

Abstract
The lipid-lowering drugs, 3-hydroxy-3-methylgulutaryl-coenzyme A (HMG-CoA) reductase inhibitors or statins, are used in the prevention and treatment of cardiovascular diseases. Recent experimental and clinical studies suggest that statins may exert vascular protective effects beyond cholesterol reduction. For example, statins improve endothelial function by cholesterol-dependent and -independent mechanisms. The cholesterol-independent or “pleiotropic” effects of statins include the upregulation and activation of endothelial NO synthase (eNOS). Because statins inhibit an early step in the cholesterol biosynthetic pathway, they also inhibit the synthesis of isoprenoids such as farnesylpyrophosphate and geranylgeranylpyrophosphate, which are important posttranslational lipid attachments for intracellular signaling molecules such as the Rho GTPases. Indeed, decrease in Rho GTPase responses as a consequence of statin treatment increases the production and bioavailability of endothelium-derived NO. The mechanism involves, in part, Rho/Rho-kinase (ROCK)-mediated changes in the actin cytoskeleton, which leads to decreases in eNOS mRNA stability. The regulation of eNOS by Rho GTPases, therefore, may be an important mechanism underlying the cardiovascular protective effect of statins.
Keywords: statin, Rho, Rho-kinase, endothelium, nitric oxide
The vascular endothelium serves as an important autocrine and paracrine organ that regulates homeostasis of the vascular wall, and impaired endothelial function is observed in a variety of pathological conditions such as hypertension, atherosclerosis, and heart failure. Endothelial dysfunction, which is characterized as the decreased synthesis, release, and/or activity of endothelial-derived nitric oxide (NO), is a strong predictor of cardiovascular disease. Indeed, hypercholesterolemia, which impairs endothelial function, is an important risk factor for vascular disease,1,2 and lipid lowering therapies have been shown to reduce atherosclerosis and cardiovascular events.3,4 For example, LDL apheresis alone can rapidly improve endothelial function.5 Similar improvements in endothelial function could be observed with 3-hydroxy-3-methylgulutaryl coenzyme A (HMG-CoA) reductase inhibitors or statins, which lower serum cholesterol levels.6,7
Because cholesterol reduction in itself improves endothelial function, it has been generally assumed that most, if not all, of the beneficial effects of statins on endothelial function are attributable to cholesterol reduction. However, one of the earliest recognizable benefits of statin therapy is the improvement in endothelial function, which in some instances occurs before significant reduction in serum cholesterol levels.8 Furthermore, a recent study showed that despite comparable modest reduction of serum cholesterol levels by ezetimibe, an intestinal inhibitor of cholesterol absorption, and statin, only the statin improved endothelial function.9 Thus, it is likely that the beneficial effects of statins on endothelial function extend beyond cholesterol reduction. Indeed, statins have been shown to reduce cardiovascular events in patients, irrespective of serum cholesterol levels.4
Inhibition of Isoprenylation of Rho GTPases by Statins
Statins inhibit HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis in the liver, which catalyzes the conversion of HMG-CoA to mevalonic acid (Figure 1). In addition to inhibiting cholesterol synthesis, statins also block the synthesis of isoprenoid intermediates such as farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP).10 Both FPP and GGPP serve as important lipid attachments for the posttranslational modification of a variety of proteins, including heterotrimeric G proteins and small GTP-binding proteins belonging to the family of Ras, Rho, Rap, and Rab GTPases.11 Isoprenylation is critical for intracellular trafficking and function of small GTP-binding proteins.12 In general, modification with FPP is necessary for proper localization of Ras family proteins, whereas GGPP is required for Rho, Rab, and Rap family proteins.11 However, some Rho GTPases require both farnesylation and geranylgeranylation for proper function and intracellular localization.
Figure 1.
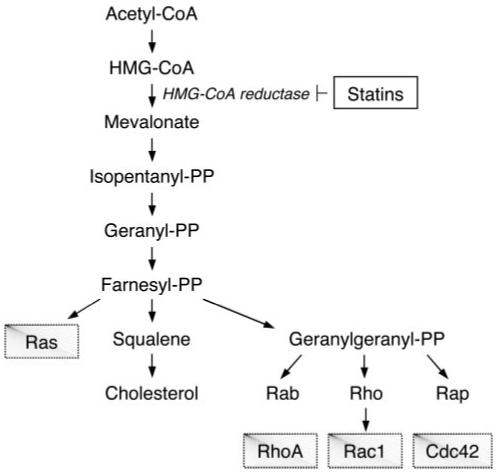
Cholesterol biosynthesis pathway and the effects of statins. Inhibition of HMG-CoA reductase by statins decreases isoprenoid intermediates such as farnesyl-PP and geranylgeranyl-PP, which leads to an inhibition of isoprenylation of small GTPases such as Ras, Rho, Rab, and Rap. Among the Rho GTPases are RhoA, Rac1, and Cdc42. CoA indicates coenzyme A; PP, pyrophosphate.
By inhibiting mevalonate synthesis, statins inhibit the synthesis of isoprenoid intermediates thereby preventing isoprenylation of small GTPases, leading to the inhibition of these signaling molecules. Interestingly, some of cholesterol-independent, or so-called “pleiotropic” effects of statins may be attributable to the ability of statins to block the synthesis of isoprenoid intermediates.
Statins and eNOS Expression
A hallmark of endothelial dysfunction is reduced bioavailability of NO, which could be caused by reduced expression of eNOS, impairment of eNOS activation, and increased inactivation of NO by oxidative stress. The ability of statins to increase eNOS expression and activation may be an important mechanism by which statins improve endothelial function in addition to cholesterol reduction (Figure 2). Indeed, statins upregulate eNOS expression by cholesterol-independent mechanism.13 The increase in eNOS expression by statins is reversed by GGPP, but not FPP, suggesting the involvement of small GTPases requiring geranylgeranylation. Indeed, transfection of endothelial cells with a dominant negative RhoA mutant, N19RhoA, leads to increase in eNOS expression.14,15 Similar effect on eNOS expression was not observed with dominant negative mutants of Rac1 or Cdc42. In agreement with these results, Shiga et al showed that inhibition of RhoA by a recombinant protein representing the Rho-binding domain of ROCK leads to the upregulation of eNOS in rabbit mesenteric artery.16 The upregulation of eNOS by statins is attributable to increase in eNOS mRNA half-life.13 For example, TNF-α, oxidized low-density lipoprotein (oxLDL), and hypoxia downregulate eNOS expression via destabilizing eNOS mRNA, and cotreatment with statins prevents eNOS downregulation by prolonging half-life of eNOS mRNA.13,17,18 The prolongation of half-life eNOS mRNA by statins is reversed by GGPP, but not FPP, suggesting the involvement of small GTPases such as Rho GTPase in this process. Indeed, inhibition of Rho and perhaps other small GTPases leads to an increase in eNOS mRNA half-life.14
Figure 2.
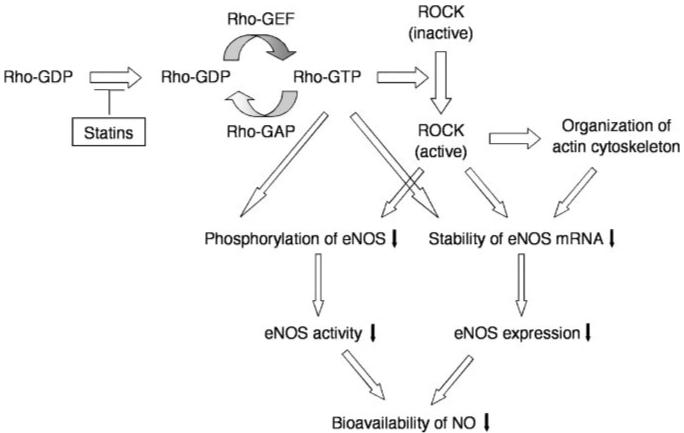
Regulation of eNOS expression and activity by statins, Rho, ROCK, and actin cytoskeleton. Statins suppress translocation of Rho by inhibiting isoprenylation of Rho. Active forms of Rho and ROCK decrease eNOS mRNA stability and eNOS phosphorylation, resulting in downregulation of eNOS expression and decrease in eNOS activity. Inhibition of Rho and ROCK activity by statins may chronically upregulate of eNOS expression and acutely stimulate eNOS activity.
An important downstream mediator of Rho is ROCK. Recent studies suggest that ROCK can also regulate eNOS mRNA stability.19,20 For example, hypoxia and thrombin, which stimulate ROCK activity, downregulate eNOS expression via destabilization of eNOS mRNA. Furthermore, direct inhibition of ROCK by ROCK inhibitors such as hydroxyfasudil and Y27632, or by overexpression of a dominant-negative mutant of ROCK, increases eNOS mRNA half-life and expression.21 Indeed, in ROCK1 knockout mice, basal eNOS expression is increased in various tissues, including the lung and kidney (Y.R. and J.K.L., unpublished data, 2005). Thus, inhibition of the Rho/ROCK pathway leading to the upregulation of eNOS may contribute to some of the cardiovascular benefits of statin therapy.
Acute Activation of eNOS by Statins
In addition to increase in eNOS expression by statins, statins can also rapidly induce the phosphorylation and activation of eNOS via the phosphatidylinositol-3 kinase (PI3K)/protein kinase Akt pathway.22 For example, treatment of cultured human endothelial cells with simvastatin rapidly increases phosphorylation of Akt and eNOS, which leads to increase angiogenesis in response to hind limb ischemia. Furthermore, the activation of Akt by statins also occurs in endothelial progenitor cells (EPC)23 and podocytes.24 The ability of Akt to phosphorylate eNOS at Ser1179 is blocked by PI3K inhibitors.25 These findings suggest that the activation of PI3K/Akt pathway mediates the rapid increase in eNOS activity by statins. Interestingly, the Rho GTPases may play a role in the activation of PI3K/Akt by statins. Inhibition of Rho or ROCK leads to the rapid phosphorylation and activation of Akt via PI3K, resulting in an increase in NO production.26 In contrast, transfection of constitutively active mutants of RhoA and ROCK leads to the inhibition of eNOS phosphorylation at Ser1177.27 This inhibition was reversed by overexpressing a constitutively-active mutant of Akt. Thus, the Rho/ROCK pathway can negatively regulate endothelial function at the level of both eNOS expression and activity via two distinct mechanisms (Figure 2).
Very recently, PTEN, a phosphatase that dephosphorylates phosphoinositide substrates, may link RhoA/ROCK with protein kinase Akt.28 RhoA/ROCK regulates the intracellular localization and phosphorylation of PTEN, and RhoA/ROCK-mediated phosphorylation of PTEN is required for the phospholipid phosphatase activity of PTEN that antagonizes PI3K-mediated Akt signaling. Therefore, inhibition of RhoA/ROCK pathway in endothelial cells may stimulate Akt activity by decreasing PTEN activity. Further experiments are needed, however, to determine whether PTEN is involved in statin-induced activation of Akt and eNOS.
Physiological Effects of Rho GTPase Inhibition by Statins
In spontaneous hypertensive stroke prone rats (SHR-SP), eNOS expression is decreased in the brain.29,30 Furthermore, the expression and activity of eNOS are decreased in the brains of mice after middle cerebral artery (MCA) occlusion. Statins confer stroke protection by increasing the expression of eNOS via inhibition of Rho-mediated actin cytoskeletal changes, leading to the stabilization of eNOS mRNA.13-15 This is associated with increase in absolute cerebral blood flow, reduction in stroke size, and improvement in neurologic function.15,31 The enhancement of cerebral blood flow by statins is absent in eNOS knockout mice. Thus, the neuroprotective effects of statins appear to be mediated, in part, by eNOS.
Similar to the effects of statins, treatment with Rho or ROCK inhibitors, such as Clostridium botulinum C3 exo-transferase, fasudil or Y27632, or actin cytoskeletal disrupter such as cytochalasin D, decreases stroke size after MCA occlusion.15,21 All of these agents upregulate eNOS expression and activity in vivo. Furthermore, the neuroprotective effects of ROCK inhibitors are absent in eNOS knockout mice, indicating the critical role of eNOS in mediating the beneficial effects of Rho/ROCK inhibition.
Summary
In addition to the lipid-lowering, statins may exert other effects on the vascular wall. The ability of statins to inhibit isoprenylation of Rho GTPase may contribute to some of their beneficial effects on improving endothelial function. However, we cannot exclude the contributions of other small GTPases such as those belonging to the Ras, Rab, Rap families whose activities are dependent on isoprenlyation and could also be inhibited by statins. Furthermore, other target molecules of Rho such as mDia, protein kinase N, and citron kinase may be inhibited by statin therapy, leading to the observed changes in endothelial function. Nevertheless, experimental and clinical studies suggest that many of the so-called pleiotropic effects of statins may be attributable to the inhibition of the Rho/ROCK pathway in the vascular wall.32,33 As such, the Rho/ROCK pathway has gained important prominence as a promising therapeutic target in cardiovascular diseases.34 It remains to be determined how Rho/ROCK regulates eNOS mRNA stability through the actin cytoskeleton. Further studies investigating the connection between actin cytoskeletal proteins and eNOS mRNA may shed more light on some of the noncholesterol benefits of statin therapy.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NS10828 and HL52233) and the American Heart Association (Bugher Foundation Award). Y.R. is a recipient of an AHA Postdoctoral Fellowship-Northeast Affiliate and the Japan Heart Foundation/Bayer-Yakuhin Research Grant Abroad.
References
- 1.Sytkowski PA, Kannel WB, D’Agostino RB. Changes in risk factors and the decline in mortality from cardiovascular disease. The Framingham Heart Study. N Engl J Med. 1990;322:1635–1641. doi: 10.1056/NEJM199006073222304. [DOI] [PubMed] [Google Scholar]
- 2.Gordon T, Kannel WB. Premature mortality from coronary heart disease. The Framingham study. JAMA. 1971;215:1617–1625. [PubMed] [Google Scholar]
- 3.Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–1389. [PubMed] [Google Scholar]
- 4.MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22. doi: 10.1016/S0140-6736(02)09327-3. [DOI] [PubMed] [Google Scholar]
- 5.Tamai O, Matsuoka H, Itabe H, Wada Y, Kohno K, Imaizumi T. Single LDL apheresis improves endothelium-dependent vasodilatation in hypercholesterolemic humans. Circulation. 1997;95:76–82. doi: 10.1161/01.cir.95.1.76. [DOI] [PubMed] [Google Scholar]
- 6.Anderson TJ, Meredith IT, Yeung AC, Frei B, Selwyn AP, Ganz P. The effect of cholesterol-lowering and antioxidant therapy on endothelium-dependent coronary vasomotion. N Engl J Med. 1995;332:488–493. doi: 10.1056/NEJM199502233320802. [DOI] [PubMed] [Google Scholar]
- 7.Treasure CB, Klein JL, Weintraub WS, Talley JD, Stillabower ME, Kosinski AS, Zhang J, Boccuzzi SJ, Cedarholm JC, Alexander RW. Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease. N Engl J Med. 1995;332:481–487. doi: 10.1056/NEJM199502233320801. [DOI] [PubMed] [Google Scholar]
- 8.O’Driscoll G, Green D, Taylor RR. Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation. 1997;95:1126–1131. doi: 10.1161/01.cir.95.5.1126. [DOI] [PubMed] [Google Scholar]
- 9.Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N, Schulz S, Manes C, Fischer D, de Groot K, Fliser D, Fauler G, Marz W, Drexler H. Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation. 2005;111:2356–2363. doi: 10.1161/01.CIR.0000164260.82417.3F. [DOI] [PubMed] [Google Scholar]
- 10.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 11.Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 12.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 13.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 14.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 15.Laufs U, Endres M, Stagliano N, Amin-Hanjani S, Chui DS, Yang SX, Simoncini T, Yamada M, Rabkin E, Allen PG, Huang PL, Bohm M, Schoen FJ, Moskowitz MA, Liao JK. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J Clin Invest. 2000;106:15–24. doi: 10.1172/JCI9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiga N, Hirano K, Hirano M, Nishimura J, Nawata H, Kanaide H. Long-term inhibition of RhoA attenuates vascular contractility by enhancing endothelial NO production in an intact rabbit mesenteric artery. Circ Res. 2005;96:1014–1021. doi: 10.1161/01.RES.0000165483.34603.91. [DOI] [PubMed] [Google Scholar]
- 17.Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem. 1997;272:31725–31729. doi: 10.1074/jbc.272.50.31725. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez-Fernandez F, Jimenez A, Lopez-Blaya A, Velasco S, Arriero MM, Celdran A, Rico L, Farre J, Casado S, Lopez-Farre A. Cerivastatin prevents tumor necrosis factor-alpha-induced downregulation of endothelial nitric oxide synthase: role of endothelial cytosolic proteins. Atherosclerosis. 2001;155:61–70. doi: 10.1016/s0021-9150(00)00535-9. [DOI] [PubMed] [Google Scholar]
- 19.Eto M, Barandier C, Rathgeb L, Kozai T, Joch H, Yang Z, Luscher TF. Thrombin suppresses endothelial nitric oxide synthase and upregulates endothelin-converting enzyme-1 expression by distinct pathways: role of Rho/ROCK and mitogen-activated protein kinase. Circ Res. 2001;89:583–590. doi: 10.1161/hh1901.097084. [DOI] [PubMed] [Google Scholar]
- 20.Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- 21.Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, Moskowitz MA, Liao JK. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke. 2005;36:2251–2257. doi: 10.1161/01.STR.0000181077.84981.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Llevadot J, Murasawa S, Kureishi Y, Uchida S, Masuda H, Kawamoto A, Walsh K, Isner JM, Asahara T. HMG-CoA reductase inhibitor mobilizes bone marrow-derived endothelial progenitor cells. J Clin Invest. 2001;108:399–405. doi: 10.1172/JCI13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bussolati B, Deregibus MC, Fonsato V, Doublier S, Spatola T, Procida S, Di Carlo F, Camussi G. Statins prevent oxidized LDL-induced injury of glomerular podocytes by activating the phosphatidylinositol 3-kinase/AKT-signaling pathway. J Am Soc Nephrol. 2005;16:1936–1947. doi: 10.1681/ASN.2004080629. [DOI] [PubMed] [Google Scholar]
- 25.Harris MB, Blackstone MA, Sood SG, Li C, Goolsby JM, Venema VJ, Kemp BE, Venema RC. Acute activation and phosphorylation of endothelial nitric oxide synthase by HMG-CoA reductase inhibitors. Am J Physiol Heart Circ Physiol. 2004;287:H560–H566. doi: 10.1152/ajpheart.00214.2004. [DOI] [PubMed] [Google Scholar]
- 26.Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, Dominiak P, Liao JK. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol. 2004;24:1842–1847. doi: 10.1161/01.ATV.0000142813.33538.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ming XF, Viswambharan H, Barandier C, Ruffieux J, Kaibuchi K, Rusconi S, Yang Z. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol. 2002;22:8467–8477. doi: 10.1128/MCB.22.24.8467-8477.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L, Wu D. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005;7:399–404. doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- 29.Kimoto-Kinoshita S, Nishida S, Tomura TT. Decrease of endothelial nitric oxide synthase in stroke-prone spontaneously hypertensive rat cerebral cortex. Neurosci Lett. 2000;288:103–106. doi: 10.1016/s0304-3940(00)01227-1. [DOI] [PubMed] [Google Scholar]
- 30.Kishi T, Hirooka Y, Mukai Y, Shimokawa H, Takeshita A. Atorvastatin causes depressor and sympatho-inhibitory effects with upregulation of nitric oxide synthases in stroke-prone spontaneously hypertensive rats. J Hypertens. 2003;21:379–386. doi: 10.1097/00004872-200302000-00030. [DOI] [PubMed] [Google Scholar]
- 31.Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, Liao JK. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1998;95:8880–8885. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laufs U, Liao JK. Isoprenoid metabolism and the pleiotropic effects of statins. Curr Atheroscler Rep. 2003;5:372–378. doi: 10.1007/s11883-003-0008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rikitake Y, Liao JK. ROCKs as therapeutic targets in cardiovascular diseases. Expert Rev Cardiovasc Ther. 2005;3:441–451. doi: 10.1586/14779072.3.3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]