

Abstract
Background and Purpose
Endothelium-derived nitric oxide (NO) plays a pivotal role in vascular protection. The Rho kinase (ROCK) inhibitor, hydroxyfasudil, prevents the downregulation of endothelial NO synthase (eNOS) under hypoxic conditions. However, it is unknown whether inhibition of ROCK can attenuate ischemia-induced endothelial dysfunction and tissue damage in vivo.
Methods
Human vascular endothelial cells were treated with increasing concentrations of hydroxyfasudil (0.1 to 100 μmol/L) and eNOS expression and activity were measured. To determine the physiological relevance of eNOS regulation by ROCK, we administered fasudil, which is metabolized to hydroxyfasudil in vivo, to mice for 2 days before subjecting them to middle cerebral artery occlusion. Cerebral blood flow, cerebral infarct size, and neurologic deficit were measured.
Results
In a concentration-dependent manner, hydroxyfasudil increased eNOS mRNA and protein expression, resulting in a 1.9- and 1.6-fold increase, respectively, at 10 μmol/L (P<0.05 for both). This correlated with a 1.5- and 2.3-fold increase in eNOS activity and NO production, respectively (P<0.05 for both). Fasudil increased cerebral blood flow to both ischemic and nonischemic brain areas, reduced cerebral infarct size by 33%, and improved neurologic deficit score by 37% (P<0.05). This correlated with inhibition of brain and vascular ROCK activity and increased eNOS expression and activity. Another ROCK inhibitor, Y-27632, also showed similar effects. The neuroprotective effects of fasudil were absent in eNOS-deficient mice.
Conclusions
These findings indicate that the neuroprotective effect of ROCK inhibition is mediated by endothelium-derived NO and suggest that ROCK may be an important therapeutic target for ischemic stroke.
Keywords: cerebral blood flow, nitric oxide, stroke
Nitric oxide (NO), constitutively produced by endothelial NO synthase (eNOS), regulates cerebral blood flow (CBF) and vascular tone and protects against ischemic stroke by increasing collateral flow to the ischemic area. Indeed, eNOS-deficient (eNOS-/-) mice develop larger cerebral infarctions after middle cerebral artery (MCA) occlusion.1 Inhibition of NOS activity by N-nitro-l-arginine methyl ester (l-NAME) decreases CBF and increases stroke size in mice lacking neuronal NO synthase.2 In contrast, augmentation of endothelial NO production by administration of the eNOS substrate, l-arginine, or HMG-CoA reductase inhibitors, statins, has been shown to decrease cerebral infarct area probably by enhancing collateral blood flow.3,4 Thus, conditions that enhance eNOS activity could have beneficial effects on cerebrovascular disease.
Recent large clinical trials suggest that statins decrease the incidence of myocardial infarctions and ischemic strokes, in part by mechanisms beyond cholesterol reduction, possibly involving the improvement and restoration of endothelial function.5-7 Many of cholesterol-independent or so-called pleiotropic effects reflect statins’ ability to block the synthesis of isoprenoid intermediates, which serve as lipid attachments for intracellular signaling molecules. The inhibition of small GTP-binding proteins Rho, Ras, and Rac, whose proper membrane localization and function are dependent on isoprenylation, may play an important role in mediating the biologic effects of statins. Statins prevent downregulation of eNOS expression and activity under hypoxia and by oxidized low-density lipoprotein through inhibition of Rho, leading to the stabilization of eNOS mRNA.8-10
Rho kinase (ROCK), one of the downstream effectors of Rho, is a serine/threonine kinase that is activated when bound to the active GTP-bound form of Rho. ROCK regulates stress fiber formation, smooth muscle contraction, and cell migration.11 Recent studies suggest that ROCK plays an important role in pathologic conditions, including cerebral and coronary vasospasm,12,13 hypertension,14 vascular inflammation and remodeling,15 and arteriosclerosis.16 Inhibition of ROCK activity may mediate some of the beneficial effects of statins. Similar to statins, ROCK inhibitors reverse the downregulation of eNOS under hypoxic conditions and in response to thrombin.17,18 However, the physiological relevance of eNOS regulation by ROCK is unknown. The aim of this study, therefore, was to investigate the effects of ROCK on eNOS in ischemic stroke.
Methods
Cell Culture
Human aortic endothelial cells (HAEC), human umbilical vein endothelial cells (HUVEC), human saphenous vein endothelial cells (HSVEC), and bovine aortic endothelial cells (BAEC) were cultured as described previously.8-10,17 Adenovirus vectors expressing dominant negative ROCK (Ad-DN-Rho-K) or β-galactosidase (Ad-LacZ) were infected as described.17
Western Blotting
Protein extraction and immunoblotting were performed as described previously.8-10,17 For measurement of ROCK activity, tissues were treated in 10% trichloroacetic acid with acetone and immunoblotting with phospho-Thr696 MYPT and MYPT polyclonal antibodies (Up-state and Santa Cruz Biotechnology) was performed, and ratio of phospho-Thr696 MYPT and MYPT was determined.
Northern Blotting
RNA extraction and northern blotting were performed as described.8-10,17 To determine eNOS mRNA stability, cells were treated with the RNA synthetase inhibitor 5,6-dichlorobenzimidazole riboside (DRB; Sigma).
Nitric Oxide Synthase Activity Assay
NOS activity was determined by measuring the conversion of [3H]-l-arginine to [3H]-l-citrulline using NOS assay kit (Calbiochem).8-10,17
Measurements of Nitric Oxide Production
Nitrite accumulation in the culture media was determined by chemiluminescence method with the use of nitric analyzer (NOA280i; Sievers Instruments, Inc). Nonspecific value was determined in the presence of 2 mmol/L of NG-monomethyl-l-arginine (l-NMMA).
Endothelial Nitric Oxide Synthase Promoter Activity Assay
BAEC (90% confluent) in 6-well plates was cotransfected with 4 μg of the [-1.8 kb] eNOS promoter linked to the luciferase reporter gene17 and 0.5 ng of pRL-CMV vector with the use of LipofectAMINE2000 reagent (Invitrogen). Luciferase activities were determined by dual-luciferase reporter assay system (Promega) with the use of Berthold L9501 luminometer.
Model of Focal Cerebral Ischemia
All experiments were conducted in accordance with National Institute of Health and Massachusetts General Hospital institutional guidelines. Mice were intraperitoneally administered with saline, fasudil (1, 3, or 10 mg/kg per day for 2 days) or Y-27632 (10 mg/kg per day for 2 days). Transient focal cerebral ischemia was induced in male wild-type or eNOS-/- mice (Taconic) as described previously.4 Both mice were on a mixed background of C57BL/6 and SV129. Littermates were used as controls. Infarct areas and neurologic deficits were determined as described.4
Cerebral Blood Flow Measurement
Regional and absolute CBF were measured using [14C]iodoantipyrine autoradiography and [14C]iodoamphetamine indicator fractionation technique, respectively.4,19
Statistical Analyses
Results are expressed as mean±standard error of mean. All data except neurologic deficit score were analyzed by means of Student t test or ANOVA followed by Fisher exact test for post hoc analyses. Neurologic deficit score was analyzed by Mann-Whitney test. A value of P<0.05 was considered statistically significant.
Results
Selectivity of Fasudil and Hydroxyfasudil for Inhibiting Protein Kinases
The inhibitory activities of fasudil and its active metabolite hydroxyfasudil on serine/threonine kinases were determined. Compared with the other kinases studied, fasudil and hydroxyfasudil were relatively more selective for ROCK1 and ROCK2, with hydroxyfasudil (IC50 value for ROCK1 and ROCK2; 0.73 and 0.72 μmol/L, respectively) being slightly more selective than fasudil (IC50 value for ROCK1 and ROCK2; 1.2 and 0.82 μmol/L, respectively). Compared with ROCKs, the IC50 value for protein kinase A (PKA) was approximately 5-fold higher for fasudil and 50-fold higher for hydroxyfasudil (IC50 value of fasudil and hydroxyfasudil; 5.3 and 37 μmol/L, respectively). The other kinases, including protein kinase C (PKC)α, have IC50 values that were >100 μmol/L for fasudil and hydroxyfasudil (data not shown). These findings suggest that the concentrations of fasudil and hydroxyfasudil used in this study were relatively selective for ROCK inhibition.
Effect of Hydroxyfasudil on Endothelial Nitric Oxide Synthase mRNA and Protein Level
Treatment with hydroxyfasudil increased eNOS mRNA to 160±10%, 156±10%, and 156±20% in HAEC, HUVEC, and HSVEC, respectively (n=3, P<0.05) (Figure 1a). In a concentration-dependent manner, hydroxyfasudil increased eNOS mRNA levels, with an EC50 value of 0.8±0.3 μmol/L (Figure 1b). Thus, the EC50 value of hydroxyfasudil for eNOS mRNA is comparable with the IC50 value of hydroxyfasudil for ROCK inhibition. Inhibition of other protein kinases such as PKC, PKA, and MLCK did not affect eNOS mRNA levels (data not shown), whereas overexpression of a dominant-negative mutant of ROCK (DN-Rho-K) increased eNOS mRNA levels compared with that of control LacZ (Figure 1c). These results indicate that inhibition of ROCK by hydroxyfasudil leads to an increase in eNOS mRNA expression.
Figure 1.
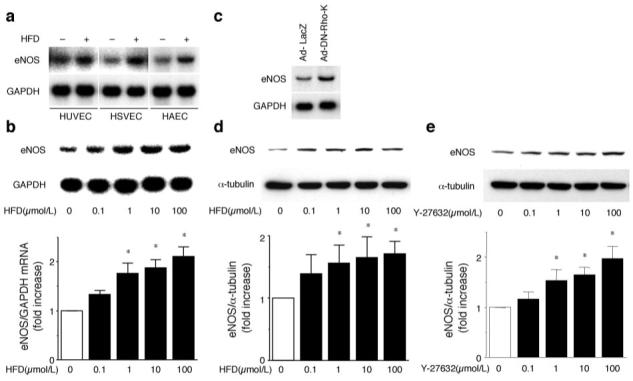
Upregulation of eNOS mRNA and protein by hydroxyfasudil (HFD). (a) HUVEC, HSVEC, and HAEC were treated with 10 μmol/L HFD for 18 hours. eNOS mRNA expression was determined by northern blotting. Blot is a representative of 3 experiments. (b) HAEC was treated with the indicated concentrations of HFD for 18 hours. eNOS mRNA levels were normalized by GAPDH mRNA expression (n=5). *P<0.05 vs basal condition. (c) HAEC was infected with Ad-LacZ or Ad-DN-Rho-K at 100 MOI. After 48 hours, eNOS mRNA expression was determined by northern blotting. (d and e) HAEC was treated with the indicated concentrations of HFD (d) or Y-27632 (e) for 96 hours. eNOS protein levels were determined by immunoblotting and normalized by α-tubulin expression (n=4). *P<0.05 vs basal condition.
Treatment with hydroxyfasudil increased eNOS protein expression in a concentration-dependent manner (Figure 1d). Similarly, another ROCK inhibitor, Y-27632, as well as overexpression of DN-Rho-K increased eNOS protein levels (Figure 1e; results not shown).
Effect of Hydroxyfasudil on Endothelial Nitric Oxide Synthase Activity and Nitric Oxide Production
In a concentration-dependent manner, treatment with hydroxyfasudil increased eNOS activity and stimulated NO production (Figure 2a). These results indicate that the increase in eNOS protein levels correlated with increases in eNOS activity and NO production.
Figure 2.
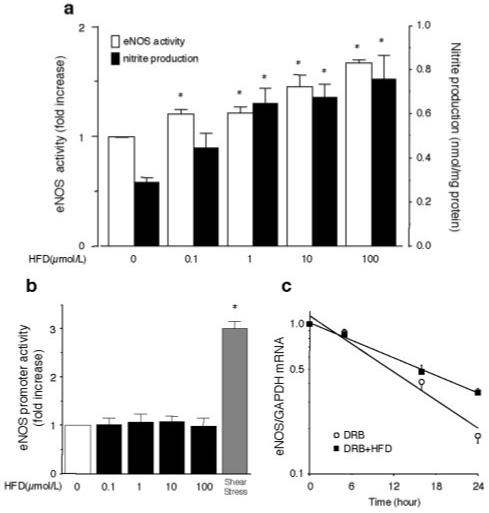
Effects of hydroxyfasudil (HFD) on eNOS activity, NO production, eNOS promoter activity, and eNOS mRNA stability. (a) HAEC was treated with the indicated concentrations of HFD for 96 hours. eNOS activity and nitrite release in culture media were measured (n=4). *P<0.05 vs basal condition. (b) Effects of hydroxyfasudil on eNOS promoter activity. BAEC were cotransfected with [-1.8 kb] eNOS promoter construct and pRL-CMV vector. Cells were harvested 48 hours after transfection and treated with the indicated conditions of HFD or exposed to laminar flow (12 dyne/cm2) (n=4). eNOS promoter activity was standardized to Renilla luciferase activity and expressed as fold induction. *P<0.05 vs basal condition. (c) Effects of HFD on eNOS mRNA stability. HAEC was treated with or without 10 μmol/L HFD in the presence of 50 μmol/L DRB (n=4).
Effect of Hydroxyfasudil on Endothelial Nitric Oxide Synthase mRNA Stability
Exposure of the cells to shear stress (12 dyne/cm2) significantly increased eNOS promoter activity (ie, 3.0-fold induction; Figure 2b). However, treatment with hydroxyfasudil (0.1 to 100 μmol/L) did not affect eNOS promoter activity. Treatment with 10 μmol/L of hydroxyfasudil increased the half-life of eNOS mRNA from 13 to 16 hours (n=4, P<0.05) (Figure 2c). These results indicate that the increase in eNOS expression by hydroxyfasudil is most likely mediated at the posttranscriptional level involving eNOS mRNA stability.
Effect of Cerebral Ischemia on ROCK Activity and Endothelial Nitric Oxide Synthase Expression
To determine whether ROCK inhibition protects against ischemic stroke, mice were administered fasudil, which is metabolized to an active metabolite hydroxyfasudil in the liver before transient MCA occlusion. After MCA occlusion, ROCK activity in the ischemic region of the brain, as measured by the Thr696 phosphorylation of myosin-binding subunit (MYPT) of myosin light chain phosphatase,11 was increased by more than 2-fold (Figure 3a). Treatment with fasudil decreased ROCK activity in the brain by 55% compared with vehicle treatment (P<0.05). Interestingly, MCA occlusion was associated with a 41% decrease in eNOS protein expression in vehicle-treated mice (Figure 3b). eNOS expression level in fasudil-treated mice after MCA occlusion was same to that in control mice.
Figure 3.
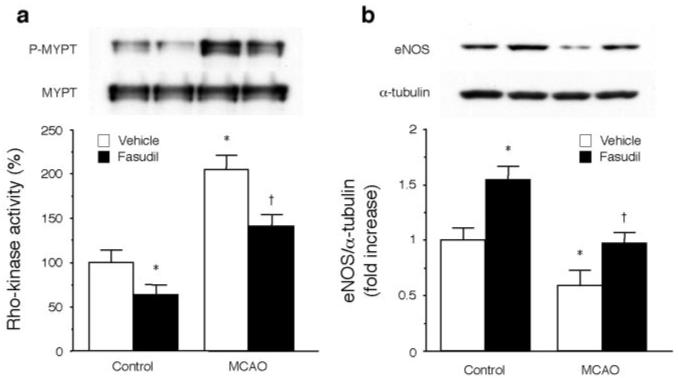
ROCK activity and eNOS expression in mouse brains. (a) ROCK activity and (b) eNOS expression in mouse brains were measured. *P<0.05 vs vehicle-treated control mice, †P<0.05 vs vehicle-treated mice subjected to MCA occlusion (MCAO) (n=10 each).
Effect of ROCK Inhibition in Ischemic Stroke
There were no significant differences in physiological parameters such as relative CBF, blood pressure, and blood gases between treatment groups (Table I available online only at http://www.strokeaha.org). The changes in relative CBF were comparable between the groups (Table, online only). In a dose-dependent manner, administration of fasudil decreased cerebral infarct volume as compared with vehicle treatment (56.6±4.9 mm3 for 10 mg/kg of fasudil versus 83.7±5.7 mm3 for vehicle; P<0.05; Figure 4a). This correlated with improvement in neurologic deficit score (1.2±0.3 versus 1.9±0.3, respectively; P<0.05). Similarly, treatment with Y-27632 also reduced stroke size and improved neurologic deficit score (Figure 4b). Y-27632 decreased ROCK activity to 67.4±1.9% (n=4, P<0.05) and increased eNOS expression to 163.3±20.3% (n=4, P<0.05) compared with vehicle treatment. These results suggest that ROCK inhibition is neuroprotective against ischemic stroke.
Figure 4.
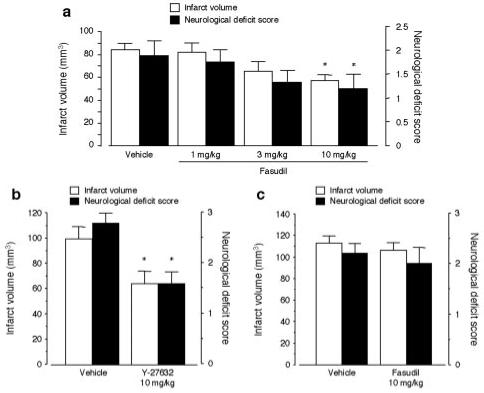
Effect of fasudil on cerebral infarct after MCA occlusion. (a) Wild-type mice were treated with vehicle (n=9) or 1, 3, or 10 mg/kg fasudil for 2 days (n=8, 9, 10, respectively). *P<0.05 vs vehicle. (b) Wild-type mice were treated with vehicle or 10 mg/kg Y-27632 for 2 days (n=5, each). *P<0.05 vs vehicle. (c) eNOS-/- mice were treated with vehicle or 10 mg/kg fasudil for 2 days (n=5, each).
Infarct volume and neurologic deficit score in eNOS-/- mice were increased compared with those of wild-type mice. Treatment with fasudil failed to reduce infarct volume and neurologic deficit score in eNOS-/- mice compared with vehicle-treated mice (Figure 4c).
Basal CBF was increased in mice treated with fasudil (10 mg/kg, 2 days) compared with that of vehicle-treated mice (536±91 versus 118±29 mL/100 g/min, n=4, P<0.05). Antipyrine autoradiography showed that there was low regional blood flow to the core infarct zone of the parietal lobe after MCA occlusion (Figure 5). In fasudil-treated mice, however, the entire core infarct zone was smaller, and within the core infarct zone and the penumbra, the blood flow was substantially higher compared with that of vehicle-treated mice. These results suggest that ROCK inhibition leads to increases in basal and regional CBF.
Figure 5.
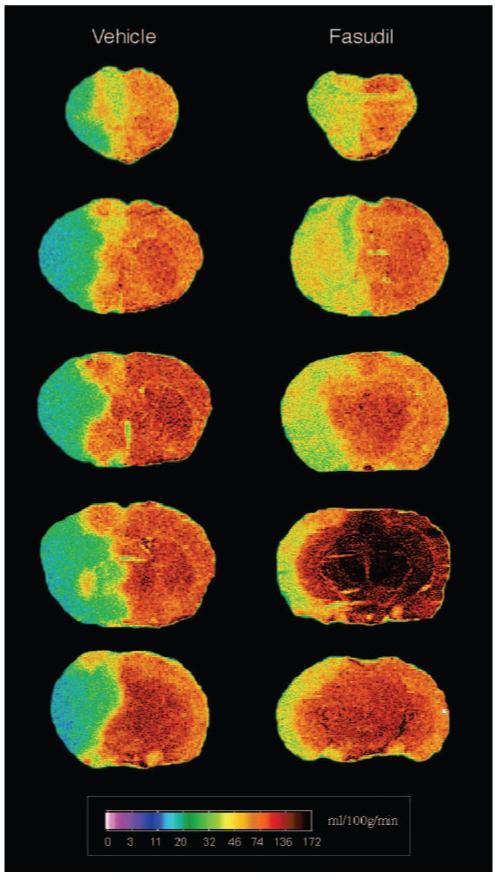
Regional cerebral blood flow. Cerebral blood flow 2 hours after MCA occlusion in mice treated with vehicle or 10 mg/kg fasudil for 2 days were measured by [14C]-iodoantipyrine autoradiography. Two independent experiments yielded similar results.
Regulation of Endothelial Nitric Oxide Synthase by ROCK Inhibition in the Vascular Wall
Despite inhibition of ROCK activity by fasudil, fasudil had no neuroprotective effects in eNOS-/- mice (Figure 6a). Compared with vehicle treatment, phosphorylation of MYPT was significantly inhibited in aortas of wild-type and eNOS-/- mice that were treated with fasudil. The inhibition of ROCK was correlated with an increase in aortic eNOS expression (Figure 6b) and NOS activity (Figure 6c). These results indicate that the neuroprotective effect of ROCK inhibition by fasudil is mediated through eNOS.
Figure 6.
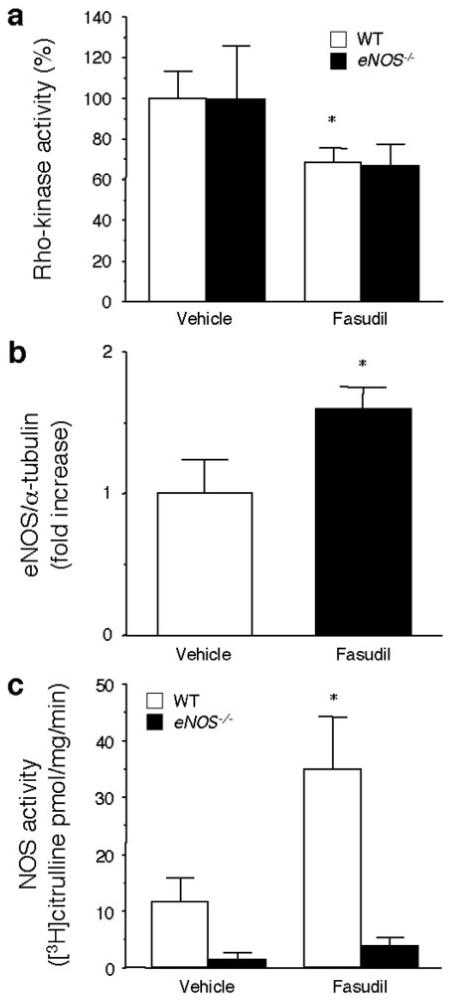
ROCK activity, eNOS expression, and NOS activity in mouse aortas. (A) ROCK activity, (B) eNOS expression, and (C) and NOS activity in mouse aortas were measured (n=10, WT; n=5, eNOS-/-). *P<0.05 vs vehicle.
Discussion
We have shown that fasudil and hydroxyfasudil, selective ROCK inhibitors, increase eNOS expression and NO production in vitro and in vivo. This leads to increased CBF, decreased cerebral infarction size, and improved neurologic deficit score after cerebral ischemia. Similarly, another ROCK inhibitor, Y-27632, was also neuroprotective against ischemic stroke. The neuroprotective effect of fasudil was mediated by eNOS because fasudil had no beneficial effect on cerebral infarct size in eNOS-/- mice despite inhibition of ROCK. These findings suggest that endothelial rather than vascular smooth muscle or leukocyte ROCK contributes to the neuroprotective effects of ROCK inhibition. Indeed, ROCK activity was increased and eNOS expression was decreased in brain tissues after cerebral ischemia. Both of these effects were reversed by treatment with fasudil.
These results are consistent with previous studies showing that inhibition of the activator of ROCK, RhoA, by statins leads to the upregulation of eNOS in vitro and in vivo.4,10 Similar to the effects of hydroxyfasudil, the increase in eNOS expression is the result of the posttranscriptional stability of eNOS mRNA.9 This is consistent with the ability of ROCK to mediate hypoxia- or thrombin-induced downregulation of eNOS expression.17,18 Because fasudil and hydroxyfasudil can also inhibit other protein kinases, albeit at higher concentrations, the reciprocal relationship between ROCK activity and eNOS expression was demonstrated by experiments using DN-Rho-K. Indeed, a recent study showed that ROCK activity is increased in the myocardium after ischemia-reperfusion injury.20 Thus, conditions, which lead to enhanced ROCK activity, could downregulate eNOS expression, making ROCK inhibition a novel therapeutic target for improving endothelial function.
Fasudil is clinically used in several countries for treatment of cerebral vasospasms after subarachnoid hemorrhage. Previous studies demonstrated that fasudil has a beneficial effect on treatment for acute cerebral ischemic stroke and vasospasm.21,22 Although the direct inhibition of ROCK in vascular smooth muscle by fasudil could contribute to the acute vasodilator response, it was unknown whether endothelial-derived NO could also mediate the neuroprotective effects of fasudil. This is especially important when we considered the more chronic effects of fasudil as shown in this study as compared with its acute vasodilator effects. Indeed, when fasudil was administered for more than 2 days, the neuroprotective effects were entirely dependent on eNOS because fasudil had no neuroprotective effects in eNOS-/- mice.
In ischemic stroke, the neurovascular unit is damaged primarily by reduction of blood flow and secondarily by ensuing inflammatory processes. Indeed, fasudil augmented NO-mediated blood flow. Improved collateral flow and microcirculation likely explain the findings. Improved collateral flow and microcirculation may also reflect other known NO-mediated cytoprotective effects such as inhibition of platelet aggregation and leukocyte adhesion. We have demonstrated that eNOS plays an essential role in neuroprotection mediated by statins.4 Thus, increasing CBF by upregulated eNOS activity is likely to play a predominant role in neuroprotection mediated by ROCK inhibition. It is possible that NO-dependent antiinflammatory effect of fasudil could be involved in the neuroprotection by fasudil. Indeed, recent studies suggest that the accumulation of leukocytes in acute cerebral infarction is correlated with brain damage.23 Postischemic inflammation in microvasculature contributes to ischemic brain injury, which could be counteracted, in part, by endothelial-derived NO. NO, which is constitutively produced by eNOS, has vascular protective effects, including inhibition of adhesion of leukocytes and monocytes to endothelium. For example, transgenic mice overexpressing eNOS show decreased leukocyte accumulation after vascular injury.24 Increased eNOS activity by estrogen and corticosteroid leads to decreased leukocyte adherence after ischemia/reperfusion injury.25,26 The augmentation or preservation of eNOS activity by fasudil treatment, therefore, could lead to beneficial antiinflammatory effects in the vascular wall. The efficacy of fasudil in ischemic stroke may be related not only to the improvement of microvascular hemodynamics, but also to inhibition of leukocyte-mediated damage.
In summary, our findings indicate acute cerebral ischemia is associated with enhanced ROCK activity and decreased eNOS expression. Inhibition of ROCK by fasudil/hydroxyfasudil restores eNOS activity and protects against cerebral ischemia. The neuroprotective effects of ROCK inhibition are absent in eNOS-/- mice, indicating the obligatory role of endothelial-derived NO in mediating these beneficial effects. There are a couple of limitations in the present study. In our experimental condition, the animals need to be treated for 2 days before ischemia. Further study regarding the therapeutic window is needed. In addition, the animals lack cerebrovascular risk factors such as hypertension and diabetes. It remains to be determined whether fasudil shows neuroprotective effects against ischemic stroke in mouse models with hypertension or diabetes. Although the mechanism by which cerebral ischemia increases ROCK activity remains to be elucidated, inhibition of ROCK appears to be a promising target for improving endothelial function and decreasing the severity of ischemic strokes.
Acknowledgments
The authors thank Kozo Kaibuchi (Nagoya University) for providing an adenovirus vector expressing a dominant-negative mutant of ROCK. This work was supported by grants from the National Institutes of Health (NS10828 and HL52233) and the American Heart Association (Bugher Foundation Award). Y. Rikitake is a recipient of an AHA Postdoctoral Fellowship-Northeast Affiliate and the Japan Heart Foundation/Bayer-Yakuhin Research Grant Abroad. H.-H. Kim is a recipient of the Post-doctoral Fellowship Program of Korea Science & Engineering Foundation (KOSEF).
References
- 1.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 2.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 3.Morikawa E, Moskowitz MA, Huang Z, Yoshida T, Irikura K, Dalkara T. L-arginine infusion promotes nitric oxide-dependent vasodilation, increases regional cerebral blood flow, and reduces infarction volume in the rat. Stroke. 1994;25:429–435. doi: 10.1161/01.str.25.2.429. [DOI] [PubMed] [Google Scholar]
- 4.Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, Liao JK. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1998;95:8880–8885. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4s) Lancet. 1994;344:1383–1389. [PubMed] [Google Scholar]
- 6.Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ, West of Scotland Coronary Prevention Study Group Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–1307. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- 7.Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, Brown L, Warnica JW, Arnold JM, Wun CC, Davis BR, Braunwald E, Cholesterol and recurrent events trial investigators The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med. 1996;335:1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- 8.Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem. 1997;272:31725–31729. doi: 10.1074/jbc.272.50.31725. [DOI] [PubMed] [Google Scholar]
- 9.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 10.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG-CoA reductase. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 11.Amano M, Fukata Y, Kaibuchi K. Regulation and functions of Rho-associated kinase. Exp Cell Res. 2000;261:44–51. doi: 10.1006/excr.2000.5046. [DOI] [PubMed] [Google Scholar]
- 12.Sato M, Tani E, Fujikawa H, Kaibuchi K. Involvement of ROCK-mediated phosphorylation of myosin light chain in enhancement of cerebral vasospasm. Circ Res. 2000;87:195–200. doi: 10.1161/01.res.87.3.195. [DOI] [PubMed] [Google Scholar]
- 13.Masumoto A, Mohri M, Shimokawa H, Urakami L, Usui M, Takeshita A. Suppression of coronary artery spasm by the ROCK inhibitor fasudil in patients with vasospastic angina. Circulation. 2002;105:1545–1547. doi: 10.1161/hc1002.105938. [DOI] [PubMed] [Google Scholar]
- 14.Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A. Possible involvement of ROCK in the pathogenesis of hypertension in humans. Hypertension. 2001;38:1307–1310. doi: 10.1161/hy1201.096541. [DOI] [PubMed] [Google Scholar]
- 15.Kataoka C, Egashira K, Inoue S, Takemoto M, Ni W, Koyanagi M, Kitamoto S, Usui M, Kaibuchi K, Shimokawa H, Takeshita A. Important role of ROCK in the pathogenesis of cardiovascular inflammation and remodeling induced by long-term blockade of nitric oxide synthesis in rats. Hypertension. 2002;39:245–250. doi: 10.1161/hy0202.103271. [DOI] [PubMed] [Google Scholar]
- 16.Miyata K, Shimokawa H, Kandabashi T, Higo T, Morishige K, Eto Y, Egashira K, Kaibuchi K, Takeshita A. ROCK is involved in macrophage-mediated formation of coronary vascular lesions in pigs in vivo. Arterioscler Thromb Vasc Biol. 2000;20:2351–2358. doi: 10.1161/01.atv.20.11.2351. [DOI] [PubMed] [Google Scholar]
- 17.Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. ROCK mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- 18.Eto M, Barandier C, Rathgeb L, Kozai T, Joch H, Yang Z, Lüscher TF. Thrombin suppresses endothelial nitric oxide synthase and upregulates endothelin-converting enzyme-1 expression by distinct pathways: role of rho/rock and mitogen-activated protein kinase. Circ Res. 2001;89:583–590. doi: 10.1161/hh1901.097084. [DOI] [PubMed] [Google Scholar]
- 19.Fujii M, Hara H, Meng W, Vonsattel JP, Huang Z, Moskowitz MA. Strain-related differences in susceptibility to transient forebrain ischemia in sv-129 and c57black/6 mice. Stroke. 1997;28:1805–1810. doi: 10.1161/01.str.28.9.1805. [DOI] [PubMed] [Google Scholar]
- 20.Bao W, Hu E, Tao L, Boyce R, Mirabile R, Thudium DT, Ma XL, Willette RN, Yue TL. Inhibition of ROCK protects the heart against ischemia/reperfusion injury. Cardiovasc Res. 2004;61:548–558. doi: 10.1016/j.cardiores.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 21.Satoh S, Ikegaki I, Suzuki Y, Asano T, Shibuya M, Hidaka H. Neuroprotective properties of a protein kinase inhibitor against ischaemia-induced neuronal damage in rats and gerbils. Br J Pharmacol. 1996;118:1592–1596. doi: 10.1111/j.1476-5381.1996.tb15579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satoh S, Suzuki Y, Harada T, Ikegaki I, Asano T, Shibuya M, Sugita K, Hidaka H. Possible prophylactic potential of HA1077, a Ca2+ channel antagonist and vasodilator, on chronic cerebral vasospasm. Eur J Pharmacol. 1992;220:243–248. doi: 10.1016/0014-2999(92)90754-r. [DOI] [PubMed] [Google Scholar]
- 23.Akopov SE, Simonian NA, Grigorian GS. Dynamics of polymorphonuclear leukocyte accumulation in acute cerebral infarction and their correlation with brain tissue damage. Stroke. 1996;27:1739–1743. doi: 10.1161/01.str.27.10.1739. [DOI] [PubMed] [Google Scholar]
- 24.Kawashima S, Yamashita T, Ozaki M, Ohashi Y, Azumi H, Inoue N, Hirata K, Hayashi Y, Itoh H, Yokoyama M. Endothelial NO synthase overexpression inhibits lesion formation in mouse model of vascular remodeling. Arterioscler Thromb Vasc Biol. 2001;21:201–207. doi: 10.1161/01.atv.21.2.201. [DOI] [PubMed] [Google Scholar]
- 25.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, Rebsamen MC, Hsieh CM, Chui DS, Thomas KL, Prorock AJ, Laubach VE, Moskowitz MA, French BA, Ley K, Liao JK. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8:473–479. doi: 10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]