine dibromide, base, and the alcohol. The reaction gave the esterified product in moderate-to-high yields (30–95%). For chiral acids, the reaction proceeded with little or no racemization. Use of a chiral alcohol in this transformation gave the ester with retention of configuration of the stereogenic center. Information is presented indicating that esterification proceeds through the intermediate generation of an acyloxyalkoxyphosphorane and where steric interactions play an important role in the energetics of the reaction.
1. Introduction
Esterification reactions are among the oldest and most often used reactions in organic chemistry.1 Numerous and diverse methods have been advanced for this transformation, assuring that most esters can be prepared. Nonetheless, a simpler, general method would facilitate future synthetic studies.
We sought a readily available esterification reagent that could convert carboxylic acids to their alkyl esters. We report herein that treating aliphatic and aromatic acids with triphenylphosphine dibromide2 (1), an excess of the primary, secondary, or tertiary alcohols, and a base provided the esterified products in a one–pot reaction. For optically active aliphatic acids, esterification proceeded with little or no racemization. Use of a chiral alcohol in this reaction gave the ester with retention of configuration of the stereogenic center. The conditions and scope of the esterification reactions provided information on the probable pathway for these transformations.
2. Results and discussion
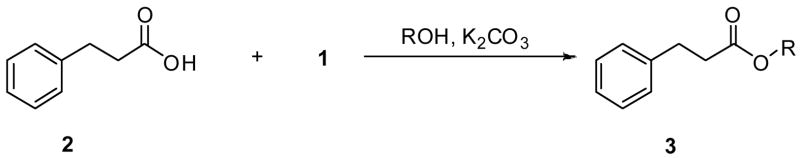
We first examined aliphatic acids and used 3-phenylpropionic acid (2) as the prototype. Treatment of 2 with 1 (5 equiv), K2CO3 (11 equiv), and excess MeOH (148 equiv) at room temperature (1 d) gave the esterified product 3a in 79% yield after purification (Table 1). Substitution of primary alcohols (EtOH, n-BuOH, allyl alcohol, 2-chloroethanol, 2-methoxyethanol) for MeOH provided the corresponding esters 3b–3f in 45–95% isolated yields. Similarly, the secondary alcohol, i-propanol, gave ester 3g in 88% yield. Finally, use of t-BuOH produced ester 3h in 63% yield. Our finding that t-butyl esters are prepared by this method is important since the acid–sensitive t-butyl group has proven to be useful in synthesis.4
Table 1.
Esterification 2 to give 3a
 | |||
|---|---|---|---|
| Entry | Alcohol | Product | Purified Yield (%)b |
| 1 | MeOH | 3ac | 79 |
| 2 | EtOH | 3bd | 95 |
| 3 | n-BuOH | 3ce | 63 |
| 4 | CH2=CHCH2OH | 3df | 53g |
| 5 | ClCH2CH2OH | 3eh | 82 |
| 6 | MeOCH2CH2OH | 3f i | 45 |
| 7 | i-PrOH | 3gd | 88 |
| 8 | t-BuOH | 3hc | 63 |
All reactions were conducted by treating 2 with an excess of alcohol (6 mL, 63–148 equiv), 1 (5 equiv), and K2CO3 (11 equiv) at room temperature for 1 d.
The products were purified by medium-pressure liquid chromatography (MPLC).
Reference 3a.
Reference 3b.
Reference 3c.
Reference 3d.
Compound 3d was purified by 2 MPLCs.
Reference 3e.
Reference 3f.
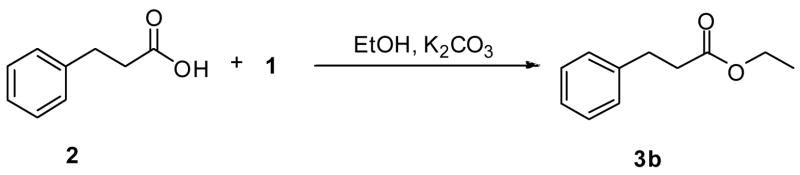
The effect of the number of equivalents of 1, K2CO3, and alcohol as well as the reaction conditions on the efficiency of the esterification process was briefly examined (Table 2). Employing the conversion of 2 to 3b as a test case, we observed lower yields of the esterified product (40–42%) compared with the 95% yield for 3b listed in Table 1 as we decreased either the number of equivalents of 1 and K2CO3 or the number of equivalents of EtOH (Table 2, entries 1 and 2). Efforts to improve the reaction efficiency by increasing the reaction temperature or reaction time (Table 2, entries 3 and 4) led to even lower yields of 3b (Table 2, entries 3 and 4).
Table 2.
Esterification conditions for the conversion of 2 to 3ba
 | |||||
|---|---|---|---|---|---|
| Entry | 1 (equiv) | K2CO3 (equiv) | Rxn Temp | Rxn Time (h) | Yield (%)b |
| 1 | 2 | 2 | 22 °C | 24 | 40 |
| 2 | 5 | 5 | 22 °C | 24 | 42 |
| 3 | 5 | 5 | 82 °C | 24 | 34 |
| 4 | 5 | 5 | 22 °C | 72 | 23 |
All reactions were run in acetonitrile using 2 (1 mmol, 1 equiv) and EtOH (5 equiv).
Isolated yields after MPLC.
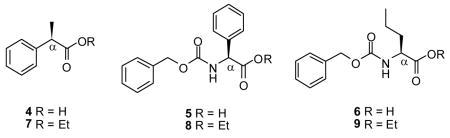
We asked the question whether the esterification procedure would proceed without racemization for optically active acids that had a chiral center at the C(α) site. To find out, we treated (R)-(−)-2-phenylpropionic acid (4), (S)-Cbz-phenylglycine (5), and (S)-Cbz-norvaline (6) with 1, EtOH, and K2CO3 (room temperature, 1 d). In all these cases, the reaction gave the optically pure esters 75 (73% yield, er ≥ 96.4%), 86 (79% yield, er ≥ 99.8 %), and 97 (77% yield, er ≥ 97.6 %).8
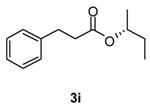
We determined if the esterification of 2 with a secondary, chiral alcohol proceeded with either retention or inversion of the stereogenic center in the alcohol. Using 2 and (R)-2-butanol we obtained 3i9 in 82% yield with complete retention of the 2-butoxy unit.
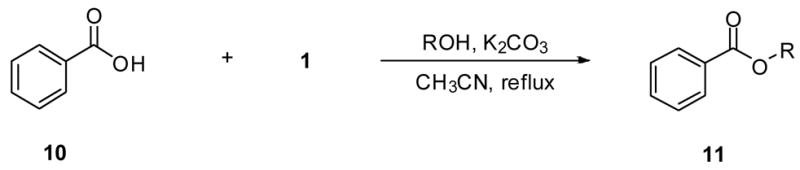
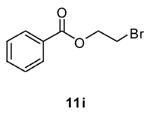
The reaction conditions needed to be modified in order to convert aromatic acids to their esters. Benzoic acid (10) served as our test substrate. Acetonitrile was used as the solvent, and reactions containing 10, 1 (5 equiv), alcohol (5 equiv), and K2CO3 (11 equiv) were heated at reflux for 3 d. Moderate yields (35–80%) were obtained for most of the purified products 11 (Table 3). For most unsubstituted alcohols the yields appeared to be largely independent of the alcohol used. However, use of t-BuOH in this procedure did not provide ester 11h. For 2-chloroethanol, we isolated a mixture of the 2-chloroethyl (11e) and 2-bromoethyl (11i) esters (NMR analysis). Similarly, when we used 2-methoxyethanol we isolated a mixture of the expected ester 11f along with the corresponding 2-bromoethyl ester 11i (NMR analysis).
Table 3.
Esterification of 10 to give 11a
Phosphorus reagents (e.g., dihalophosphoranes, dialkoxyphosphoranes, Wittig reagents, trihalophosphines) have been used extensively in synthesis. In particular, treating carboxylic acids with 1 in aprotic solvents provides the acid halides.11–13 Likewise, the dialkoxyphosphoranes and other similarly disubstituted reagents (e.g., bis(2,2,2-trifluoroethoxy)phosphorane,14–16 2,2,2-triphenyl-4,5-(2′,2″-biphenylene)-1,3,2-dioxaphospholane,17 triphenylphosphine thiocyanogen,18–22 triphenylphosphine ditriflate23) have been used to prepare carboxylic acid derivatives.
In Scheme 1, we propose a pathway for the observed esterification reactions. In the initial step, 1 undergoes rapid halide exchange leading either to the monoalkoxy 12 or dialkoxy phosphorane, 24–26 which can possibly exist in equilibrium with the phosphonium salt 13. The subsequent attack of 12 by the acid carboxylate provides intermediate 14,27 which can either react with the alcohol in solution (pathway a) or with the appended phosphorane alkoxy unit by an intramolecular route after pseudorotation to give 14′ to provide the esterified product 16 (pathway b). The pathway in Scheme 1 is similar, in part, to that proposed for the Mitsunobu reaction.28 Unlike most Mitsunobu transformations, we observed retention of the stereogenic center in 3i.9 We also discovered that 2 was converted to ester 3h with t-BuOH (Table 1). Recently, Anders and coworkers have conducted a density functional analysis of the Mitsunobu reaction providing a rationale for both the typical inverted product and the atypical retention adduct.30 Significantly, these investigators suggested that the retention product stems from an acyloxyalkoxyphosphorane species similar to 14 that undergoes Berry pseudorotation and intramolecular substitution.
Scheme 1.

Proposal Pathway for 1-Mediated Esterification Processes
The pathway outlined in Scheme 1 is consistent with the reported ease with which dihalophosphoranes react with alcohols,24–26 and phosphorane-mediated mechanisms that activate carboxylic acid derivatives toward nucleophilic substitution reactions.11–13,31,32 Several experimental observations supported the proposed pathway and provided insights into the controlling factors that governed this transformation. First, the yields for 3c were independent of the order in which reagents were added. For example, we obtained 3c in 63% yield after we added 1 to a reaction mixture consisting of 2, n-BuOH, and K2CO3. Similarly, 3c was produced in 70% yield after we premixed 1, n-BuOH, and K2CO3 for 20 min before adding 2. These results are consistent with the initial reaction of 1 with the alcohol rather than with the carboxylate. Second, we tested whether dialkoxyphosphoranes, similar to intermediate 12, could convert carboxylic acids to the corresponding ester. Treatment of 2 with diethoxyphosphorane (DTPP, 18) gave 3b (85%), thus demonstrating that 18 and similar compounds can serve as esterification reagents. Indeed, the Evans laboratory reported that 18-treated 19 provided the esterified product 20.33 The utility of 18 for ethyl esterification was not further explored. Third, we observed that 2 was converted to ester 3i9 with retention of the stereogenic center in the (R)-2-butanoxy group, and that 2 gave ester 3h with t-butanol. These collective findings suggested that the reaction proceeded through intermediate 14.
The different reactivities of 2 and 10 with 1 prompted us to
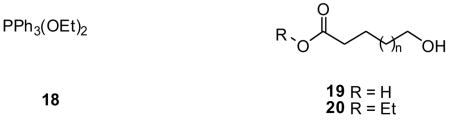
determine if either electronic or steric factors governed the esterification process. We esterified trans-cinnamic acid (21), 3-(3-pyridyl)propionic acid (22), and nicotinic acid (23) under the same conditions employed for either 2 or 10. The α,β–unsaturated acid 21 readily reacted with EtOH under the mild conditions used for the aliphatic acid 2 (room temperature, 1 d) to give 2434 (57%). The 3-pyridyl-substituted aliphatic acid 22 was converted to the ethyl 25a35 (76%), i-propyl 25b (75%), and t-butyl 25c (30%) esters at room temperature (1 d). Esterification of nicotinic acid (23) with EtOH and i-propanol required the use of acetonitrile and heat (reflux, 3 d) to give the ethyl 26a36a (40%) and i-propyl 26b36b (65%) esters, respectively. The esterification yields of 23 were similar to those of 10 (Table 3), demonstrating that substituting the phenyl group in 10 by the electron-deficient pyridine ring in 23 did not markedly affect this transformation. Collectively, these results showed that electronic effects produced by the R-substituent in carboxylic acids (RCO2H) could not account for the different conditions needed for 2 or 10 esterification. Consistent with this notion, is the finding that the pKa values37 for 2 and 10 fall within a narrow range (pKa 4.2–4.4) indicating that under the mild basic conditions of our reactions both carboxylic acids are converted to the carboxylates.
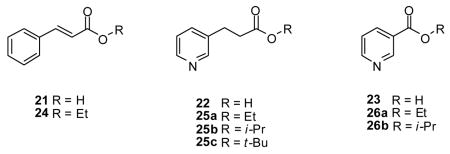
We then asked whether steric factors affected esterification of 2 and 10. In our proposed pathway (Scheme 1), 14 serves as the central intermediate. This species places the carboxylic acid R-subsituent (alkyl, aryl) in proximity to 1 of the 3 phenyl groups in the proposed trigonal–bipyramidal intermediate 14. X-ray crystallographic studies of disubstituted triphenylphosphoranes25,38 show that the 3 phenyl groups are canted with respect to one another. If the esterification reaction proceeds through 14 then the phenyl R-substituent in 10 is likely to undergo an adverse steric interaction with 1 of the 3 phenyl groups in the triphenylphosphorane compared with the aliphatic R-substituent in 2. We have modeled the 14 intermediate for the 2, 10, and 23 transformations using SPARTAN ‘06 semi-empiric AM1 equilibrium geometry calculations and we found that the energy–minimized values for the central phosphorane intermediate for 10 and 23 to be appreciably higher than 2 (data not shown). To test this hypothesis further, we treated the tertiary–substituted aliphatic acid, 2-methyl-2-phenylpropionic acid (27) with 1 and EtOH and compared the ease of esterification of 27 with 4. We found that 4 converted to the ethyl ester 7 (73%) under mild conditions (room temperature, 1 d) while 27 required acetonitrile and heat (60 °C, 3 d) to provide its corresponding ethyl ester 2839 (66%). Molecular–modeling studies indicated that the energy minimized value for the intermediate 14 generated from 27 was higher than predicted from either 2 or 4. These results, taken together, support the proposed pathway for esterification (Scheme 1) and provide evidence that the energetics for the formation of 14 governs the conditions required for esterification.40
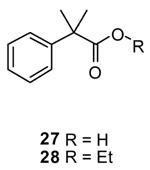
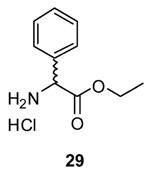
The efficiency of the 1-mediated esterification reaction was compared with established procedures. We chose (S)-Cbz-phenylglycine (5) as a test substrate since it contained a commonly used protecting group (Cbz) and a stereogenic center prone to racemization. First, using Fischer esterification conditions41 (EtOH (115 equiv), HCl (20 equiv), 78 °C, 16 h) we obtained partially racemized ester 2942 in near quantitative yield and where the Cbz-group was removed. Second, when 5 was esterified with EtOH using the mixed anhydride procedure43 (i, isobutyl chloroformate (1.2 equiv), 4-methylmorpholine (1.2 equiv), −78 °C; ii, EtOH (5 equiv), −78 °C–22 °C, 4 h) we obtained partially racemized 8 in 61% yield. Third, addition of thionyl chloride (15 equiv) to a dichoromethane solution containing 5 and a catalytic amount of DMF (40 °C, 2 h), followed by concentration of the reaction mixture and addition of EtOH44 led to a complex product mixture. Finally, treatment of 5 with thionyl chloride (2 equiv) and EtOH (35 equiv) at 22 °C (16 h)45 gave optically pure 8 in 93% yield with little or no racemization.
The proposed pathway for esterification of carboxylic acids (Scheme 1) suggests that similar treatment of esters with 1 and alcohols would provide the trans-esterified products. We briefly explored this using acetonitrile as a solvent and a temperature maintained at reflux for 24 h. Treatment of 3a with 1, K2CO3 (6 equiv) and EtOH (5 equiv) gave 3b (70%) along with the recovered methyl ester 3a (<10%). When we substituted n-BuOH for EtOH we obtained a mixture of methyl (3a), and n-butyl (3c) esters in a 1:1.5 ratio, respectively, and an ~60% yield for 3c.
3. Conclusions
In summary, we report a simple, one–pot experimental protocol for converting alkyl and aromatic acids to their alkyl esters using 1 and that proceeds with little or no racemization. Reagent 1 is commercially available and can be prepared from triphenylphosphine and Br2 in near–quantitative yields.25 The method relies on in situ conversion of 1 to an alkoxyphosphorane, which serves as the esterification agent. The described one–pot protocol has several important advantages over the conventional two-step procedure which entails converting the acid to the acid halide with 1,11–13 then treating the isolated acid halide with the alcohol. These advantages include the procedure’s simplicity as well as eliminating the need to handle and isolate acid halides which are moisture sensitive and are lachrymators,11–13 or the use of aprotic solvents to generate the acid halide. Our studies indicate that the reaction is governed, in part, by the energetics that form the initial pentavalent intermediate 14 containing the carboxylate and in which steric interactions have an important role. Finally, the different reactions reported for 1 among functional groups will affect the scope of this esterification process with respect to the permissible R–substituents in the starting acid.
4. Experimental
4.1. General procedures
Unless otherwise noted, all materials were used as received from a commercial supplier without further purification. All reactions were monitored by analytical thin layer chromatography (TLC) plates (Aldrich, Cat # Z12272-6) and analyzed with 254 nm UV light. The reactions were purified by MPLC (CombiFlash Rf) with self packed columns (silica gel from Dynamic Adsorbents Inc., Cat # 02826-25). Known compounds were characterized by 1H NMR, 13C NMR and MS (when possible) and compared to their literature values. Melting points were determined with a Thomas–Hoover melting point apparatus and are uncorrected. Optical rotations were obtained on a Jasco P-1030 polarimeter. Proton (1H NMR, 300 MHz) and carbon (13C NMR, 75 MHz) nuclear magnetic resonance spectra were taken on a Varian Gemini 2000 spectrometer in CDCl3. Chemical shifts (δ) were reported in ppm relative to TMS as an internal reference. Coupling constants (J) were reported in Hertz (Hz). Peak multiplicity is indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), br (broad), sept (septet) and m (multiplet). The high–resolution mass spectrum was performed on a Bruker Apex-Q 12 Tesla FTICR by Dr. M. Crowe at the University of North Carolina–Chapel Hill, and the low–resolution mass spectra were done on a BioToF-II-Bruker Daltonics spectrometer by Dr. S. Habibi. Microanalysis were provided by Atlantic Microlab, Inc. (Norcross, GA). The yields refer to isolated yields of compounds estimated to be ≥ 95% pure as determined by 1H NMR.
4.2. General Procedure for the Esterification of Aliphatic Acids 2 and α,β–Unsaturated Acid 21
Reagent 1 (2.20 g, 5 mmol) was added to an alcoholic solution (6 mL) of K2CO3 (1.60 g, 11 mmol) and the acid (1 mmol). The reaction mixture was stirred at room temperature (24 h). The volatiles were removed and H2O (25 mL) and CH2Cl2 (25 mL) were added to the residue. The organic layer was removed and the aqueous layer was washed with CH2Cl2 (25 mL). The organic layers were combined and concentrated in vacuo. The residue was purified by Medium Pressure Liquid Chromatography (MPLC) with hexanes as the eluent to obtain a colorless oil. The compounds prepared by this method were consistent with those published. Representatives examples follow:
4.2.1. i-Propyl 3-(Pyridin-3-yl)propanoate (25b)
Rf = 0.37 (50% EtOAc in hexanes); IR (neat) 2779, 2392, 1731, 1579, 1429, 1376, 1186, 1035, 858, 802, 715 cm−1; 1H NMR (CDCl3): δ 1.20 (d, J = 6.3 Hz, 2 CH3), 2.61 (t, J = 7.5 Hz, CH2), 2.95 (t, J = 7.5 Hz, CH2), 5.00 (sept, J = 6.3 Hz, OCH), 7.22 (dd, J = 4.8, 7.8 Hz, H5), 7.54 (dt, J = 2.1, 7.8 Hz, H4), 8.45–8.49 (m, H2 and H6); 13C NMR (CDCl3): δ 21.7 (2 CH3), 28.1 (CH2), 35.7 (CH2), 67.9 (CHO), 123.3, 135.8, 147.7, 149.9 (C5H4N), 171.8 (C(O)), the remaining pyridine resonance was not detected and is believed to overlap with nearby peaks; MS (M+H+)(ESI+) 194.2 [M + H+] (calcd for C12H17NO2H+ 194.1); Anal. Calcd. for C11H15NO2: C, 68.37; H, 7.82; N, 7.25. Found: C, 68.23; H, 7.88; N, 7.26.
4.2.2. t-Butyl 3-(Pyridin-3-yl)propanoate (25c)
Rf = 0.38 (50% EtOAc in hexanes); IR (neat) 2924, 2860, 1727, 1456, 1369, 1254, 1152, 1031, 847, 801, 714 cm−1; 1H NMR (CDCl3): δ 1.41 (s, C(CH3)3), 2.56 (t, J = 7.8 Hz, CH2), 2.91 (t, J = 7.8 Hz, CH2), 7.21 (dd, J = 4.8, 7.8 Hz, H5), 7.53 (br d, J = 7.8 Hz, H4), 8.45–8.49 (m, H2 and H6); 13C NMR (CDCl3): δ 28.0 (C(CH3)3), 28.2 (CH2), 36.5 (CH2), 80.7 (OC(CH3)3), 123.3, 135.8, 147.7, 149.9 (C5H4N), 171.6 (C(O)), the remaining pyridine resonance was not detected and is believed to overlap with nearby peaks; MS (M+H+)(ESI+) 208.2 [M + H+] (calcd for C12H17NO2H+ 208.1); Anal. Calcd. for C12H17NO2·0.07H2O: C, 69.09; H, 8.29; N, 6.71. Found: C, 69.21; H, 8.20; N, 6.32.
4.3. General Procedure for the Esterification of Chiral Acids
Reagent 1 (2.20 g, 5 mmol) was added to an alcoholic solution (6 mL) of K2CO3 (0.76 g, 5.5 mmol) and the acid (1 mmol). The reaction mixture was stirred at room temperature (24 h). The volatiles were removed and H2O (25 mL) and CH2Cl2 (25 mL) were added to the residue. The organic layer was separated and the aqueous layer was washed with CH2Cl2 (25 mL). The organic layers were combined and concentrated in vacuo. The residue was purified by MPLC with hexanes as the eluent to obtain the esters. The compounds prepared by this method were consistent with those published.
4.4. General Procedure for the Esterification of Aromatic Acids 10, and Acids 21 and 23
K2CO3 (1.60 g, 11 mmol) was added to a CH3CN solution (6 mL) containing the acid (1 mmol). The mixture was stirred at room temperature (20 min) and 1 (2.20 g, 5 mmol) and the alcohol (5 mmol) were added. The reaction mixture was stirred at reflux (72 h). The volatiles were removed and H2O (25 mL) and CH2Cl2 (25 mL) were added to the residue. The organic layer was separated and the aqueous layer was washed with CH2Cl2 (25 mL). The organic layers were combined and concentrated in vacuo. The residue was purified by MPLC with hexanes as the eluent to obtain a colorless oil. The compounds prepared by this method were consistent with those published.
4.5. General Procedure for the Esterification of Aliphatic Acids 2 with DTPP (18)
DTPP (18) (1.76 g, 5 mmol) was added to an alcoholic solution (6 mL) of the acid (1 mmol), and the reaction mixture was stirred at room temperature (24 h). The volatiles were removed and the residue was purified by MPLC with hexanes as the eluent to obtain a colorless oil.
Supplementary Material
Spectroscopic data (1H and 13C NMR) of all purified products, and experimental details on the use of 18. Supplementary data associated with this article can be found, in the online version, at.
Acknowledgments
We thank Mr. Pierre Morieux and Ms. Amber M. King (University of North Carolina-Chapel Hill) for helpful discussions and advice, and Professor Michel Gagne (University of North Carolina-Chapel Hill) for his help in the energy minimization studies. The project was supported by grant RO1N5054112 (HK) from the National Institute of Neurological Disorders and Stroke.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Otera J. Esterification Methods. Reactions and Applications. Wiley-VCH; Weinheim, Germany: 2003. [Google Scholar]
- 2.Paquette LA. Encyclopedia of Reagents for Organic Synthesis. Vol. 8. John Wiley; Chichester, England: 1995. pp. 5370–5375. [Google Scholar]
- 3.(a) Black PJ, Edwards MG, Williams JMJ. Eur J Org Chem. 2006:4367–4378. [Google Scholar]; (b) Hayashi M, Kawabata H, Yoshimoto K, Tanaka T. Phosphorus, Sulfur, and Silicon. 2007;182:433–445. [Google Scholar]; (c) Horiguchi H, Tsurugi H, Satoh T, Miura M. J Org Chem. 2008;73:1590–1592. doi: 10.1021/jo702546t. [DOI] [PubMed] [Google Scholar]; (d) Crosignani S, White PD, Linclau B. J Org Chem. 2004;69:5897–5905. doi: 10.1021/jo049239e. [DOI] [PubMed] [Google Scholar]; (e) Mukaiyama T, Ichikawa J, Asami M. Chem Letters. 1983:293–296. [Google Scholar]; (f) Takahashi S, Cohen LA, Miller HK, Peake EG. J Org Chem. 1971;36(9):1205–1209. [Google Scholar]
- 4.Stanton MG, Gagne MR. J Org Chem. 1997;62:8240–8242. doi: 10.1021/jo971138b. [DOI] [PubMed] [Google Scholar]
- 5.Yang H, Henke E, Bornscheuer UT. J Org Chem. 1999;64:1709–1712. doi: 10.1021/jo981780l. [DOI] [PubMed] [Google Scholar]
- 6.(a) Hang J, Tian SK, Tang L, Deng L. J Am Chem Soc. 2001;123:12696–12697. doi: 10.1021/ja011936q. [DOI] [PubMed] [Google Scholar]; (b) Deng L, Hang J, Tang L. US patent 2003/0166963. 2003;A1 [Google Scholar]
- 7.Moriniere JL, Danree B, Lemoine J, Guy A. Syn Commun. 1988;18:441–444. [Google Scholar]
- 8.Use of 11 equivalents of K2CO3 instead of 5 led to no detectable loss in the optical purity of 7.
- 9.Lin YY, Palmer DN, Jones JB. Can J Chem. 1974;52:469–476. [Google Scholar]
- 10.(a) Li LC, Ren J, Liao TG, Jiang JX, Zhu HJ. Eur J Org Chem. 2007:1026–1030. [Google Scholar]; (b) Pouchert C, Behnke J. Aldrich FT-NMR Chemical Co. Aldrich Chemical Co.; Milwaukee, WI: 1993. [Google Scholar]; (c) Faler CA, Joullié MM. Tetrahedron Lett. 2006:7229–7231. [Google Scholar]; (d) Guo Q, Miyaji T, Hara R, Shen B, Takahashi T. Tetrahedron. 2002;58:7327–7334. [Google Scholar]; (e) Wiseman RL, Johnson SM, Kelker MS, Foss T, Wilson IA, Kelly JW. J Am Chem Soc. 2005;127:5540–5551. doi: 10.1021/ja042929f. [DOI] [PubMed] [Google Scholar]; (f) Hans JJ, Driver RW, Burke SD. J Org Chem. 2000;65:2114–2121. doi: 10.1021/jo991711m. [DOI] [PubMed] [Google Scholar]; (g) Chen CT, Kuo JH, Pawar VD, Munot YS, Weng SS, Ku CH, Liu CY. J Org Chem. 2005;70:1188–1197. doi: 10.1021/jo048363v. [DOI] [PubMed] [Google Scholar]
- 11.Bestmann HJ, Mott L. Liebigs Ann. 1966;693:132–133. [Google Scholar]
- 12.Naora H, Ohnuki T, Nakamura A. Chem Letters. 1988:143–144. [Google Scholar]
- 13.Aizpurua JM, Palomo C. Synthesis. 1982:684–687. [Google Scholar]
- 14.Kubota T, Kitazume T, Ishikawa N. Chem Letters. 1978:889–892. [Google Scholar]
- 15.Kubota T, Miyashita S, Kitazume T, Ishikawa N. Chem Letters. 1979:845–846. [Google Scholar]
- 16.Kubota T, Miyashita S, Kitazume T, Ishikawa N. J Org Chem. 1980;45:5052–5057. [Google Scholar]
- 17.Kelly JW, Robinson PL, Evans SA., Jr J Org Chem. 1986;51:4473–4475. [Google Scholar]
- 18.Tamura Y, Kawasaki T, Yasuda H, Gohda N, Kita Y. J Chem Soc Perkin I. 1981:1577–1581. [Google Scholar]
- 19.Iranpoor N, Firouzabadi H, Shaterian HR. J Chem Res (S) 1999:676–677. [Google Scholar]
- 20.Iranpoor N, Firouzabadi H, Shaterian HR. Tetrahedron Lett. 2002;43:3439–3441. [Google Scholar]
- 21.Iranpoor N, Firouzabadi H, Shaterian HR. Syn Commun. 2002;32:3653–3657. [Google Scholar]
- 22.Pritchard KM, Al-Rawi JM, Hughes AB. Syn Commun. 2005;35:1601–1611. [Google Scholar]
- 23.Hendrickson JB, Schwartzman SM. Tetrahedron Lett. 1975;4:277–280. [Google Scholar]
- 24.Herr ME, Johnson RA. J Org Chem. 1972;37:310–312. [Google Scholar]
- 25.Mathieu-Pelta I, Evans SA., Jr J Org Chem. 1994;59:2234–2237. [Google Scholar]
- 26.Wiley GA, Rein BM, Hershkowitz RL. Tetrahedron Lett. 1964;36:2509–2513. [Google Scholar]
- 27.(a) Burton DJ, Koppes WM. J Org Chem. 1975;40:3026–3032. [Google Scholar]; (b) Shintou T, Mukaiyama T. J Am Chem Soc. 2004;126:7359–7367. doi: 10.1021/ja0487877. [DOI] [PubMed] [Google Scholar]
- 28.(a) Hughes DL. In: Organic Reactions. Beak P, et al., editors. Vol. 42. John Wiley & Sons, Inc.; New York: 1992. pp. 335–656. [Google Scholar]; (b) But TYS, Toy PH. Chem Asian J. 2007;2:1340–1355. doi: 10.1002/asia.200700182. [DOI] [PubMed] [Google Scholar]
- 29.For other examples of Mitsunobu reactions proceeding with retention of the alcohol stereogenic center, see: Dinsmore CJ, Mercer SP. Org Lett. 2004;6:2885–2888. doi: 10.1021/ol0491080.Liao X, Wu Y, De Brabander JK. Angew Chem Int Ed. 2003;42:1648–1652. doi: 10.1002/anie.200351145.Smith AB, III, Safanov IG, Corbett RM. J Am Chem Soc. 2002;124:11102–11113. doi: 10.1021/ja020635t.Ahn C, Correia R, DeShong P. J Org Chem. 2002;67:1751–1753. doi: 10.1021/jo001590m.Ahn C, DeShong P. J Org Chem. 2002;67:1754–1759. doi: 10.1021/jo001525c.
- 30.Schenk S, Weston J, Anders E. J Am Chem Soc. 2005;127:12566–12576. doi: 10.1021/ja052362i. [DOI] [PubMed] [Google Scholar]
- 31.Burton DJ, Koppes WM. J Chem Soc, Chem Commun. 1973:425. [Google Scholar]
- 32.(a) Smissman EE, Alkaysi HN, Creese MW. J Org Chem. 1975;40:1640–1641. [Google Scholar]; (b) Chaturvedi D, Mishra N, Mishra V. Tetrahedron Lett. 2007;48:5043–5045. [Google Scholar]
- 33.Kelly JW. PhD Dissertation. University of North Carolina; Chapel Hill: 1986. [Google Scholar]
- 34.Chandrasekhar S, Reddy GPK, Nagesh C, Reddy CR. Tetrahedron Lett. 2007;48:1269–1271. [Google Scholar]
- 35.Amatore M, Gosmini C, Périchon J. J Org Chem. 2006;71:6130–6134. doi: 10.1021/jo060855f. [DOI] [PubMed] [Google Scholar]
- 36.(a) Sambrook MR, Beer PD, Wisner JA, Paul RL, Cowley AR, Szemes F, Drew MGB. J Am Chem Soc. 2005;127:2292–2302. doi: 10.1021/ja046278z. [DOI] [PubMed] [Google Scholar]; (b) Srinivasan R, Srinivasa Rao K, Jayachitra G, Ralte SL. Syn Commun. 2006;36:2883–2886. [Google Scholar]
- 37.Weast Robert C. Handbook of Chemistry and Physics. Vol. 54. CRC Press; Cleveland, Ohio: 1974. pp. D-129–D-130. [Google Scholar]
- 38.Barnes NA, Godfrey MS, Halton RTA, Law S, Pritchard RG. Angew Chem Int Ed. 2006;45:1272–1275. doi: 10.1002/anie.200503335. [DOI] [PubMed] [Google Scholar]
- 39.Raber DJ, Gariano P, Jr, Brod AO, Gariano A, Guida WC, Guida AR, Herbst MD. J Org Chem. 1979;44:1149–1154. [Google Scholar]
- 40.A similar adverse steric interaction may exist after Berry psuedorotation of 14 to 14′ prior to intramolecular attack to give the esterified product.
- 41.Fischer E, Speier A. Chem Ber. 1895;28:3252–3258. [Google Scholar]
- 42.(a) Duhamel L, et al. Tetrahedron. 1988;44:5495–5506. [Google Scholar]; (b) O’Donnell MJ, Bennett WD, Jacobsen WN, Ma Y, Huffman JC. Tetrahedron Lett. 1989;30:3909–3912. [Google Scholar]
- 43.Anderson GW, Zimmerman JE, Callahan FM. J Am Chem Soc. 1967;89:5012–5017. doi: 10.1021/ja00995a032. [DOI] [PubMed] [Google Scholar]
- 44.Bennacer B, Rivalle C, Grierson DS. Eur J Org Chem. 2003:4569–4574. [Google Scholar]
- 45.Brenner M, Huber W. Helv Chim Acta. 1953;36:1109–1115. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectroscopic data (1H and 13C NMR) of all purified products, and experimental details on the use of 18. Supplementary data associated with this article can be found, in the online version, at.