

Abstract
Purpose
SP4 is a transcription factor abundantly expressed in retina that binds to the GC promoter region of photoreceptor signal transduction genes. We have previously shown that SP4 may be involved in the transcriptional activation of these genes alone or together with other transcription factors such as SP1, neural retina leucine zipper protein (NRL), and cone-rod homeobox gene (CRX). Since mutations in NRL and CRX are involved in inherited retinal degenerations, SP4 was considered a good candidate for mutation screening in patients with this type of diseases. The purpose of this work, therefore, was to investigate possible mutations in SP4 in a cohort of patients affected with different forms of retinal degenerations.
Methods
270 unrelated probands with various forms of retinal degeneration including autosomal dominant and autosomal recessive retinitis pigmentosa (RP), autosomal dominant and autosomal recessive cone-rod dystrophy (CRD), and Leber's congenital amaurosis (LCA), were screened for mutations in the SP4 gene. Single strand conformation polymorphism (SSCP) analysis was performed on the six SP4 gene exons including flanking regions followed by direct sequencing of SSCP variants.
Results
Nine different sequence variants were found in 29 patients, four in introns and five in exons. Many of the probands were previously screened for mutations in the genes encoding the α-, β- and γ-subunits of rod-specific cGMP phosphodiesterase (PDE6A, PDE6B, PDE6G), the β-subunit of rod-specific transducin (GNB1), and peripherin/rds (RDS). One group of seven probands of Hispanic background that included five with arRP, one with RP of unknown inheritance (isolate) and 1 with arCRD carried an Asn306Ser mutation in SP4. Of the seven, the isolate case was homozygous and the other 6 heterozygous for the variant. Two arRP and the arCRD probands carried an additional intronic GNB1 variant. DNA from the family members of the arCRD proband could not be obtained, but for the other two families, all affected members and none of the unaffected carried both the SP4 Asn306Ser allele and the GNB1 intronic variant.
Conclusions
If mutations in SP4 do cause retinal degenerative disease, their frequency would be low. While digenic disease with the SP4 Asn306Ser and the GNB1 intronic variant alleles has not been established, neither has it been ruled out. This leaves open the possibility of a cooperative involvement of SP4 and GNB1 in the normal function of the retina.
Introduction
Transcription factors have been shown to play an important role not only in the biology of photoreceptors and other retinal cells, but also as sites of mutations causing degenerative disease. For example, mutations in CRX and NRL cause various forms of progressive retinal dystrophies [1-4]. The SP family of transcription factors (SP1-5) is formed by a group of proteins that selectively bind to the "GC box" in the promoter region of many genes [5-7] via three putative zinc finger domains of the C2H2 type [8]. Based on sequence similarities, SP1, SP3, and SP4 are more closely related to each other than to SP2 and SP5 [6,9] and the three have similar affinity for the GC box [10,11]. While SP1 and SP3 are ubiquitously expressed, SP4 is most abundant in the developing nervous system, particularly in the hippocampus [12] and retina [13], although it is also expressed in other tissues [10,14]. Sp1 and Sp3 knockout mice all die after E10 or at birth, respectively. Sp4 knockout mice appear to develop normally to birth, but after birth, many pups die by P28 and those that survive are small and have other abnormalities [12]. In the last few years, we have studied extensively the SP4 transcription factor and have found that it is present in all retinal layers, interacts with CRX and NRL and activates transcription of several rod specific genes including PDE6B and RHO, encoding the β-subunit of cGMP-phosphodiesterase (β-PDE) and rod opsin, respectively [13,15,16]. Because of its specific involvement in transcription of rod genes and because of the history of transcription factor mutations causing retinal degeneration, we considered SP4 a good candidate gene to screen for missense mutations in patients with various forms of retinal degeneration. To this end, we screened the 6 exons of the SP4 gene in a group of 270 patients with various forms of retinal degeneration that had been screened previously for a number of photoreceptor genes [17-21]. Although we could not establish that mutations in the SP4 gene cause retinal degenerative disease, neither could we rule this possibility out because the families of two patients in which an SP4 missense mutation and a GNB1 intron 2 variant were present, segregated with disease. Interestingly, the inherited retinal degeneration of the Rd4 mouse is caused by an inversion of mouse chromosome 4, and the site of the telomeric breakpoint is precisely on intron 2 of the Gnb1 gene [22].
Methods
Patients
270 patient probands of mixed ethnicities (56% European, 17% Asian, 13% Black, 14% Hispanic) were screened for variants in the six exons of the SP4 gene, including 49 with autosomal dominant retinitis pigmentosa (adRP), 103 with autosomal recessive retinitis pigmentosa (arRP), 26 with autosomal dominant cone-rod dystrophy (adCRD), 52 with autosomal recessive cone-rod dystrophy (arCRD), and 40 with Leber's congenital amaurosis (LCA). Many of the above patients had been previously screened for mutations in the genes encoding rod- αPDE, βPDE, γPDE, rod β-transducin and RDS-peripherin. 95 controls with a similar ethnic distribution (58% European, 16% Asian, 13% Black, 13% Hispanic) were screened for each of the above genes and SP4 as well. Written informed consent was obtained in compliance with the tenets of the declaration of Helsinki and with the approval of the office of Human Research Protection of the School of Medicine, University of California, Los Angeles.
Polymerase chain reaction
Blood was drawn in 10-20 ml aliquots and DNA was extracted from the leukocytes by standard methods. Initial screening was done by SSCP as described previously [17-21]. The exons of SP4 were amplified by polymerase chain reaction (PCR) directly from genomic DNAs with appropriate primers pairs. Each PCR amplicon included 50-150 nt of intronic flanking sequence on each side of the exon. The PCR protocol was 94 °C for 3 min followed by 30 cycles of 94 °C for 45 s, 55-60 °C for 45 s and 72 °C for 45 s, followed by 5 min at 72 °C. The sequences of primer pairs are presented in Table 1.
Table 1. Primer sequences used for SP4 and GNB1 screening.
| Exon 1 |
Sp4 |
1F: |
5’-TGTGCCAGCTACAGCCTCCT |
|
| Sp4 |
1R: |
5’-CCCCTTAAAGCATCTCAGCG |
||
| Exon 2 |
Sp4 |
2F: |
5’-GCCCGCGCTCGCGGGTTTATGGAGATTA |
|
| Sp4 |
2R: |
5’-GGCGGCCAAAGGGCATAACTCCCTCTC |
||
| Exon 3 |
1 |
Sp4 |
3AF: |
5’-GCAACTACTGGCCTCCACTA |
| Sp4 |
3AR: |
5’-GACCAGGGGTGGAAGAATT |
||
| 2 |
Sp4 |
3BF: |
5’-ACTTGTTGCCTCCACTCCT |
|
| Sp4 |
3BR: |
5’-GTAGAGATGAACTACTAGTTGG |
||
| 3 |
Sp4 |
3CF: |
5’-GCCCCCGCCGAAACTTCAGACAGTGGAAGGTC |
|
| Sp4 |
3CR: |
5’-CTGGCAAAGCTAGAGTCACT |
||
| 4 |
Sp4 |
3DF: |
5’-GCCCCCGCCGAGGCTCAAGTTGTAACAACC |
|
| Sp4 |
3DR: |
5’-ATTGATCCTGTGCATTCTGC |
||
| 5 |
Sp4 |
3EF: |
5’-CTACTGAGTCTGAAGCCCA |
|
| Sp4 |
3ER: |
5’-CGGGCGGGGGGTTGGGATAACCCAGCATTC |
||
| 6 |
Sp4 |
3FF: |
5’-TTCAGGGCAAATCAGTTGGC |
|
| Sp4 |
3FR: |
5’-CAGAGGTCTACGTCAAACC |
||
| Exon 4 |
Sp4 |
4F: |
5’-GCCATTAACCCCTTAGTTTTTG |
|
| Sp4 |
4R: |
5’-CCGGGCCCGGCCTGCTGTTTTATGCCTTCCAA |
||
| Exon 5 |
Sp4 |
5F: |
5’-CAAACCTATTCAGAGAAAGCATC |
|
| Sp4 |
5R: |
5’-GCCTTCTCCTTCATTTGCAT |
||
| Exon 6 |
Sp4 |
6F: |
5’-TATAATGTGACCAGAAGGGAG |
|
| Sp4 |
6R: |
5’-TAATCCACTTGACCAGCCCA |
||
|
GNB1 |
||||
| Exon 3 |
Gnb1 |
3F: |
5’-GATCTCCTGACCTCGTGATCT |
|
| Gnb1 | 3R: | 5’-TAACATYCTAACCTGAAACTC |
In each primer pair of this table F indicates the forward primer annealing to the DNA in the 5'-3' direction and R indicates the reverse primer, annealing to the DNA in the 3'-5' direction. Since exon three of SP4 is long, six sets of primer pairs were used for its amplification.
Single strand conformation polymorphism
Amplicons were separated by electrophoresis in 7% acrylamide gels and analyzed by standard P32 autoradiography or silver staining methods to reveal polymorphisms as described previously [17-21].
Sequencing
Amplicons carrying polymorphisms were purified using the QIA QUICK PCR purification kit (Qiagen, Valencia, CA) and sequenced using the Dyenamic ET Terminator cycle sequencing kit (Amersham, Piscataway, NJ).
Results
SSCP screening of the SP4 gene showed 9 sequence variants in 29 patients, five present in exons (Table 2). The heterozygous Leu241Val and Pro286Ala missense variants, both present in exon 3 of arRP probands did not segregate with disease in the corresponding families. An Asn306Ser missense variant in exon 3 was present in both alleles of one isolate RP proband, in one allele of five arRP probands and in one arCRD proband. This missense variant was also present in 1/95 controls. Interestingly, although only 14% of the 270 patients and 13% of the 95 controls were Hispanic, all seven patients and the 1 control that carried Asn306Ser were Hispanic. Thus, 18.4% (7/38) of the Hispanic patients carried Asn306Ser while none of the other patients did, including 0/151 patients of European origin. The other 2 coding region variants were both silent. Neither was present in 95 controls. The remaining 4 intronic sequence variants were present in patients and absent from controls with the exception of -121 A to C which was present in one control (Table 2).
Table 2. Sequence variants detected in the screening of the SP4 gene.
| Variant |
Exon/
Intron |
Frequency in patients |
Frequency in controls |
| Leu241Val |
Exon 3 |
1/270 |
0/95 |
| Pro286Ala |
Exon 3 |
1/270 |
0/95 |
| Asn306Ser |
Exon 3 |
7/270 |
1/95 |
| Ala276Ala |
Exon 3 |
1/270 |
0/95 |
| Gln451Gln |
Exon 3 |
5/270 |
0/95 |
| -121A→C |
Intron 1 |
4/270 |
1/95 |
| +52T→C |
Intron 3 |
5/270 |
0/95 |
| +70T→C |
Intron 5 |
4/270 |
0/95 |
| -37, 38Δ2C | Intron 2 | 5/270 | 0/95 |
All sequence variants present in the coding region of SP4 were found in exon three. The only SP4 variant that segregated with disease was Asn306Ser.
Table 3 shows the results of previous screenings of several photoreceptor genes in the six probands with the Asn306Ser mutation in one allele. Patient 856 has arCRD while the other 5 patients have arRP. Three of the six probands also carried an A-G variant in intron 2 of the GNB1 gene. DNAs of the family members of one of these probands could not be obtained (family 2177). However, in the families of the other two probands, the SP4 missense and the GNB1 intronic variant segregated with disease (Figure 1A,B). None of the other variants in the screened genes segregated with disease. We found no additional variants in the genes encoding α-, β- and γ-cGMP-phosphodiesterase, RDS/peripherin or the β-subunit of transducin in the RP isolate patient homozygous for Asn306Ser.
Table 3. Sequence variants in five genes of six patients with the heterozygous SP4 Asn306Ser mutation.
| Patient |
PDE6A |
PDE6B |
PDE6G |
GNB1 |
RDS |
| 449 |
none |
none |
none |
none |
none |
| 485 |
Exon 14, Phe597Phe |
none |
none |
Intron 2, A to G, 103rd bp from 3’ splice site |
none |
| Intron 18, A to C, 21st bp from 3’ splice site |
|||||
| Exon 20, Phe779Phe |
|||||
| 856 |
not done |
not done |
not done |
none |
not done |
| 1526 |
Intron 18, A to C, 21st be from 3’ splice site |
Exon 6, Ile330Ile |
Intron 3, C to G, 9th bp of 3’ splice site |
Intron 2, A to G, 103rd bp from 3’ splice site |
none |
| Exon 20, Phe779Phe |
|||||
| 1824 |
none |
none |
none |
none |
none |
| 2177 | none | none | none | Intron 2, A to G, 103 rdbp from 3’ splice site | none |
The probands of three families (485, 1526, and 2177) had an intronic sequence variant in GNB1 in addition to SP4 Asn306Ser. These sequence variants segregated with disease in families 485 and 1526. We were unable to obtain the DNA of members of family 2177. The sequence variant in intron 18 of PDE6A did not segregate with disease in families 485 and 1526.
Figure 1.
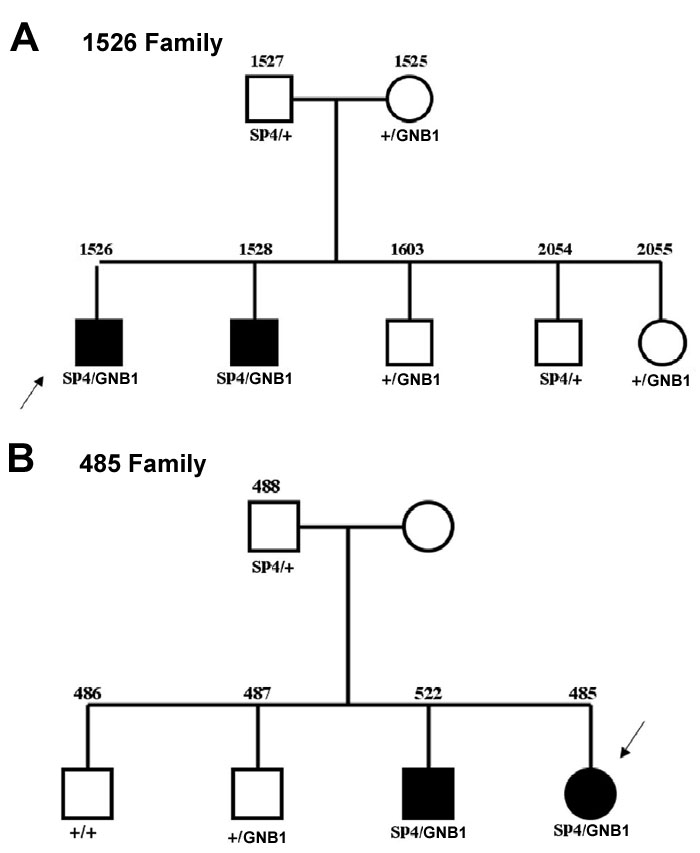
Pedigrees number 485 and number 1526 segregate the SP4/GNB1 alleles. Filled symbols indicate individuals with retinitis pigmentosa. Probands are indicated by arrows. The allele designation SP4 denotes the Asn306Ser allele, GNB1 denotes the A-G substitution in intron 2 of the GNB1 gene, + denotes normal alleles.
Discussion
Even though 2/3 of mice born with the Sp4 gene deleted die within the first four weeks of life [14], the surviving mice have many abnormalities including severe retinal degeneration (our data, not shown). Therefore, we considered the human SP4 gene a good candidate for the site of missense mutations causing retinal degeneration, given its involvement in the transcription of several photoreceptor genes including PDE6B [13,15,16] and the phenotype of the Sp4 knockout mouse.
We found nine unique sequence variants in the SP4 gene of 29 patients affected with different types of inherited retinal degenerative disease. Of these, five variants were in one of the six exons of SP4. Two of the variants coded for the same amino acid (Ala276Ala and Gln451Gln). Two more variants predicted amino acid changes in the SP4 protein, Leu241Val and Pro286Ala, but neither segregated with disease in the corresponding families. The fifth missense Asn306Ser mutation was present in seven probands. One of the probands that had the homozygous Asn306Ser mutation was an isolate with RP, so we could not tell if the two Asn306Ser alleles were causing disease. In family 449, an affected sibling of the proband did not carry the Asn306Ser allele; for the probands of families 856 (arCRD) and 1824, neither a second variant SP4 allele nor a variant in any of the other genes previously screened could be identified. The three remaining probands all carried Asn306Ser and an intronic A-G substitution 103 bp upstream of the 3' splice site of intron 2 of the GNB1 gene. We could not obtain DNAs from the family of one of the probands (2177), but in the families of the other two probands (485 and 1526) only the two affecteds in each pedigree carried both alleles (Figure 1A,B). Both of these families had arRP. All seven families carrying Asn306Ser were of Hispanic background and so was the 1 control carrying the same mutation. Thus, 18.4% of the Hispanic patients carried this allele while none of the patients from other backgrounds did (0/151 Europeans, 0/35 Blacks and 0/46 Asians). The higher frequency of Asn306Ser in Hispanic patients (p<0.001 applying the Fisher's exact test) compared to a relatively low frequency in Hispanic controls (1/12=8.3%) suggests that this variant may be pathogenic.
There are several additional reasons that implicate SP4 Asn306Ser as a mutant allele that may contribute to autosomal recessive disease. (1) We have no family history for the proband that carried two alleles of Asn306Ser. Therefore, the two Asn306Ser alleles together could be the cause of that isolate patient's disease. (2) In each of the three families where the Asn306Ser allele did not segregate with disease, two mutant alleles in other genes may have rendered the presence of the heterozygous Asn306Ser coincidental and unrelated to disease. However, this does not rule out the possibility that two Asn306Ser alleles could cause disease. (3) For the two families where only the affecteds carried both the Asn306Ser allele and the GNB1 variant intronic allele, pathogenesis is possible at least genetically. Furthermore, GNB1 has three GC boxes in its promoter and SP4 interacts with GC boxes. Thus, digenic disease may be plausible. To answer the question of pathogenicity of Asn306Ser, functional assays of the protein carrying this variant would have to be conducted. Nevertheless, the possibility that Asn306Ser may be pathogenic is supported by the fact that Asn306Ser is in the transactivation domain of the SP4 protein and in one of six glycosylation sites (N-X-S/T; Figure 2). Changing asparagine to serine eliminates this site of posttranslational modification and this may affect the function of the protein. Interestingly, at position 306 there is an asparagine only in the human SP4 sequence while in the mouse, rat, dog and cow there is a threonine. Therefore, asparagine is not a conserved residue. With regard to the GNB1 intronic variant, it is not in a splice site or a consensus branch point, but it may be in a heretofore-unknown regulatory region.
Figure 2.
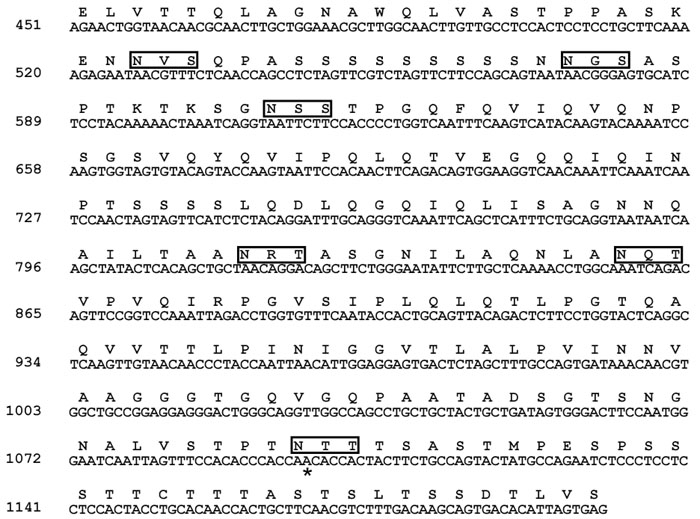
Partial nucleotide sequence of the cDNA encoding human SP4 and the corresponding predicted amino acid sequence. Boxes indicate potential N-glycosylation sites (N-X-S/T). Asterisk (*) indicates the location of the A to G mutation (AAC to AGC causes the change of Asn306 to Ser).
For the intronic GNB1 variant, a DNA fragment including exon 2, intron 2 (carrying the variant), and exon 3 would have to be expressed to determine if this variant caused a splicing problem. However, the sequences adjacent to the A-G substitution do not correspond to consensus branch point sequences. Another possibility is that the A-G substitution would disrupt an enhancer or a repressor sequence causing altered expression of the GNB1 gene. Although there is no direct evidence that the Asn306Ser mutation in the SP4 gene and the intron 2 A-G variant of the GNB1gene together are responsible for disease in the affected individuals, digenic disease cannot be ruled out without further testing the pathogenicity of the alleles. It is certainly plausible that the protein products of a phototransduction gene like GNB1 and a transcription factor that may influence its expression like SP4 can together cause digenic disease when one allele of each carries a mutation.
Acknowledgements
This work was done with the support of a grant from the Foundation Fighting Blindness and grant EY02651 from NIH to DBF.
References
- 1.Freund CL, Gregory-Evans CY, Furukawa T, Papaioannou M, Looser J, Ploder L, Bellingham J, Ng D, Herbrick JA, Duncan A, Scherer SW, Tsui LC, Loutradis-Anagnostou A, Jacobson SG, Cepko CL, Bhattacharya SS, McInnes RR. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell. 1997;91:543–53. doi: 10.1016/s0092-8674(00)80440-7. [DOI] [PubMed] [Google Scholar]
- 2.Swain PK, Chen S, Wang QL, Affatigato LM, Coats CL, Brady KD, Fishman GA, Jacobson SG, Swaroop A, Stone E, Sieving PA, Zack DJ. Mutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degeneration. Neuron. 1997;19:1329–36. doi: 10.1016/s0896-6273(00)80423-7. [DOI] [PubMed] [Google Scholar]
- 3.Bessant DA, Payne AM, Mitton KP, Wang QL, Swain PK, Plant C, Bird AC, Zack DJ, Swaroop A, Bhattacharya SS. A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat Genet. 1999;21:355–6. doi: 10.1038/7678. [DOI] [PubMed] [Google Scholar]
- 4.Martinez-Gimeno M, Maseras M, Baiget M, Beneito M, Antinolo G, Ayuso C, Carballo M. Mutations P51U and G122E in retinal transcription factor NRL associated with autosomal dominant and sporadic retinitis pigmentosa. Hum Mutat. 2001;17:520. doi: 10.1002/humu.1135. [DOI] [PubMed] [Google Scholar]
- 5.Harrison SM, Houzelstein D, Dunwoodie SL, Beddington RS. Sp5, a new member of the Sp1 family, is dynamically expressed during development and genetically interacts with Brachyury. Dev Biol. 2000;227:358–72. doi: 10.1006/dbio.2000.9878. [DOI] [PubMed] [Google Scholar]
- 6.Philipsen S, Suske G. A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res. 1999;27:2991–3000. doi: 10.1093/nar/27.15.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Treichel D, Becker MB, Gruss P. The novel transcription factor gene Sp5 exhibits a dynamic and highly restricted expression pattern during mouse embryogenesis. Mech Dev. 2001;101:175–9. doi: 10.1016/s0925-4773(00)00544-x. [DOI] [PubMed] [Google Scholar]
- 8.Lania L, Majello B, De Luca P. Transcriptional regulation by the Sp family proteins. Int J Biochem Cell Biol. 1997;29:1313–23. doi: 10.1016/s1357-2725(97)00094-0. [DOI] [PubMed] [Google Scholar]
- 9.Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 10.Hagen G, Muller S, Beato M, Suske G. Cloning by recognition site screening of two novel GT box binding proteins: a family of Sp1 related genes. Nucleic Acids Res. 1992;20:5519–25. doi: 10.1093/nar/20.21.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hagen G, Muller S, Beato M, Suske G. Sp1-mediated transcriptional activation is repressed by Sp3. EMBO J. 1994;13:3843–51. doi: 10.1002/j.1460-2075.1994.tb06695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou X, Qyang Y, Kelsoe JR, Masliah E, Geyer MA. Impaired postnatal development of hippocampal dentate gyrus in Sp4 null mutant mice. Genes Brain Behav. 2006 doi: 10.1111/j.1601-183X.2006.00256.x. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 13.Lerner LE, Gribanova YE, Ji M, Knox BE, Farber DB. Nrl and Sp nuclear proteins mediate transcription of rod-specific cGMP-phosphodiesterase beta-subunit gene: involvement of multiple response elements. J Biol Chem. 2001;276:34999–5007. doi: 10.1074/jbc.M103301200. [DOI] [PubMed] [Google Scholar]
- 14.Gollner H, Bouwman P, Mangold M, Karis A, Braun H, Rohner I, Del Rey A, Besedovsky HO, Meinhardt A, van den Broek M, Cutforth T, Grosveld F, Philipsen S, Suske G. Complex phenotype of mice homozygous for a null mutation in the Sp4 transcription factor gene. Genes Cells. 2001;6:689–97. doi: 10.1046/j.1365-2443.2001.00455.x. [DOI] [PubMed] [Google Scholar]
- 15.Lerner LE, Gribanova YE, Whitaker L, Knox BE, Farber DB. The rod cGMP-phosphodiesterase beta-subunit promoter is a specific target for Sp4 and is not activated by other Sp proteins or CRX. J Biol Chem. 2002;277:25877–83. doi: 10.1074/jbc.M201407200. [DOI] [PubMed] [Google Scholar]
- 16.Lerner LE, Peng GH, Gribanova YE, Chen S, Farber DB. Sp4 is expressed in retinal neurons, activates transcription of photoreceptor-specific genes, and synergizes with Crx. J Biol Chem. 2005;280:20642–50. doi: 10.1074/jbc.M500957200. [DOI] [PubMed] [Google Scholar]
- 17.Danciger M, Blaney J, Gao YQ, Zhao DY, Heckenlively JR, Jacobson SG, Farber DB. Mutations in the PDE6B gene in autosomal recessive retinitis pigmentosa. Genomics. 1995;30:1–7. doi: 10.1006/geno.1995.0001. [DOI] [PubMed] [Google Scholar]
- 18.Gao YQ, Danciger M, Zhao DY, Blaney J, Piriev NI, Shih J, Jacobson SG, Heckenlively JH, Farber DB. Screening of the PDE6B gene in patients with autosomal dominant retinitis pigmentosa. Exp Eye Res. 1996;62:149–54. doi: 10.1006/exer.1996.0019. [DOI] [PubMed] [Google Scholar]
- 19.Gao YQ, Danciger M, Akhmedov NB, Zhao DY, Heckenlively JR, Fishman GA, Weleber RG, Jacobson SG, Farber DB. Exon screening of the genes encoding the beta- and gamma-subunits of cone transducin in patients with inherited retinal disease. Mol Vis. 1998;4:16. http://www.molvis.org/molvis/v4/a16/ [PubMed] [Google Scholar]
- 20.Gao YQ, Danciger M, Longmuir R, Piriev NI, Zhao DY, Heckenlively JR, Fishman GA, Weleber RG, Jacobson SG, Stone EM, Farber DB. Screening of the gene encoding the alpha'-subunit of cone cGMP-PDE in patients with retinal degenerations. Invest Ophthalmol Vis Sci. 1999;40:1818–22. [PubMed] [Google Scholar]
- 21.Piri N, Gao YQ, Danciger M, Mendoza E, Fishman GA, Farber DB. A substitution of G to C in the cone cGMP-phosphodiesterase gamma subunit gene found in a distinctive form of cone dystrophy. Ophthalmology. 2005;112:159–66. doi: 10.1016/j.ophtha.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 22.Kitamura E, Danciger M, Yamashita C, Rao NP, Nusinowitz S, Chang B, Farber DB. Disruption of the gene encoding the beta1-subunit of transducin in the Rd4/+ mouse. Invest Ophthalmol Vis Sci. 2006;47:1293–301. doi: 10.1167/iovs.05-1164. [DOI] [PubMed] [Google Scholar]