

Abstract
Objectives
Parenteral nutrition-associated liver disease (PNALD) is a major problem with prolonged TPN administration. Our laboratory previously demonstrated significant changes in the expression of mdr1 and mdr2, two hepatocyte transporters, in a TPN mouse model. Our present study hypothesized that these changes would lead to functional changes in the liver, and would contribute to the development of liver dysfunction.
Methods
Mice received either intravenous saline and standard chow or TPN with or without intravenous lipids. Functional assays were performed after 7 days of infusion.
Results
TPN with lipids led to a significant increase in serum bile acid levels, consistent with an early state of PNALD. Use of TPN without lipids prevented elevation in bile acid levels. In both TPN groups, mdr2 expression was significantly (68%) lower than controls; and bile phosphatidylcholine content, a functional measure of mdr2, was 40% less than controls. Mdr1 expression in the TPN with lipid group was 31% higher than controls; while in the TPN without lipids mice there was no significant change. Hepatocyte extrusion of rhodamine dye, a measure of mdr1 function, declined only in TPN with lipids. PPAR-α expression decreased in both TPN groups. Fenofibrate given with TPN resulted in an increased expression of mdr1 and mdr2, and functionally increased hepatocyte rhodamine extrusion and presence of bile phosphatidylcholine in the TPN with lipid group.
Conclusions
The study shows that TPN led to alterations in the function of mdr1 and mdr2 expressed proteins. The changes help in the understanding of the mechanisms leading to PNALD; and suggest that fibrate administration may palliate these changes.
Keywords: Total parenteral nutrition, Parenteral nutritional-associated liver disease (PNALD), Cholestasis, Multidrug resistance gene, P-glycoprotein, Lipid, peroxisome proliferator-activated receptors (PPARs), Fibrate
Introduction
Liver dysfunction in patients who receive total parenteral nutrition (TPN) is one of its most devastating complications (1, 2). The development of parenteral nutrition-associated liver disease (PNALD) predisposes patients to an increased incidence of sepsis, higher mortality rates and the potential to develop irreversible liver injury (3-7). Despite the severity of this process, the etiology of PNALD has not been determined (1). This disorder may be seen at all ages, but is most prevalent in premature infants (3, 8). The condition of the liver can further deteriorate with the development of sepsis (3, 8).
Over the past decade the etiology of several forms of cholestasis has been attributed to mutations in members of proteins in the ATP-binding cassette (ABC) transporter family (9, 10) (11). These genes encode a number of proteins which transport various components of bile from hepatocytes into the bile canaliculi (9, 12, 13); and include the bile salt transporter (bile salt export pump; bsep), the multidrug resistance 2 (mdr2) which is responsible for the transport of phosphatidylcholine (PC) into the bile canaliculi, the canalicular anionic conjugated transporter (cmoat; also referred as mrp2), and multidrug resistance 1 (mdr1) which transports chemotherapy drugs and potentially other toxins into bile (14). mdr1 and mdr2 encode P-glycoprotein (P-gp) related proteins, and defects or disruption in the function of these proteins may result in a number of pathologic processes. As an example, individuals with the Dubin-Johnson syndrome have mutations in human MRP2 (ABCC2) which leads to a disruption in the transport of bilirubin glucuronide conjugates (15). Progressive familial intrahepatic cholestasis (PFIC) is an autosomally recessively inherited disorder in which two of the three known causative genes are ABC transporters (11). PFIC Type 2 is due to a mutation in the human BSEP gene (ABCB11), and those with PFIC Type 3 have a mutation in human MDR3 (ABCB4) (16). A genetic model of this disease has been developed in mice by disrupting the (mdr2) gene (equivalent to human MDR3) (17). Mice with a homozygous disruption of the mdr2 gene develop a pronounced inflammatory non-suppurative cholangitis and fibrosis, which results in death within the first six months of life (18). The pathologic description of the liver and bile composition in these mdr2 gene-disrupted mice have some similarities to that seen in experimental models of PNALD and neonates with this disorder (17, 19). Namely, bile in both animals on TPN and with a disrupted mdr2 gene have diminished levels of PC in their bile (20). Although these two processes have their distinct differences, the pathologic changes in both conditions include: bile duct proliferation, periportal inflammation and eventually fibrosis. Based on these factors, we had investigated the expression of mdr1 and mdr2 in a mouse model of TPN (21). In our previous work, we showed that mdr2 expression decreased with TPN administration, whereas mdr1 expression increased (21). This suggested that TPN administration resulted in a dysregulation of transporter expression. To investigate further into the details of these altered hepatic canalicular transporters, this current study examines the subsequent functional relevance of these changes in this mouse TPN model.
Materials and Methods
Ethics
The current study conformed to the guidelines for the care and use of laboratory animals established by the University Committee on the Use and Care of Animals (UCUCA) at the University of Michigan and all protocols received UCUCA approval.
Animals
Adult mice
C57BL/6 male, 10-12-week-old, specific pathogen-free mice, (Jackson Laboratories, Bar Harbor, ME) weighing 23-25 g were housed in metabolic cages and subjected to an acclimatization period of one week. They were fed standard chow and had access to tap water ad libitum. The mice were maintained in a 12:12-hr day-night rhythm at a constant temperature of 23°C and a relative humidity of 40% to 60%.
Intravenous Hyperalimentation
Mice were anesthetized with ketamine (87 mg/kg) and rompun (13 mg/kg) given together intramuscularly. Catheterization and administration of TPN was similar to that described previously (22, 23). On the first day all mice received intravenous crystalloid solution (dextrose 5% in 0.45% NS with 20 mEq KCl/L) at a rate of 9.6 mL/day. After 24 hours animals were randomized into three groups. The control group received the same intravenous physiologic saline at 7 mL/24 hrs, in addition to standard laboratory mouse chow and water ad libitum. The TPN+lipid group received a standard TPN solution intravenously at 7 mL/24 hrs with no enteral nutrition. This TPN solution contained a balanced mixture of amino acids, fats and dextrose in addition to electrolytes, trace elements and vitamins as previously described (24). In a separate group of mice, TPN was administered without intravenous fats (TPN w/o lipids), similar to that previously described (25). To allow for a matched amount of caloric delivery between groups, dextrose delivery was increased. For the TPN w/o lipids group, mice received a mixture of 2.44 grams of amino acids and 15.3 grams dextrose in 50 ml of water, in addition to other standard additives. This led to an essentially isocaloric mixture compared to the TPN+lipid group (i.e., ∼235 Kcal/kg/day; 1.65 grams of amino acids, 9.6 grams dextrose, and 1.8 grams of lipids (Liposyn 20% Abbott Laboratories, Chicago, ILL.) in 50-ml of water). Caloric delivery was based on caloric intake measurements from previous investigators, and food was weighed daily to insure that caloric intake in the study groups were closely matched (26, 27).
After 7 days of total continuous infusion, body weights were measured, blood was collected for bile acids and mice were sacrificed using CO2. Whole liver samples were harvested. After removing all visible fatty and vascular tissue, the samples were sliced into approximately equal quantities and stored at -70°C until examination.
Serum Bile Acids
To show the status of cholestasis in our models, mouse serum bile acid levels were measured according to manufacturer's instructions by the enzymatic determination kit (Sigma-Aldrich Diagnostics, St. Louis, MO).
Canalicular Transport Protein Gene Expression
Isolation of Liver Total RNA and DNA
Morsellated 30 mg liver samples were washed with phosphate-buffered saline (PBS) and homogenized directly into 3 ml TRIzol reagent (Gibco BRL, Gaithersburg, MD) on ice, according to the manufacturer's instructions.
Reverse Transcriptase Reaction (RT) and Polymerase Chain Reaction (PCR)
RT- PCR was performed as previously described (21). To insure that the DNA product was examined at the exponential portion of the PCR curve, the adequate number of cycles was used for each transporter (21). β-Actin expression (equal number of PCR cycles to the respective transporter gene) was also generated for each sample as a control.
Mdr1, mdr2 and β-actin primers were designed and used for RT-PCR as same as used in our previous paper (21). Other primers of canalicular transporters used in the PCR reactions were: bsep (GeneBank accession number AB003303) Forward: 5′ - CTT GAG TAC ACA GCA AGA CT - 3′ and Reverse: 5′ - GGT AAG TTA GAA CTA CCA GTT G - 3′; and cmoat (GeneBank accession number AF227274) Forward: 5′ - GCT AGC GGA CTA GAA GAA CA -3′ and Reverse: 5′- GCT TGA GCC TTA GAG TTT GA - 3′.
PPAR primers used were: PPAR-alpha (accession number: NM 011144; Forward: 5′ -CCT CAG GGT ACC ACT ACG GA – 3′, and Reverse: 5′- CCG AAT CTT TCA GGT CGT GT -3′) and PPAR-gamma (accession number: NM 011146; Forward: 5′- TTT TCA AGG GTG CCA GTT TC -3′ and Reverse: 5′- AAT CCT TGG CCC TCT GAG AT -3′).
Functional Expression of P-glycoprotein
MDR1 function
The function of mdr1 in hepatocytes was assessed by the ability of these cells to actively transport the vital dye Rhodamine 123 using previously described methods (28). Mouse hepatocytes were isolated according the modified protocol of Invitrogen's Hepatocyte Product line (Gibco BRL Division of Invitrogen, Life Technologies Inc. Grand Island, NY). Briefly, under anesthesia, a 24-gauge catheter was placed into portal vein and the liver was perfused with Liver Perfusion Medium followed by Liver Digest Medium. After that, the liver was removed and gently morcellated in Liver Transport Medium. The cell suspension was spun down at 50g for 5min, resuspended in cold Hepatocyte Wash Medium, filtered through 100μm nylon mesh (Millipore) and centrifuge again at 50g for 5min. The isolated hepatocytes were incubated with 5ml Rhodamine 123 (0.2mM, Sigma-Aldrich) with 1ml HBSS and 10% bovine serum albumin. Hepatocytes were incubated for 10 minutes at 37°C in the dark, followed by 3 successive washes, and resuspension in HBSS/BSA. Samples were then incubated for the assessment of active extrusion of dye for 30 minutes at 37°C. Following this, cells were immediately washed and resuspended at 4°C, and remaining intracellular Rhodamine 123 was assessed by flow cytometry. Controls included the following: negative extrusion control (cells washed and suspended in cold HBSS/BSA without incubation) and an inhibition of mdr1 function with simultaneous incubation with verapamil (10μg/ml). Expression of extrusion directly correlated with mdr1 function, and is expressed as the ratio of rhodamine remaining over baseline intracellular levels of rhodamine.
Mdr2 function
Mdr2 encoded P-gp is responsible for the transport of phosphatidylcholine (PC) across the bile canalicular membrane. Phospholipid content of bile was measured in both control and TPN groups. On day 7, hepatic and total bile phosphate and phospholipid levels were determined during a one-hour un-stimulated bile collection period. In the detail, selected mice (N = 5 per group) from each group were re-anesthetized after 7 days of treatment. The common bile duct was ligated with a 5-0 silk suture and a silastic catheter (0.25 mm i.d.) was placed into the gallbladder and held in place with a suture ligature. Bile samples were immediately frozen and stored at -70°C under nitrogen gas until assayed. Total lipids were extracted using a chloroform/methanol method. Lipid extracts were dried under nitrogen gas, dissolved in chloroform and the total phospholipid content determined as phospholipid phosphorus (29, 30). Extracted phospholipids were separated by thin-layer chromatography on silica gel 60 plates using chloroform/methanol/7N NH3OH (60:35:5). Plates with individual phospholipids were placed in 8% (wt/vol) CuSO4 phentahydrate in a water/methanol/concentration of either H3PO4 (60: 32: 8) or 0.1% (wt/vol). Ninhydrin in ethanol was used to visualize individual lipids after autoclaving (150°C, 15 min, 110°C, 10 min, respectively). Plates were imaged with a Kodak 1D system and density images were recorded for each phospholipid. PC concentrations were determined using known amounts of egg-yolk and commercially obtained PC (Sigma-Aldrich, St. Louis, MO) as standards. The percentage of PC from the chromatographic analysis was used to calculate the total PC content in the phospholipid extract. Additional phospholipids were determined both in the liver as well as the bile. Each band was identified based on commercially obtained standards (Sigma-Aldrich), including phosphatidylglycine (PG), phosphatidylethanolamine (PE), sphingomyelin and lysophosphatidylcholine (LPC). The relative level of each of these was determined relative to the expression of PC.
Fibrate Administration on the TPN-treated Models
Fibrates have been reported to increase P-glycoprotein (P-gp) expression, including both mdr1 and mdr2 (31); and this increase appears to be dependent on the expression of PPAR-α (32-34). Because of the decreased P-gp expression in our TPN-treated model (21), we next hypothesized that the administration of fibrate would increase the expression of P-gp, and PPARs in this TPN-treated model. Fenofibrate administration (100mg/kg/day) was gently administered to mice using a blunt gastro-feeding needle starting at 48 hours after catheterization, and was continued daily till mice were euthanized.
Endotoxin Analysis
Immediately after sacrificing mice, liver homogenates were placed in endotoxin-free tubes and a standard Limulus assay (Sigma-Aldrich) detected levels of endotoxin. All samples were assayed together and were compared to both negative and positive controls to ensure test accuracy.
Statistical Analysis
Results were expressed as the mean ± SD. The statistical analysis of the data was performed using unpaired t tests, or when more than one group was studied, one-way analysis of variance (when comparing control versus TPN with or without lipids), with P < .05 considered significant. Post hoc intergroup analysis was performed using Bonferroni t tests.
Results
General Description of Mice
The survival rate was 95% in control group, 72% in TPN+lipids group, and 85% in TPN w/o lipids group. Body weights trended lower in TPN mice; however the differences were not significant (P>0.05) between TPN+lipid (body weight at start and end of study): (23.4 ± 2.2g and 21.3 ± 2.2g) or TPN w/o lipid (24.1 ± 2.0g and 22.1 ± 2.4g) and the control (22.2 ± 1.4g, versus 24.6 ± 2.7g) groups.
Serum Bile Acids
Serum bile acids levels are known to be elevated with cholestasis (35), and were used to examine for a state of early PNALD with TPN. Serum total bile acid (TBA) levels in mice receiving TPN+lipids was significantly higher than the levels in control mice (81.4 ±29.7 μmol/l vs. 33.3 ±24.0 μmol/l, respectively) (Figure 1). As elevated levels of bile acids are consistently seen in states of cholestasis, this suggests that TPN with lipids is associated with an early state of cholestasis. Interestingly, serum bile acid values were not significantly different between the mice in the TPN w/o lipids group (46.1 ±21.5 μmol/l) and the control group.
Figure 1.
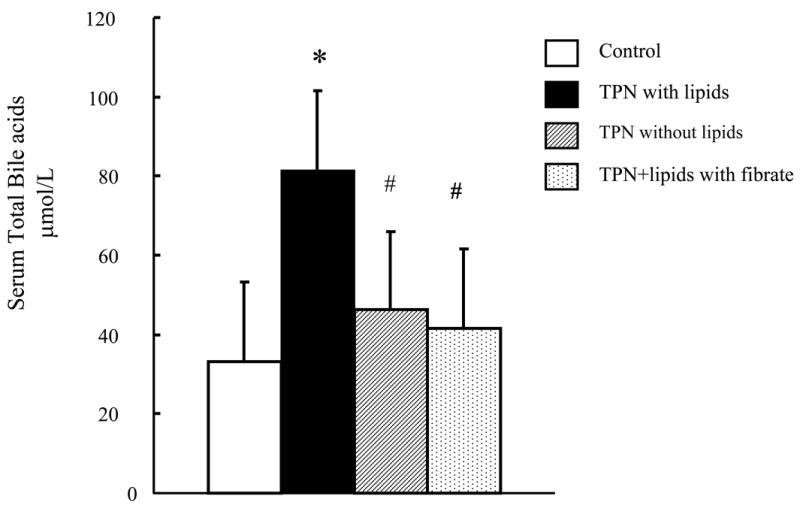
Serum total bile acid levels are shown for each study group. Note the significant rise in bile acids in the TPN with lipid group.
* P < 0.05 versus Control. #P < 0.05 versus TPN with or TPN without lipids.
Expression of Canalicular Transport Proteins
TPN with lipids led to a 32% decline in mdr2 mRNA expression compared to control mice (Table 1; P<0.05). The TPN+lipid group also had a 31% increase in mdr1 mRNA expression compared with controls (P<0.05). In the TPN w/o lipids group, mdr2 mRNA expression also decreased compared with controls; while there was no significant difference in mdr1 mRNA expression (P>0.05) compared with controls. Both cmoat and bsep expression failed to show any significant changes in expression in all three study groups, although there was a non-significant decline in bsep in the TPN+lipid group compared to controls (Table 1).
Table 1.
mRNA expression of canalicular transport proteins. Results are expressed as a ratio to β-actin expression.
| Group | mdr2 | mdr1** | cmoat | bsep |
|---|---|---|---|---|
| Control | 0.94±0.24 | 0.37±0.09 | 0.86±0.20 | 1.46(0.25 |
| TPN + lipids | 0.63±0.27* | 0.63±0.14* | 0.80±0.15 | 1.14(0.20 |
| TPN without lipids | 0.56 ±0.18 | 0.36 ±0.14 | 0.79(0.16 | 1.32(0.22 |
P < 0.05 control versus TPN.
mdr1 detected both mdr1a and mdr1b isoforms. N= minimum of 10 per group.
Phospholipid Content of Liver and Bile
mdr2 encoded P-gp is responsible for PC transport across the bile cannaliculi, and phospholipids content was measured to assess the functional role of this encoded protein in each study group.
Liver
Phospholipid contents of liver and bile were measured after 7-days for each group (Tables 2 and 3). The total phosphate level in the liver was used as a control for lipid expression, and was noted to decrease slightly (P>0.05) between controls and the TPN+lipid group. Phosphatidylcholine (PC) and lysophosphatidylcholine (LPC) levels in the liver were not significantly different (P>0.05) between control and standard TPN (TPN+lipids) groups (Table 2).
Table 2.
Phospholipid content of liver using thin-layer chromatography (TLC). Results of phosphotidylcholine (PC), lysophosphatidylcholine (LPC) contents were expressed as a ratio of PC or LPC content to total liver phosphate content.
| Group | Total phosphate in liver
(nmol /g wet weight) |
PC in liver
(μg/ nmol total phosphate) |
LPC in liver
(μg/ nmol total phosphate) |
|---|---|---|---|
| Control | 30.6 ( 5.4 | 1.84 ( 0.29 | 0.082 ( 0.0 |
| TPN+lipids | 24.7 ( 3.5 * | 1.64 ( 0.13 | 0.088 ( 0.0 |
P < 0.05 Control versus TPN. N= minimum of 10 per group.
Table 3.
Summary of the major findings of bile flow, bile acid content and phospholipid content in bile using thin-layer chromatography (TLC). Note the decline in the phosphatidylcholine levels in TPN-treated groups (with and without lipids), and a return to control levels with fenofibrate. N= minimum of 10 per group.
| Group | Bile flow
(μl/h) |
Bile acid
(mmol/μl bile) |
T-P
(nmol/μl bile) |
PC
(μg/nmol T-P) |
|---|---|---|---|---|
| Control | 21.3 ± 12.4 | 60.5 ± 38.5 | 13.3 ± 4.9 | 0.41 ± 0.12 |
| TPN + lipids | 25.2 ± 11.0* | 51.6 ± 28.3 | 16.1 ± 4.3 | 0.25 ± 0.09* |
| TPN without lipids | 16.0 ± 10.3 | 60.9 ± 30.1 | 18.4 ± 0.8 | 0.27 ± 0.08* |
| TPN+lipids with fibrate | 26.6 ± 16.6* | 54.9 ± 28.7 | 27.7 ± 18.3*# | 0.39 ± 0.10# |
P < 0.05 Control versus TPN+lipids.
P < 0.05 versus TPN with or without lipids.
Abbreviations: T-P : Total phosphate, PC : phosphatidylcholine,
Bile
Bile flow measurements showed a slight increase in the TPN with lipid group (Table 3). Interestingly, bile flow significantly declined in the TPN w/o lipid group. The total intrahepatic bile acid levels were similar in all groups. As an internal control, total phosphate measurements in the bile were not significantly different (P>0.05) between control and TPN groups both with lipid and without lipids. PC content in the bile, however, significantly (P<0.05) declined in mice receiving TPN for both the group with lipids and without lipids, compared to control values.
The ratio of total phospholipids to bile acids in bile samples (Figure 2) was significantly lower in the TPN+lipids group (0.25 ±0.09) compared to control values (0.43 ±0.30). The decline for the TPN w/o lipid group (0.39 ±0.22) was lower than controls, but the difference was not significant. As the encoded P-gp of mdr2 functions to transport PC from the hepatocyte to the bile canaliculi, it suggests that the observed decline in mdr2 mRNA and its encoded P-gp had a physiologic impact in the actual formation of bile, with a reduction in bile PC content.
Figure 2.
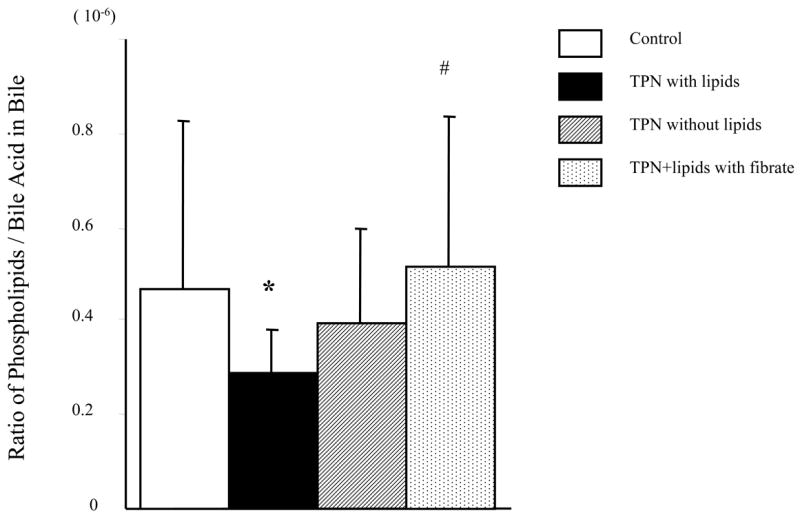
Ratio of total phospholipids to bile acid levels in collected bile samples using thin layer chromatography. Note that there is a significant decline in the ratio in the TPN with lipid group; whereas removal of lipids, or administration of fenofibrate to mice receiving TPN and lipids resulted in a rise in this ratio, with levels equaling control values in the fibrate group.
* P < 0.05 versus Control. #P < 0.05 versus TPN with or TPN without lipids.
Alteration of mdr1 function
Because we previously observed an increase in mdr1 expression in the TPN+lipid group (21), we next examined the functional characteristics of the mdr1 encoded P-gp. This was addressed by the ability of hepatocyte mdr1 encoded P-gp to actively transport rhodamine from an intracellular to an extracellular location (Figure 3). Uptake of Rhodamine 123 dye was similar in TPN and control groups, showing that the loading of dye and membrane integrity were not different. Active transport of rhodamine, however, was significantly lower in the TPN with lipid group compared to control mice. This suggests that despite an increased expression of mdr1 encoded P-gp, the function of the mdr1 transporter was actually depressed with TPN administration. In contrast, in the mice receiving lipid-free TPN, rhodamine transport was not significantly different from control mice; suggesting that the removal of lipids resulted in improved mdr1 encoded P-gp function.
Figure 3.
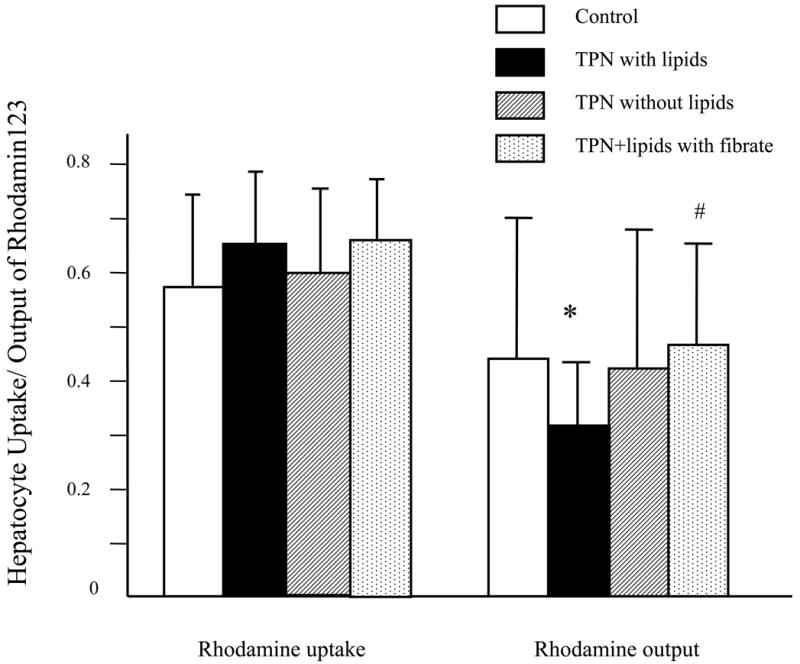
Hepatocyte transport of Rhodamine 123. Functional transport of rhodamine out of hepatocytes was assessed by the loss of fluorescent activity (due to an active transport of intracellular rhodamine). Note that uptake of rhodamine was similar in all treatment groups. However, there was a significant decline in rhodamine transport in the TPN with lipid group. Removal of lipids from TPN resulted in an improvement of rhodamine transport, and administration of fenofibrate to TPN+lipids mice also resulted in transport function that was not significantly different from control values.
* P < 0.05 versus Control. #P < 0.05 versus TPN with or TPN without lipids.
PPAR Expression
Expression of PPAR mRNA is shown in Table 4 and Figure 4. In both TPN-treated models, PPAR-alpha expression decreased (TPN w/ lipids; 0.80±0.11, TPN w/o lipids 0.79±0.08) significantly, compared with the control group (1.02±0.17). However PPAR-gamma expression did not change significantly between groups.
Table 4.
PPAR-α and PPAR-γ mRNA expression are shown. In TPN-treated models, PPAR- α expression decreased significantly; whereas administration of fenofibrate resulted prevented this decline.
| PPAR- α | PPAR -γ | |
|---|---|---|
| Control | 1.02 ± 0.17 | 0.44 ± 0.12 |
| TPN + lipids | 0.80 ± 0.11* | 0.43 ± 0.12 |
| TPN without lipids | 0.79 ± 0.08* | 0.58 ± 0.10 |
| TPN+lipids with fibrate | 1.05 ± 0.23# | 0.55 ± 0.11 |
P < 0.05 versus Control.
P < 0.05 versus TPN with or without lipids.
Figure 4.
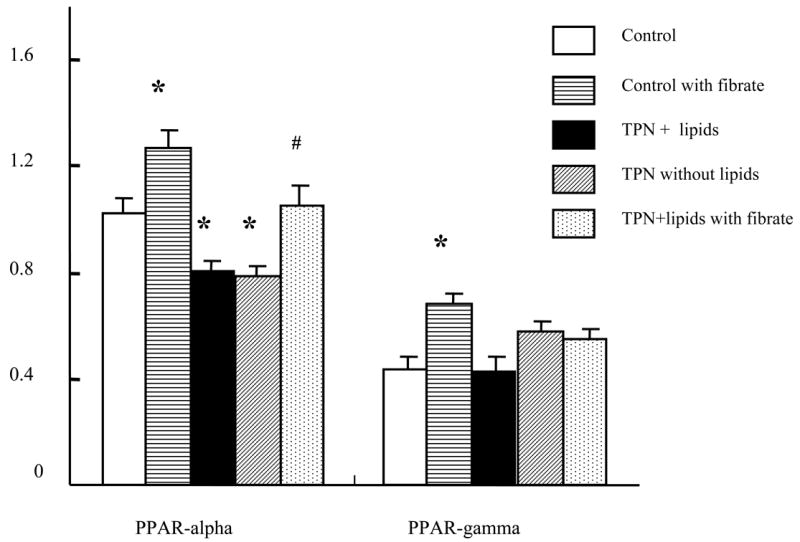
PPAR-alpha and PPAR-gamma mRNA expression are shown. In TPN-treated models (with or without lipids), PPAR-alpha expression decreased significantly. Administration of fenofibrate increased control values of PPAR-alpha and PPAR-gamma. Fibrates also prevented the observed decline in PPAR-alpha with TPN administration.
* P < 0.05 versus Control. #P < 0.05 versus TPN with/without lipids.
Fibrate administration and TPN
Fibrates have been shown to up-regulate both P-gp and PPAR-alpha expression, and were studied to examine whether administration could influence transporter function. TPN+lipids mice treated with fenofibrate led to an increase in mdr2 mRNA expression (0.69±0.13; P>0.05 compared to untreated TPN+lipid group) and mdr1 mRNA expression (0.75 ± 0.05; P<0.05 compared to TPN+lipid group) (Figure 5). Functionally, this led to a significant increase in the ratio of total phospholipids to bile acids (0.52 ± 0.31) (Figure 2). Table 3 shows that fenfibrate given to the TPN+lipid group resulted in a marked increase in the amount of bile phosphatidylcholine content; and the ratio of phospholipids to bile acids rose to control levels (Figure 2). This suggests that fenofibrate led to an up-regulation of mdr2 encoded P-gp transporter function. Additionally, fenofibrate administration to TPN+lipids mice increased the amount of rhodamine output (0.21± 0.15 vs. 0.44±0.18) (Figure 3 and Figure 5); and levels were not significantly different that those of controls. Fenofibrate administration significantly increased PPAR-alpha mRNA expression (1.05±0.08; Table 4 and Figure 4); where as there was no significant change in PPAR-gamma mRNA expression. Interestingly, coincident with these changes in the function of mdr1 and mdr2, the level of bile acids in TPN+lipid mice given fibrates (41.6 ±15.0 μmol/l) was not significantly difference with control.
Figure 5.
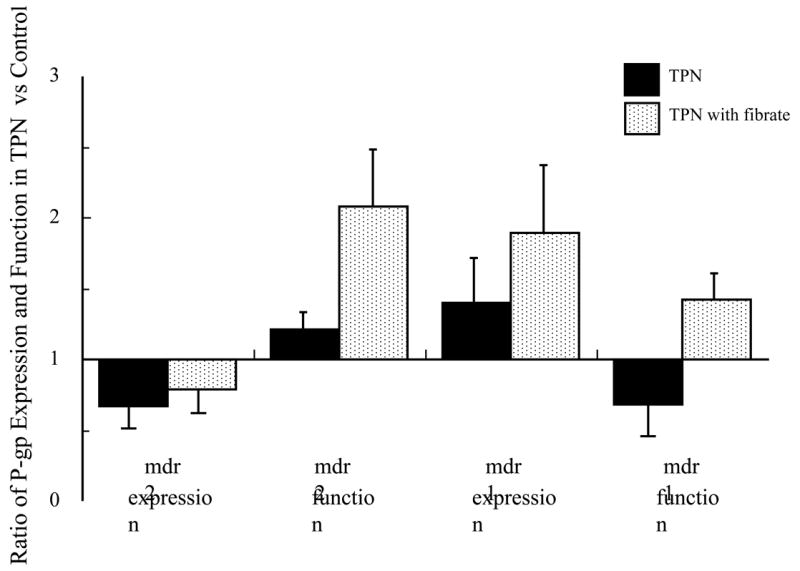
Summary of the effects of fenofibrate administration with mice receiving TPN and intravenous fats. Note the decline in mdr2 and rise in mdr1 expression with TPN+lipids; Functionally, a decline in both mdr1 and mdr2 was observed. Administration of fenofibrate to TPN mice normalized mdr1 and mdr2 expression and function.
Hepatic Endotoxin Content
As endotoxin has been shown to alter the expression of a number of bile canalicular transport proteins (36-38), we assessed endotoxin content in isolated liver homogenates freshly obtained from both control and TPN-treated groups (N = 6 per group). Endotoxin levels were not detectable in either TPN groups or in the controls.
Discussion
Since the very early reports of PNALD (39), a number of potential associated risk factors have been identified, including an inverse relationship with gestational age and birth weight, and a direct relationship with length of TPN administration and number of septic episodes (1, 3, 6, 40, 41). This suggests that the etiology of PNALD may be due to a number of different etiologies. Despite the advancement of multiple theories, and several attempts at therapeutic interventions (42, 43), no definitive etiology or proven therapy has yet been established for this disorder (7).
Over the past decade a number of cholestatic conditions including Dubin Johnson syndrome (cmoat), progressive intrahepatic cholestasis types 1 and 2 (bsep) and progressive intrahepatic cholestasis type 3 (Mdr3) have been attributed to defects in the ATP-binding cassette (ABC) transporter family of proteins (9, 14). Each of these proteins actively transports a component of bile across the bile canalicular membrane. Descriptive terminology varies considerably among species. In humans, two homologous P-glycoprotein (P-gp) isoforms (MDR1 and MDR3) exist that are encoded by individual genes (44, 45), whereas mice possess three separately encoded homologous isoforms (mdr1a, mdr1b and mdr2) (46, 47). MDR1 in humans, and mdr1a and mdr1b in mice appear to have identical function (28). Human MDR3 P-gp, based on homology and function, is almost identical to mdr2 in mice (48). The gene mdr1 encodes a P-gp, which can transport a wide variety of drugs (including chemotherapy agents) and substrates (including toxins) across biological membranes (including the bile canalicular membrane) in a temperature-sensitive and ATP-dependent manner (49). Unlike mdr1, the mdr2 gene encodes a P-gp transporter that is responsible for the transport of phosphatidylcholine (PC) across the bile canalicular membrane (17, 48). Interestingly, mdr2-deficient mice are subject to fatal liver disease before six months of age. Although there are distinct differences from the clinical condition of PNALD, in the absence of mdr2-encoded P-gp, the bile contains a disproportionately high concentration of cholesterol and potentially hepatotoxic bile acids, which may induce bile sludging and cholestatic liver injury (17). There is some evidence to suggest that changes in mdr2 may play a critical role in the early mediation of PNALD. First, the histological appearance of cholestasis in mdr2-deficient mice is similar to that observed with PNALD. Specifically, bile duct proliferation is noted in both mdr2-/- mice and with TPN administration in humans. Also noted in both are periportal inflammation, fibrosis and degeneration of hepatocytes. Secondly, the alteration in bile composition in mdr2-deficient mice is also similar to changes observed in the bile composition of animal models of TPN; namely bile in both mdr2-deficient mice and mice on TPN show a rise in the relative percentage of bile acids, and a decline in the total amount of phospholipids (20, 50). Phosphatidylcholine, which is the predominate phospholipid in bile, has been shown to be decreased in a rabbit model of TPN (20), as well as observed in the present study.
Our study examined several known bile canalicular transport proteins, and showed that mdr2 mRNA declined with TPN administration. The mRNA expression of mdr1 increased with TPN and the remaining transporter genes, cmoat and bsep failed to show any significant change. In our previous study, mdr1 expression was similarly noted to be elevated, and mdr2 was noted to decline with TPN (21). This current study examined the functional relevance of these changes. Our present study showed that the decline in mdr2 expression was associated with a decline in bile phosphatidylcholine content. We also showed that despite an observed increased expression of mdr1, hepatocyte extrusion of rhodamine dye, as a measure of mdr1 function, declined with TPN with lipids. The functional relevance of these changes may be quite important, as a loss of PC in the bile, may increase the relative concentration of bile acids which may be injurious to the liver. Further, the decline in mdr1 function may allow for the build up of toxins within hepatocytes, which may also lead to liver injury. Importantly, these changes in mdr occurred prior to any histologic evidence of cholestasis, suggesting that they may be early mediators of these changes (21).
The etiology behind these changes observed in this study is unknown. Our TPN model is associated with a moderate mortality rate; most like due to a high rate of bacterial translocation (51). This translocation may result in increased levels of endotoxin, and other investigators have shown that endotoxin can decrease the expression of mdr2 (52). Endotoxin can also down-regulate other hepatic transport proteins and may help explain the worsening of a jaundiced state with sepsis, as well as act as a contributor to hepatocyte injury (9, 38, 53). However, we were unable to detect any appreciable endotoxin levels in any of our study groups. Further work will be needed to understand the mechanisms involved. We did note that TPN therapy in mice resulted in a loss of body weight. It is uncertain if the changes in weight may account for any of the observed changes in the liver, although the weight changes between groups were small and were not significant.
Lipid administration has been implicated into the etiology of PNALD by several investigators (54-58). The current studies on mice receiving TPN without lipids suggest that some of the functional action of these hepatocyte transporters were shown to have been affected by the presence or absence of lipids. Our finding that serum bile acid levels were lower in lipid-free TPN mice supports the contention of other authors that lipids have a role in the formation of TPN-associated liver disease. It was, however, important to note that neither mdr2 expression, nor PC content in bile, failed to change with the removal of lipids. This suggests that the observed changes in mdr2 expression may well not be the main determinant of early cholestatic changes with TPN administration. However, PNALD may well be a multi-factorial process (59, 60); and as such, the changes in mdr2 expression, and decline in bile phosphatidylcholine expression may still be a contributing factor to the development of PNALD. The removal of lipids from TPN did, however, lead to decreased expression of mdr1, and a return to the normal ability of Rhodamine 123 to be transported out of hepatocytes. This suggests that this alteration in mdr1 function may be one mechanism by which lipids contribute to the development of PNALD.
The peroxisome proliferator activated receptors (PPAR) are one of the first characterized members of the nuclear hormone receptor protein family. PPARs have been shown to have an important role in glucose and lipid metabolism. PPARs have also been shown to be activated by fibrates which act as lipid lowering agent compounds (34, 61). Further, fibrates have been reported to increase P-gp expression in the liver, most likely by an increase in PPAR-α expression (31) (62, 63). PPARα and PPARγ are known to be factors which can modulate P-gp expression within hepatocytes (32, 64, 65). Our previous study showed that administration of intravenous fats with TPN resulted in significant alterations in lipid metabolism and hepatocyte survival (21). We therefore hypothesized that the decline in mdr2 expression might be correlated with changes in PPAR expression. Our present findings of a decline in PPAR-α with TPN, and a prevention of this decline with the co-administration of fibrates was coincident with an increase of PC in the bile. This suggests that fibrates are acting to up-regulate mdr2 function, and the use of fibrates may have a beneficial role during TPN administration. The improvement in mdr1 function with fibrate administration may also be mediated by an increased expression of PPAR-alpha, and may well be beneficial in removing hepatotoxins from the liver. Future studies will need to be done to further determine the relevance of PPAR-α and changes in mdr1 and mdr2 function.
Recently, in a rodent model of hepatic steatosis, it was shown that mediation of liver injury occurred through matrix metalloproteinase inhibitor-mediated up-regulation of IL-6 and PPAR-alpha (66); and repair of these changes may occur with use of matrix metalloproteinase 8 activity (67). As similar association of liver injury and IL-6 has been seen with bile duct ligation models (68). As PPARs are a major transcription factors for lipid metabolism, and may contribute to cytokine-mediate liver injury (69), this pathway may have a very influential role. Future work in exploring the influence of cytokines and MMP activity may well be important to further understand the mechanisms responsible for PNALD, and its relation to lipid administration. One limitation of our work is that the mouse model, as with any animal model, may not perfectly reflect PNAC in a clinical setting. It is possible that TPN in a rat may be a better model, however, both rodent models show decreased bile flow, and each have potential advantages (70).
In conclusion, administration of TPN in a mouse model resulted in a decline in mdr2 functional as shown by decreased PC in the bile of mice receiving TPN. TPN also led to a decline in the function of mdr1, as shown by a reduction in hepatocyte transport of rhodamine. Removal of lipids from TPN resulted in a normalization of mdr1 expression and function; however, removal of lipids did not change mdr2 expression or function. Fenofibrate administration to mice receiving TPN with lipids resulted in a decline in serum bile acid levels. Additionally, fibrates resulted in an improvement in both mdr1 and mdr2 functionality; and might help to reduce the incidence of PNALD.
References
- 1.Teitelbaum D. Parenteral Nutrition-Associated Cholestasis. Current Opinion in Pediatrics. 1997;9:270–5. doi: 10.1097/00008480-199706000-00016. [DOI] [PubMed] [Google Scholar]
- 2.Karrer F, Bensard D. Neonatal cholestasis. Seminars in Pediatric Surgery. 2000;9:166–9. doi: 10.1053/spsu.2000.18847. [DOI] [PubMed] [Google Scholar]
- 3.Beath S, Davies P, Papadpoulou A, Khan A, Buick R, Corkery J, et al. Parenteral nutrition-related cholestasis in postsurgical neonates: Multivariate analysis of risk factors. J Pediatr Surg. 1996;31(4):604–6. doi: 10.1016/s0022-3468(96)90507-2. [DOI] [PubMed] [Google Scholar]
- 4.Ginn-Pease M, Pantalos D, King D. TPN_associated hyperbilirubinemia: A common problem in newborn surgical patients. J Pediatr Surg. 1985;20:436–9. doi: 10.1016/s0022-3468(85)80236-0. [DOI] [PubMed] [Google Scholar]
- 5.Farrell MK, Balistreri WF. Parenteral nutrition and hepatobiliary dysfunction. Clin Perinatol. 1986;13(1):197–212. [PubMed] [Google Scholar]
- 6.Merritt RJ. Cholestasis associated with total parenteral nutrition. J Pediatr Gastroenterol Nutr. 1986;5(1):9–22. doi: 10.1097/00005176-198601000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Moss RL, Amii LA. New approaches to understanding the etiology and treatment of total parenteral nutrition-associated cholestasis. Seminars in Pediatric Surgery. 1999;8(3):140–7. doi: 10.1016/s1055-8586(99)70015-6. Review 101 refs. [DOI] [PubMed] [Google Scholar]
- 8.Drongowski RA, Coran AG. An analysis of factors contributing to the development of total parenteral nutrition-induced cholestasis. JPEN J Parenter Enteral Nutr. 1989;13(6):586–9. doi: 10.1177/0148607189013006586. [DOI] [PubMed] [Google Scholar]
- 9.Trauner M. Molecular alterations of canalicular transport systems in epxerimental models of cholestasis: Possible functional correlations. Yale J Biol Med. 1998;70:365–78. [PMC free article] [PubMed] [Google Scholar]
- 10.Balistreri W, Bezerra J, Jansen P, Karpen S, B L, Suchy F. Intrahepatic cholestasis: summary of an American Association for the Study of Liver Diseases single-topic conference. Hepatology. 2005;42(1):222–35. doi: 10.1002/hep.20729. [DOI] [PubMed] [Google Scholar]
- 11.Trauner M, wagner M, Fickert P, Zollner G. Molecular regulation of hepatobiliary transport systems: Clinical implications for understanding and treating cholestasis. J Clin Gastroenterol. 2005;39 2:S111–S24. doi: 10.1097/01.mcg.0000155551.37266.26. [DOI] [PubMed] [Google Scholar]
- 12.Muller M, Roelofsen H, Jansen PL. Secretion of organic anions by hepatocytes: involvement of homologues of the multidrug resistance protein. Semin Liver Dis. 1996;16(2):211–20. doi: 10.1055/s-2007-1007233. [DOI] [PubMed] [Google Scholar]
- 13.Oude Elferink R, Meijer D, Kuipers F, Jansen P, Growen A, Groothuis G. Hepatobiliary secretion of organic compounds: molecular mechanisms of membrane transport. Biochim Biophys Acta. 1995;1241:215–8. doi: 10.1016/0304-4157(95)00006-d. [DOI] [PubMed] [Google Scholar]
- 14.Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–66. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 15.Keppler D, Leier I, Jedlitschky G, Mayer R, Buchler M. The function of the multidrug resistance proteins (MRP and cMRP) in drug conjugate transport and hepatobiliary excretion. Adv Enzyme Regul. 1996;36:17–29. doi: 10.1016/0065-2571(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 16.Carlton V, Pawlikowska L, Bull L. Molecular basis of intrahepatic cholestasis. Ann Med. 2004;36(8):606–17. doi: 10.1080/07853890410018916. [DOI] [PubMed] [Google Scholar]
- 17.Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75(3):451–62. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 18.Van Nieuwkerk CM, Elferink RP, Groen AK, Ottenhoff R, Tytgat GN, Dingemans KP, et al. Effects of Ursodeoxycholate and cholate feeding on liver disease in FVB mice with a disrupted mdr2 P-glycoprotein gene. Gastroenterology. 1996;111(1):165–71. doi: 10.1053/gast.1996.v111.pm8698195. [DOI] [PubMed] [Google Scholar]
- 19.Duerksen DR, Van Aerde JE, Chan G, Thomson AB, Jewell LJ, Clandinin MT. Total parenteral nutrition impairs bile flow and alters bile composition in newborn piglet. Dig Dis Sci. 1996;41(9):1864–70. doi: 10.1007/BF02088759. [DOI] [PubMed] [Google Scholar]
- 20.Das J, Poulos N, Ansari G. Biliary lipid composititon and bile acid profiles during and after enteral fast of total parenteral nutrition in the rabbit. J Pediatr Gastro Nutr. 1996;22(1):85–91. doi: 10.1097/00005176-199601000-00014. [DOI] [PubMed] [Google Scholar]
- 21.Tazuke Y, Kiristioglu I, Heidelberger KP, Eisenbraun MD, Teitelbaum DH. Hepatic P-glycoprotein changes with total parenteral nutrition administration. Jpen: Journal of Parenteral & Enteral Nutrition. 2004;28(1):1–6. doi: 10.1177/014860710402800101. see comment. [DOI] [PubMed] [Google Scholar]
- 22.Yang H, Antony PA, Wildhaber BE, Teitelbaum DH. Intestinal intraepithelial lymphocyte gammadelta-T cell-derived keratinocyte growth factor modulates epithelial growth in the mouse. J Immunol. 2004;172(7):4151–8. doi: 10.4049/jimmunol.172.7.4151. [DOI] [PubMed] [Google Scholar]
- 23.Yang H, Finaly R, Teitelbaum DH. Alteration in epithelial permeability and ion transport in a mouse model of total parenteral nutrition. Critical Care Medicine. 2003;31(4):1118–25. doi: 10.1097/01.CCM.0000053523.73064.8A. [DOI] [PubMed] [Google Scholar]
- 24.Kiristioglu I, Teitelbaum DH. Alteration of the intestinal intraepithelial lymphocytes during total parenteral nutrition. Journal of Surgical Research. 1998;79(2):91–6. doi: 10.1006/jsre.1998.5408. [DOI] [PubMed] [Google Scholar]
- 25.Tazuke Y, Drongowski RA, Btaiche I, Coran AG, Teitelbaum DH. Effects of lipid administration on liver apoptotic signals in a mouse model of total parenteral nutrition (TPN) Pediatr Surg Int. 2004 doi: 10.1007/s00383-003-1115-1. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Kudsk KA, Gocinski B, Dent D, Glezer J, Langkamp-Henken B. Effects of parenteral and enteral nutrition on gut-associated lymphoid tissue. The Journal of Trauma. 1995;39(1):44–52. doi: 10.1097/00005373-199507000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Wildhaber B, Yang H, Spencer A, Drongowski R, Teitelbaum D. Lack of Enteral Nutrition—Effects on the Intestinal Immune System. J Surg Res. 2005;123:8–16. doi: 10.1016/j.jss.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 28.Eisenbraun M, Mosley R, Teitelbaum D, Miller R. Altered development of intestinal intraepithelial lymphocytes in P-glycoprotein-deficient mice. Dev Comp Immunol. 2000;24:783–95. doi: 10.1016/s0145-305x(00)00029-x. [DOI] [PubMed] [Google Scholar]
- 29.Weinhold P, Ville C. Phospholipid metabolism in the liver and lung of rats during development. Biochim Biophys Acta. 1965;196:540–50. doi: 10.1016/0005-2760(65)90070-6. [DOI] [PubMed] [Google Scholar]
- 30.Abe A, Gregory S, Lee L. Reduction of globotrioaosyleramide Fabry disease mice by substrate deprivation. J Clin Invest. 2000;105:1563–71. doi: 10.1172/JCI9711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chianale J, Vollrath V, Wielandt AM, Amigo L, Rigotti A, Nervi F, et al. Fibrates induce mdr2 gene expression and biliary phospholipid secretion in the mouse. Biochem J. 1996;314(Pt 3):781–6. doi: 10.1042/bj3140781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kok T, Bloks V, Wolters H, Havinga R, Jansen P, Staels B, et al. Peroxisome proliferator-activated receptor alpha (PPARalpha)-mediated regulation of multidrug resistance 2 (Mdr2) expression and function in mice. J Biochem. 2003;369:539–47. doi: 10.1042/BJ20020981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aoyama T. Altered constitutive expression of fatty acid- metabolizing enzymes in mice lacking the peroxysome proliferator- activated receptor-alpha (PPAR-a) J Biol Chem. 1998;273:5678–84. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- 34.Yamazaki K, Kuromitsu J, Tanaka I. Microarray analysis of gene expression changes in mouse liver induced by peroxisome proliferator- activated receptor alpha agonists. Biochem Biophys Res Commun. 2002;290(3):1114–22. doi: 10.1006/bbrc.2001.6319. [DOI] [PubMed] [Google Scholar]
- 35.Boyer J. Nuclear receptor ligands: rational and effective therapy for chronic cholestatic liver disease? Gastroenterology. 2005;129(2):735–40. doi: 10.1016/j.gastro.2005.06.053. [DOI] [PubMed] [Google Scholar]
- 36.Green RM, Beier D, Gollan JL. Regulation of hepatocyte bile salt transporters by endotoxin and inflammatory cytokines in rodents. Gastroenterology. 1996;111(1):193–8. doi: 10.1053/gast.1996.v111.pm8698199. [DOI] [PubMed] [Google Scholar]
- 37.Trauner M, Arrese M, Lee H, Boyer J, Karpen S. Endotoxin downregulates rat hepatic ntcp gene expression via decreased activity of critical transcription factors. J Clin Invest. 1998;101(10):2092–100. doi: 10.1172/JCI1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vos T, Hooiveld G, Koning H, Childs S, Meijer D, Moshage H, et al. Up-regulation of the multidrug resistance genes, mrp1 and mdr1b, and down-regulation of the organic anion transporter mrp2, and bile salt transporter, spgp, in endotoxemic rat liver. Hepatology. 1998;28(6):1637–44. doi: 10.1002/hep.510280625. [DOI] [PubMed] [Google Scholar]
- 39.Peden VH, Witzleben CL, Skelton MA. Total parenteral nutrition. J Pediatr. 1971;78(1):180–1. doi: 10.1016/s0022-3476(71)80289-5. [DOI] [PubMed] [Google Scholar]
- 40.Sax HC, Bower RH. Hepatic complications of total parenteral nutrition. Journal of Parenteral and Enteral Nutrition. 1988;12(No 6):615–8. doi: 10.1177/0148607188012006615. [DOI] [PubMed] [Google Scholar]
- 41.Bell RL, Ferry GD, Smith EO, Shulman RJ, Christensen BL, Labarthe DR, et al. Total parenteral nutrition-related cholestasis in infants. JPEN J Parenter Enteral Nutr. 1986;10(4):356–9. doi: 10.1177/0148607186010004356. [DOI] [PubMed] [Google Scholar]
- 42.Teitelbaum DH, Tracy TJ, Aouthmany M, Llanos A, Brown M, Yu S, et al. Use of Cholecsytokinin-Octapeptide for the Prevention of Parenteral Nutrition-Associated Cholestasis. Pediatrics. 2005;115(5):1332–40. doi: 10.1542/peds.2004-1014. [DOI] [PubMed] [Google Scholar]
- 43.Heubi JE, Wiechmann DA, Creutzinger V, Setchell KD, Squires RJ, Couser R, et al. Tauroursodeoxycholic acid (TUDCA) in the prevention of total parenteral nutrition-associated liver disease. Journal of Pediatrics. 2002;141(2):237–42. doi: 10.1067/mpd.2002.125802. see comment. [DOI] [PubMed] [Google Scholar]
- 44.Roninson I, Chin J, KG C. Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc Natl Aacd Sci USA. 1986;83:4538–42. doi: 10.1073/pnas.83.12.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ueda K, Cardarelli C, Gottesman M. Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Aacd Sci USA. 1987;84:3004–8. doi: 10.1073/pnas.84.9.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gross P, Ben Neriah Y, Croop J. Isolation and expression of a complementary DNA that confers multidrug resistance. Nature. 1986;323:728–31. doi: 10.1038/323728a0. [DOI] [PubMed] [Google Scholar]
- 47.Raymond M, Gros P. Cell-specific activity of cis-acting regulatory elements in the promoter of the mouse multidrug resistance gene mdr1. Mol Cell Biol. 1990;10:6036–40. doi: 10.1128/mcb.10.11.6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crawford A, Smith A, Hatch V. Hepatic secretion of phospholipid vesicles in the mouse critically depends on mdr2 or MDR3 P-glycoprotein expression. Visualization by electron microscopy. J Clin Invest. 1997;100:2562–7. doi: 10.1172/JCI119799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gottesman M, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 50.Dawes L, Greiner M, Joehl R. Altered gallbladder bile acidification with long-term total parenteral nutrition. J Surg Res. 1999;81:21–6. doi: 10.1006/jsre.1998.5492. [DOI] [PubMed] [Google Scholar]
- 51.Sun X, Yang H, Nose K, Nose S, Haxhija EQ, Koga H, et al. Decline in intestinal mucosal IL-10 expression and decreased intestinal barrier function in a mouse model of total parenteral nutrition. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G139–47. doi: 10.1152/ajpgi.00386.2007. [DOI] [PubMed] [Google Scholar]
- 52.Hartmann G, Kim H, Piquette-Miller Reguration of the hepatic multidrug resistance gene expression by endotokin and inflammatory cytokines in mice. Internat Immunopath. 2001;1:189–99. doi: 10.1016/s0162-3109(00)00271-x. [DOI] [PubMed] [Google Scholar]
- 53.Tracy TF, Jr, Fox ES. CD14-lipopolysaccharide receptor activity in hepatic macrophages after cholestatic liver injury. Surgery. 1995;118(2):371–7. doi: 10.1016/s0039-6060(05)80347-2. [DOI] [PubMed] [Google Scholar]
- 54.Gura K, Duggan C, Collier S, Jennings R, Folkman J, Bistrian B, et al. Reversal of parenteral nutrition-associated liver disease in two infants with short bowel syndrome using parenteral fish oil: implications for future management. Pediatrics. 2006;118(1):e197–201. doi: 10.1542/peds.2005-2662. [DOI] [PubMed] [Google Scholar]
- 55.Clayton PT, Whitfield P, Iyer K. The role of phytosterols in the pathogenesis of liver complications of pediatric parenteral nutrition. Nutrition. 1998;14:158–64. doi: 10.1016/s0899-9007(97)00233-5. [DOI] [PubMed] [Google Scholar]
- 56.Iyer K, Spitz L, Clayton P. New insight into mechanims of parenteral nutrition -associated cholestasis: Role of plant sterols. J Pediatr Surg. 1998;33(1):1–6. doi: 10.1016/s0022-3468(98)90349-9. [DOI] [PubMed] [Google Scholar]
- 57.Iyer KR, Spitz L, Clayton P. BAPS prize lecture: New insight into mechanisms of parenteral nutrition-associated cholestasis: role of plant sterols. British Association of Paediatric Surgeons. Journal of Pediatric Surgery. 1998;33(1):1–6. doi: 10.1016/s0022-3468(98)90349-9. [DOI] [PubMed] [Google Scholar]
- 58.Colomb V, Jobert-Giraud A, Lacaille F, Goulet O, Fournet JC, Ricour C. Role of lipid emulsions in cholestasis associated with long-term parenteral nutrition in children. Journal of Parenteral & Enteral Nutrition. 2000;24(6):345–50. doi: 10.1177/0148607100024006345. [DOI] [PubMed] [Google Scholar]
- 59.Teitelbaum DH, Tracy T. Parenteral nutrition-associated cholestasis. Seminars in Pediatric Surgery. 2001;10(2):72–80. doi: 10.1053/spsu.2001.22386. [DOI] [PubMed] [Google Scholar]
- 60.Btaiche IF, Khalidi N. Parenteral nutrition-associated liver complications in children. Pharmacotherapy. 2002;22:188–211. doi: 10.1592/phco.22.3.188.33553. [DOI] [PubMed] [Google Scholar]
- 61.Xu C, Li C, AN K. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. 2005;28(3):249–68. doi: 10.1007/BF02977789. [DOI] [PubMed] [Google Scholar]
- 62.Nishioka T, Hyogo H, Numata Y, Yamaguchi A, Kobuke T, Komichi D, et al. A nuclear receptor-mediated choleretic action of fibrates is associated with enhanced canalicular membrane fluidity and transporter activity mediating bile acid-independent bile secretion. J Atheroscler Thromb. 2005;12(4):211–7. doi: 10.5551/jat.12.211. [DOI] [PubMed] [Google Scholar]
- 63.Kok T, Bloks V, Wolters H, Havinga R, Jansen P, Staels B, et al. Peroxisome proliferator-activated receptor alpha (PPARalpha)-mediated regulation of multidrug resistance 2 (Mdr2) expression and function in mice. Biochem J. 2003;369:539–47. doi: 10.1042/BJ20020981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Accatino L, Pizarro M, Solis N, Koenig CS, Vollrath V, Chianale J. Modulation of hepatic content and biliary excretion of P- glycoproteins in hepatocellular and obstructive cholestasis in the rat. J Hepatol. 1996;25(3):349–61. doi: 10.1016/s0168-8278(96)80122-x. [DOI] [PubMed] [Google Scholar]
- 65.Chieli E, Romiti N, Cervelli F, Tongiani R. Effects of flavonols on P-glycoprotein activity in cultured rat hepatocytes. Life Sci. 1995;57(19):1741–51. doi: 10.1016/0024-3205(95)02152-9. [DOI] [PubMed] [Google Scholar]
- 66.Alwayn I, Andersson C, Lee S, Arsenault D, Bistrian B, Gura K, et al. Inhibition of matrix metalloproteinases increases PPAR-alpha and IL-6 and prevents dietary-induced hepatic steatosis and injury in a murine model. Am J Physiol Gastrointest Liver Physiol. 2006;291(6):G1011–9. doi: 10.1152/ajpgi.00047.2006. [DOI] [PubMed] [Google Scholar]
- 67.Harty M, Huddleston H, Papa E, Puthawala T, Tracy A, Ramm G, et al. Repair after cholestatic liver injury correlates with neutrophil infiltration and matrix metalloproteinase 8 activity. Surgery. 2005;138(2):313–20. doi: 10.1016/j.surg.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 68.Gehring S, Dickson E, San Martin M, van Rooijen N, Papa E, Harty M, et al. Kupffer cells abrogate cholestatic liver injury in mice. Gastroenterology. 2006;130(3):810–22. doi: 10.1053/j.gastro.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 69.Kim M, Sweeney T, Shigenaga J, Chui L, Moser A, Grunfeld C, et al. Tumor necrosis factor and interleukin 1 decrease RXRalpha, PPARalpha, PPARgamma, LXRalpha, and the coactivators SRC-1, PGC-1alpha, and PGC-1beta in liver cells. Metabolism. 2007;56(2):267–79. doi: 10.1016/j.metabol.2006.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Belli D, Albrecht R, La Scala G, Desjeux J, Pelissier M. Homocysteine prevents total parenteral nutrition (TPN)-induced cholestasis without changes in hepatic oxidative stress in the rat. J Pediatr Gastroenterol Nutr. 2003;36(2):200–5. doi: 10.1097/00005176-200302000-00008. [DOI] [PubMed] [Google Scholar]