

Abstract
MHC class II molecules are expressed on the surface of antigen presenting cells and are loaded with peptides processed from the phagosomal compartment of these cells. Such complexes interact with the CD4 positive T lymphocyte receptor for antigen and a strong interaction is followed by T cell activation and proliferation. As class II expression is critical for antigen specific immunity its expression mostly restricted to a few cell types but can be induced on others in response to interferon γ. This expansion of antigen presenting ability plays a role in increasing the duration and intensity of the immune response. Nitric oxide and antioxidants attenuate this class II induction through negative effects on the induction of class II transactivator protein expression and on the binding of transcription factor NF-Y to the class II promoter.
THE BIOLOGICAL IMPORTANCE OF MHC CLASS II
In vertebrates, the adaptive immune system has evolved to tailor the anti-microbial response to the challenges posed by particular micro-organisms. This system is highly effective, and once an infection has been successfully dealt with, re-exposure (even many years later) is met with a rapid re-activation of the response, leading to eradication of the microbe with no symptoms of disease. Adaptive immunity is provided by T and B lymphocyte recognition of non-self peptides (antigens) and the T lymphocytes are the key effector cells. CD8 positive T cells are cytotoxic to infected host cells, but CD4 T cells (T helper, Th cells) have a wider role, secreting cytokines that are required for the microbicidal activity of macrophages or eosinophils, and the maturation and proliferation of immunoglobulin-producing B cells. T cells cannot recognize antigenic peptides unless these have been cleaved from the organism by phagocytic antigen-presenting cells (APC) and loaded onto major histocompatibility complex (MHC) molecules on the APC cell surface [1]. MHC class I molecules present proteins found in the cytosol of APC to CD8 T cells. Cytosolic antigens are usually viral (viruses gain entry to the cytosol either by receptor-mediated endocytosis followed by lysosomal escape, or by direct fusion of their lipid bilayer coat to the cell surface), and lead to a cytotoxic T cell response to eliminate infected cells. Because all cell types are potential targets for virus infection, class I is ubiquitously expressed. In contrast, class II molecules present antigens derived from endocytic vesicles in the APC, where they are found after the phagocytosis of the micro-organism. MHC class II molecules are heterodimers comprising glycoprotein α (32 kDa) and β (26 kDa) chains [2]. Each chain has two extracellular domains, the most exterior of which interact with eachother to form a peptide binding groove. Recognition of class II bound peptide antigens is restricted to CD4 T cells. These CD4 cells are then activated to secrete cytokines that co-ordinate either a cell mediated or humoral immune response as appropriate for efficient clearance of the particular microbe in question. Thus, class II is crucial for immune function, and patients with a rare genetic deficiency in class II expression (Bare Lymphocyte Syndrome, BLS) exhibit recurrent and severe bacterial, fungal and viral infections [3]. The class II genes are highly polymorphic, so most people are heterozygotic at each allele [4]. In addition, many individuals have two copies of the β chain gene at the DR locus (see below), so an APC can express as many as sixteen different class II heterodimers (although not all α and β chain combinations are stable). An individual’s combination of MHC encoding genes is known as a haplotype. This broad class II repertoire will allow for the efficient loading and presentation of antigenic peptides of widely differing structures. The MHC haplotype is instrumental in shaping the individual’s T cell repertoire: the T cell receptor for MHC/antigenic peptide (the TCR) is highly variable, due to somatic mutation, and developing T cells bearing a TCR that cannot interact with the MHC available in that individual are deleted in the thymus (positive selection) [5]. APC load self and non-self peptides onto MHC and thus can present self peptides to T cells via class II. However, a strong interaction between TCR and self peptide also leads to T cell deletion (negative selection) [6]. Nevertheless, potentially auto-reactive T lymphocytes can be detected in the periphery, and so the predisposition to mount an immune response against self antigen is determined in large part by MHC. This is illustrated by the observation that the presence of certain MHC molecules do predispose individuals to the development of defined auto-immune diseases [7]. Class II plays an important role in organ allograft rejection (the origin of the name MHC comes from the observation that matching donor class I and II haplotype prevents rejection). Firstly, donor (graft) APC can migrate to recipient lymph nodes where T cells can be activated by donor mismatched class II (direct allorecognition). Secondly, mismatched donor class II proteins can be processed and presented to recipient T cells as antigen on class II by recipient (host) APC (indirect allorecognition).
MHC class II expression is tightly regulated. Indeed, it is absent on most cell types. Expression is constitutive on the so-called “professional” APC: dendritic cells (DC), monocytes, macrophages, B cells and thymic epithelial cells. Class II is regulated on these cells in a maturation-dependent manner, so that B lymphocytes lose expression when they mature to plasma cells, whereas DC expression increases with maturation. In addition to expression on professional APC, it can also be induced on most cell types, usually in the context of chronic inflammation, under the influence of the cytokine interferon γ (IFNγ). Class II expression confers the ability to present antigen and activate antigen-specific T cells. This is thought to be important in the establishment of chronicity in immune-driven inflammatory responses. For instance, endothelial cell class II expression was one of the earliest defining characteristics of “endothelial activation.” [8].
NO AND ROS GENERATION WITHIN APC
Generation of reactive oxygen species (ROS) occurs within APC as a by-product of mitochondrial electron transport, as it does in all cells [9]. In addition, the NADPH oxidase holoenzyme, first described in phagocytes, is capable on induction of producing high levels of the superoxide anion. This, and other ROS derived from it after its conversion by superoxide dismutase (SOD) to hydrogen peroxide, play a vital role in the microbicidal capability of these cells. There is also some evidence that oxidation contributes to the unfolding of antigenic proteins, aiding in their processing and presentation on class II [10]. In bone marrow-derived APC of macrophage/monocyte lineage and B lymphocytes, the NADPH oxidase is comprised of five components: gp91, p22, p40, p47 and p67. Semi-professional APC (i.e. cells which do not normally express class II but can be induced by IFNγ to do so) such as fibroblast-derived cells and endothelial cells (EC) also express inducible NADPH oxidase. In some cell types the gp91 homolog Nox1 is expressed as well as or instead of gp91 phox [11]. In addition to their role in the lysosomal compartment of the cell, ROS can react with cytosolic and nuclear proteins, phospholipids and nucleic acid, often with deleterious consequences to the cell when these reactions are uncontrolled. These damaging effects can be mitigated by cellular antioxidants, catalase and glutathione peroxidase among them. The healthy cell is thus able to tightly control redox tone, and an expanding variety of intracellular signaling mechanisms have been shown to be sensitive to it in terms of signal modulation. In this way, ROS are able to influence gene transcription programs and well-described examples include the transcription factor families NFκB and AP-1 [12].
Nitric oxide (NO), like ROS, is a diffusible small molecule capable of covalent modification of protein and DNA. It is synthesized from arginine by nitric oxide synthase (NOS), the inducible form of which (iNOS) generates large amounts of NO in activated phagocytes, which leads to microbial killing by direct nitrosative reactions, and this can also cause damage to host cellular machinery if uncontrolled [13]. NO reacts rapidly with ROS, which, depending on the context, can result in successful ROS scavenging, or the formation of even more damaging oxidative and nitrosative intermediates such as peroxynitrite. Endothelial NOS (eNOS) tonically elaborates low levels of NO that activate guanyl cyclase in vascular smooth muscle cells, leading to cGMP-mediated relaxation of blood vessel tone [14]. NO modulates the function of several transcription factors, including NFκB and AP-1 [15]. Depending on the context, NO can synergize with or antagonize the effects of ROS on these pathways. In this review we address the evidence for an effect of NO and ROS on the level of MHC class II expression by APC.
EFFECT OF NO AND ROS ON CLASS II EXPRESSION
Studies on elicited mouse macrophages and the murine macrophage cell line RAW264.7 have shown that the NO donor SNAP inhibits IFNγ-induced class II expression at the protein and message level [16]. Since NO exerts various anti-inflammatory effects including the attenuation of endothelial-leukocyte adhesion, it appears likely that NO could also regulate class II expression [17]. Indeed, in our studies on human saphenous vein EC, we found inhibition of class II upregulation after IFNγ by the NO donor GSNO, which inhibited rate of gene transcription as well as activity of a promoter-reporter construct comprising the proximal 300 bp of the class II promoter [18]. These observations reproduced the effect of another NO donor, SNP, on IFNγ-stimulated rat astrocyte cultures [19]. In contrast, an opposite effect was seen in T67 astrocytoma cells, where IFNγ-induced class II expression was inhibited by the NOS inhibitor L-NAME [20]. In a further study, on stimulated bovine mononuclear cells, there was no effect on class II expression by either an NO donor, GSNO, or a NOS inhibitor, NMLA [21]. This variability may be explained in terms of diversity in species, cell type and treatment conditions studied. Evidence for downregulation of class II by NO in vivo came in a study on the effect of the NO donor SIN-1 in experimental allergic encephalitis (EAE) [22]. Clinical outcome in rats with this immune mediated demyel-inating inflammatory model was improved by SIN-1 and this correlated with a four-fold downregulation of class II expression on peripheral blood mononuclear cells 14 days after disease induction. Studies from our laboratory showed that ROS have the opposite effect to NO on class II [18]. Treatment of HSVEC by H2O2 or IFNγ generated ROS as shown by DCF immunofluorescence, and IFNγ induction of class II was inhibited by the antioxidants PEG SOD, PEG catalase, n-acetyl cytseine (NAC) and pyrrolidine dithiocar-bamate (PDTC). These treatments reduced class II message transcription and class II promoter activity. Animal studies have recapitulated these findings. In a renal allograft model in rats, treatment with the antioxidant 21-aminosteroid attenuated histological markers of rejection as well as class II expression [23]. Finally, a study on human monocyte/macrophages showed that incubation with the antioxidant drug Celastrol, a plant-derived triterpene, inhibited the induced level of class II expression [24].
TRANSCRIPTIONAL CONTROL OF MHC II
The class II region is on chromosome 6 in humans and chromosome 17 in mice [25]. There are separate genes for the α and β chains of the class II heterodimer and these are adjacent except in HLA-DO. Humans have three classical class II genes, HLA-DP, DQ and DR, the latter two being analogous to the mouse genes H-2A and E [4]. In addition, situated between the HLA-DP region and the region for DQ and DR, are genes for the non-classical molecules HLA-DO and DM (H-2O and M in mice). DM facilitates peptide loading of MHC class II. The role of DO is less clear, possibly it acts to inhibit the action of DM in B lymphocytes. Between the DM region and the gene for DOβ lies the gene for the TAP peptide transporter and the genes for some proteosome components. These proteins have a role in antigen processing and peptide loading. The class II region is highly polymorphic and subject to considerable linkage dysequilibrium, with some haplotypes occurring at much greater frequency in the population than others.
Our current understanding of class II transcriptional control comes mainly from investigation into the Bare Lymphocyte Syndrome (BLS) [26]. APC from BLS patients lack the expression of class II and also of HLA-DM. Expression of the invariant chain (Ii), which stabilizes the class II heterodimer prior to peptide loading, is reduced. However, familial genetic studies in BLS revealed that the disease locus does not co-segregate with the class II region itself. Furthermore, when intact class II promoter region-reporter constructs were transfected into cells from BLS patients they produced no reporter activity. At this point it was hypothesized that a trans-acting factor may be necessary for class II expression, and that the gene encoding it was defective in BLS. With the establishment of cell lines from each BLS lineage, and also the identification of class II deficient lines, it became possible to undertake somatic cell fusions between these lines. Some fusions resulted in the restoration of class II expression, while others did not. This lead to the understanding that BLS was a heterogenous disorder, and on this basis the cell lines were classified into four complementation groups, A to D. DNA-protein interaction studies using the class II promoter region had identified a DNA-binding factor that binds to the X-box region of the promoter. This factor could not be detected in complementation groups B, C and D, and was termed regulatory factor X (RFX). When purified it was found to comprise 3 subunits, of size 75, 36 and 33 kDa. Peptide sequences from within the latter two sub-units lead to EST searches for the genes, and the genes for both were successfully identified and termed rfxap (for RFX associated protein) and rfxank (so called because the protein contains ankyrin repeats, also known as RFXB), respectively [27, 28]. Mutations in these genes were found to explain BLS in complementation groups D and B. The 75 kDa protein was identified by screening of a cDNA library for the ability to restore class II expression in cell lines from group C. It was termed RFX5 as it was the fifth protein discovered in a family of X-box binding proteins [29]. Using a similar complementation approach, the defect in group A was found to be located in the mhciita gene on chromosome 16 (16p13) [30]. The protein it encodes, CIITA (class II trans-activating factor) is the master-regulator of MHC class II but it does not bind directly to DNA. Its transfection into many cell types leads to inducible class II expression [31]. Unlike RFX, which is ubiquitous, its expression is closely correlated with class II expression on both a cell-specific and IFNγ-inducible basis, and the mhciita -/- mouse lacks that expression (except for a low level on lymph node and thymic dendritic cells) as well as expression of H2-M and reduced Ii expression [32].
MHC class II promoter elements contain three motifs (see Fig. 1): the W (or S) box, the X box (X1 and X2) and the Y box nearest to the start site. RFX binds both to the X1 and the W/S motifs, possibly as a dimer [33, 34]. RFX5 has a DNA binding domain and also interacts with NF-Y (see below) through its C-terminal. RFXANK is able to sustain protein-protein interaction that assembles RFX and mediates its interaction with CIITA through its ankyrin repeats. The C-terminal of RFXAP is the only essential domain for class II expression, although the exact requirement differs for HLA-DQ, DR and DP [35]. Three proteins have been described as X2 box binding factors, AP1, X2BP and CREB [36-38]. The Y box binds the NF-Y heterotrimer that consists of A, B and C sub-units [39]. The interaction of RFX with NF-Y may stabilize the DNA binding of one or both factors. CIITA interacts with all of these proteins, via its N-terminal region that contains an acidic domain, a PST domain (rich in proline, serine and threonine) and a GTP binding domain [31]. It is binds to the MHC II transcriptosome via its interaction with RFX and NF-Y. It recruits histone acetyl transferases CBP and pCAF, as well as TFIID components and it is required for gene transcription. Control of class II expression is in the main mediated through regulation of CIITA transcription. The mhciita promoter region is unusual in that there are three alternative functional start sites, giving rise to transcripts of different lengths for the first exon and thus different N-termini for the protein [40]. P1 is the most upstream of exon 2 and is constitutively active in immature DC and gives rise to a high surface expression of class II on these cells [41]. The second promoter, P3, is constitutively active in B cells, this function mapping to two proximal elements, ARE-1 and -2 [42]. The distal P3 region is responsible for IFNγ induced upregulation in monocytes and macrophages. P4 promoter drives class II expression in thymic epithelial cells and is required for the IFNγ-responsive expression in all APC of non-hematopoietic lineage, such as EC and fibroblasts. It contains three transcription factor binding elements, GAS (γ activated sequence), E-box (binds upstream stimulatory factor 1, USF-1) and IRF-1 (binds IFN regulatory factor 1) [43].
Fig. (1).
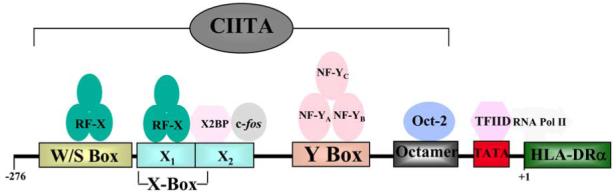
Transcription factor binding to MHC class II promoter. MHC class II promoter elements contain three motifs: the W/S box, the X box (X1 and X2) and the Y box nearest to the start site. The RFX heterotrimer binds both to the X1 and the W/S motifs, possibly as a dimer. X2 box binding factors include AP1 (c-jun and c-fos) and X2BP. The Y box binds the NF-Y heterotrimer that consists of A, B and C subunits. CIITA interacts with all of these proteins but does not bind DNA directly. It is necessary for the deacetylation of histone and the recruitment of TFIID components
MECHANISMS OF NO AND ROS MODULATION OF CLASS II TRANSCRIPTION
Due to the close correlation of class II and CIITA expression it is likely that most factors that regulate class II do so via control of CIITA transcription. This is in fact the case for LPS, IL-1 and IL-4 (upregulation) and IFNβ, IL-10 and TGFβ (downregulation) (3). However, this is not the only level of regulation. For instance, PGE2 increases cAMP level, which causes a PKA-induced phosphorylation of CIITA protein that reduces transcription of class II [44]. In murine macrophages, the NO-induced inhibition in class II transcription seems to be correlated with a reduction in steady state level of CIITA mRNA [16]. What are the possible mechanisms for the modulation of IFNγ induction of CIITA transcription by NO? IFNγ binds to a membrane bound heterodimeric receptor, IFNGR, which then oligomerizes and transphosphorylation takes place between the receptor and the Janus kinases Jak1 and 2. This leads to the recruitment and activation by phosphorylation of the transcription factor Stat1, which dimerizes and translocates to the nucleus where it binds to GAS elements in gene promoters including the CIITA P3 promoter [45]. This pathway is modulated by downregulatory proteins, such as PIAS (protein inactivator of Stat1), which represses trans-activation, and SOCS family proteins (suppressor of cytokine signaling), which bind to and inactivate Jak [46-49]. Jak proteins are also dephosphorylated and inactivated by SHP1 and 2 (SH2 containing phosphatases), and there is evidence that at least in rat smooth muscle cells, NO activates SHP2 [50, 51]. The IFNγ/Jak/Stat cascade interacts with other signaling networks, such as the Src family kinase Fyn, Rap1 (Ras GTPase activating protein 1) and the MAPK family members ERK1/2 [52]. Downstream, both Ras and ERK can activate Stat1 and these pathways might potentially be modulated by NO [53]. We have found that NO does inhibit Stat1 DNA binding in HSVEC (unpublished data) but we have also found that GSNO and L-NAME have no discernible effect on CIITA mRNA accumulation [18].
Is there an effect of ROS on the IFNγ induced transcription of CIITA that underlies the observed increase in class II expression? ROS have been shown to activate the Jak/Stat pathway: in Rat-1 fibroblasts, H2O2 stimulated Stat1 DNA binding and this was inhibited by NAC, PDTC and glutathione [54]. ROS have also been shown to modulate the accessory pathways affecting Stat1 activation, SHP1/2, Ras and ERK1/2, in a manner converse to that described for NO [55, 56]. In our hands, antioxidants inhibited IFNγ-induced Stat1 DNA binding in HSVEC (unpublished data). However, we could detect no effect of antioxidants downstream of this, on the accumulation of CIITA mRNA. In summary, while there is every reason to believe that in certain cell-specific contexts, both NO and antioxidants could reduce IFNγ mediated CIITA transcription, our findings lead us to conclude that in EC at least, class II transcription is being modified at another level. This might be the binding of AP-1 to the X2 box sequence of the class II promoter - AP-1 is a redox- and NO-sensitive transcription factor [57, 58]. Our studies have focused on the binding of the NF-Y heterotrimer to the Y box in the class II promoter. Binding of transcription factors to the Y box can be induced by H2O2 and inhibited by NO [18, 59]. This can be postulated to be an important part of the general redox-sensitive transcriptional response, as analyses of the human and mouse catalase gene promoter showed that transcription was critically dependent on NF-Y, and catalase is clearly an important part of the homeostatic response to oxidative stress [60, 61]. The way in which ROS and NO regulate NF-Y is unknown, and certainly there are no cysteine residues in NF-Y proteins for ROS or NO to react with directly. We have found that NO inhibits the nuclear localization of NF-Y, but the mechanism for this remains unknown.
SUMMARY
MHC class II molecules present self and non-self peptides to T cells and thereby shape the T cell repertoire. Oxidative stress is often an accompaniment of cellular activation, and this is certainly the case for antigen presenting cells. From the limited amount of data available to date, ROS appear to enhance class II expression on activated APC by increasing the DNA binding of the transcription factor NF-Y. A resultant increase in class II expression may help amplify the T cell response to infection and bring about successful clearance of the microbe. On the other hand, an oxidative milieu might lower the threshold for APC to activate auto-reactive T cells and trigger an auto-immune response. Oxidative stress might also facilitate class II expression on cells that do not normally function as APC, increasing T cell-APC interaction and perpetuating immune-mediated inflammatory responses to the detriment of host tissue. NO seems to reduce class II expression, either by downregulating IFNγ-induced expression of class II transactivator (CIITA), or by inhibiting DNA binding of NF-Y at the class II promoter Y box. This effect can be viewed as counteracting that of ROS, and thereby limiting the propensity to develop chronic or self-directed immune responses.
ACKNOWLEDGEMENTS
This work was supported by grants from the National Institutes of Health (HL52233 and HL48743) and the Wellcome Trust UK.
REFERENCES
References 62-64 are related articles recently published in Current Pharmaceutical Design.
- [1].Benacerraf B. Role of MHC gene products in immune regulation. Science. 1981;212(4500):1229–38. doi: 10.1126/science.6165083. [DOI] [PubMed] [Google Scholar]
- [2].Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu Rev Immunol. 1994;12:259–93. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- [3].Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu Rev Immunol. 2001;19:331–73. doi: 10.1146/annurev.immunol.19.1.331. [DOI] [PubMed] [Google Scholar]
- [4].Davies DA, Manstone AJ, Viza DC, Colombani J, Dausset J. Human transplantation antigens: the HL-A (Hu-1) system and its homology with the mouse H-2 system. Transplantation. 1968;6(4):571–86. [PubMed] [Google Scholar]
- [5].Kisielow P, Teh HS, Bluthmann H, von Boehmer H. Positive selection of antigen-specific T cells in thymus by restricting MHC molecules. Nature. 1988;335(6192):730–3. doi: 10.1038/335730a0. [DOI] [PubMed] [Google Scholar]
- [6].Zepp F, Staerz UD. Thymic selection process induced by hybrid antibodies. Nature. 1988;336(6198):473–5. doi: 10.1038/336473a0. [DOI] [PubMed] [Google Scholar]
- [7].McDevitt HO. The role of MHC class II molecules in susceptibility and resistance to autoimmunity. Curr Opin Immunol. 1998;10(6):677–81. doi: 10.1016/s0952-7915(98)80088-5. [DOI] [PubMed] [Google Scholar]
- [8].Pober JS, Collins T, Gimbrone MA, Jr, Cotran RS, Gitlin JD, Fiers W, et al. Lymphocytes recognize human vascular endothelial and dermal fibroblast Ia antigens induced by recombinant immune interferon. Nature. 1983;305(5936):726–9. doi: 10.1038/305726a0. [DOI] [PubMed] [Google Scholar]
- [9].Soberman RJ. The expanding network of redox signaling: new observations, complexities, and perspectives. J Clin Invest. 2003;111(5):571–4. doi: 10.1172/JCI18099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Carrasco-Marin E, Paz-Miguel JE, Lopez-Mato P, Alvarez-Dominguez C, Leyva-Cobian F. Oxidation of defined antigens allows protein unfolding and increases both proteolytic processing and exposes peptide epitopes which are recognized by specific T cells. Immunology. 1998;95(3):314–21. doi: 10.1046/j.1365-2567.1998.00618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Van Heerebeek L, Meischl C, Stooker W, Meijer CJ, Niessen HW, Roos D. NADPH oxidase(s): new source(s) of reactive oxygen species in the vascular system? J Clin Pathol. 2002;55(8):561–8. doi: 10.1136/jcp.55.8.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166(12 Pt 2):S4–8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- [13].Nathan C. Inducible nitric oxide synthase: what difference does it make? J Clin Invest. 1997;100(10):2417–23. doi: 10.1172/JCI119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rapoport RM, Murad F. Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ Res. 1983;52(3):352–7. doi: 10.1161/01.res.52.3.352. [DOI] [PubMed] [Google Scholar]
- [15].Pfeilschifter J, Eberhardt W, Beck KF. Regulation of gene expression by nitric oxide. Pflugers Arch. 2001;442(4):479–86. doi: 10.1007/s004240100586. [DOI] [PubMed] [Google Scholar]
- [16].Kielar ML, Sicher SC, Penfield JG, Jeyarajah DR, Lu CY. Nitric oxide inhibits INFgamma-induced increases in CIITA mRNA abundance and activation of CIITA dependent genes--class II MHC, Ii and H-2M. Class II TransActivator. Inflammation. 2000;24(5):431–45. doi: 10.1023/a:1007012128392. [DOI] [PubMed] [Google Scholar]
- [17].De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Jr, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96(1):60–8. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Grimm M, Spiecker M, De Caterina R, Shin WS, Liao JK. Inhibition of major histocompatibility complex class II gene transcription by nitric oxide and antioxidants. J Biol Chem. 2002;277(29):26460–7. doi: 10.1074/jbc.M110538200. [DOI] [PubMed] [Google Scholar]
- [19].Heuschling P. Nitric oxide modulates gamma-interferon-induced MHC class II antigen expression on rat astrocytes. J Neuroimmunol. 1995;57(12):63–9. doi: 10.1016/0165-5728(94)00162-h. [DOI] [PubMed] [Google Scholar]
- [20].Colasanti M, Mollace V, Cundari E, Massoud R, Nistico G, Lauro GM. The generation of nitric oxide participates in gamma IFN-induced MHC class II antigen expression by cultured astrocytoma cells. Int J Immunopharmacol. 1993;15(6):763–71. doi: 10.1016/0192-0561(93)90150-w. [DOI] [PubMed] [Google Scholar]
- [21].Schuberth HJ, Hendricks A, Leibold W. There is no regulatory role for induced nitric oxide in the regulation of the in vitro proliferative response of bovine mononuclear cells to mitogens, alloantigens or superantigens. Immunobiology. 1998;198(4):439–50. doi: 10.1016/S0171-2985(98)80051-X. [DOI] [PubMed] [Google Scholar]
- [22].Xu LY, Yang JS, Link H, Xiao BG. SIN-1, a nitric oxide donor, ameliorates experimental allergic encephalomyelitis in Lewis rats in the incipient phase: the importance of the time window. J Immunol. 2001;166(9):5810–6. doi: 10.4049/jimmunol.166.9.5810. [DOI] [PubMed] [Google Scholar]
- [23].Salahudeen A, Wang C, McDaniel O, Lagoo-Denadyalan S, Bigler S, Barber H. Antioxidant lazaroid U-74006F improves renal function and reduces the expression of cytokines, inducible nitric oxide synthase, and MHC antigens in a syngeneic renal transplant model. Partial support for the response-to-injury hypothesis. Transplantation. 1996;62(11):1628–33. doi: 10.1097/00007890-199612150-00017. [DOI] [PubMed] [Google Scholar]
- [24].Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25(7):1341–57. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- [25].Ting JP, Trowsdale J. Genetic control of MHC class II expression. Cell. 2002;109(Suppl):S21–33. doi: 10.1016/s0092-8674(02)00696-7. [DOI] [PubMed] [Google Scholar]
- [26].Waldburger JM, Masternak K, Muhlethaler-Mottet A, Villard J, Peretti M, Landmann S, et al. Lessons from the bare lymphocyte syndrome: molecular mechanisms regulating MHC class II expression. Immunol Rev. 2000;178:148–65. doi: 10.1034/j.1600-065x.2000.17813.x. [DOI] [PubMed] [Google Scholar]
- [27].Durand B, Sperisen P, Emery P, Barras E, Zufferey M, Mach B, et al. RFXAP, a novel subunit of the RFX DNA binding complex is mutated in MHC class II deficiency. EMBO J. 1997;16(5):1045–55. doi: 10.1093/emboj/16.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Masternak K, Barras E, Zufferey M, Conrad B, Corthals G, Aebersold R, et al. A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat Genet. 1998;20(3):273–7. doi: 10.1038/3081. [DOI] [PubMed] [Google Scholar]
- [29].Steimle V, Mach B. Complementation cloning of mammalian transcriptional regulators: the example of MHC class II gene regulators. Curr Opin Genet Dev. 1995;5(5):646–51. doi: 10.1016/0959-437x(95)80034-4. [DOI] [PubMed] [Google Scholar]
- [30].Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome) Cell. 1993;75(1):135–46. [PubMed] [Google Scholar]
- [31].Harton JA, Ting JP. Class II transactivator: mastering the art of major histocompatibility complex expression. Mol Cell Biol. 2000;20(17):6185–94. doi: 10.1128/mcb.20.17.6185-6194.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chang CH, Guerder S, Hong SC, van Ewijk W, Flavell RA. Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity. 1996;4(2):167–78. doi: 10.1016/s1074-7613(00)80681-0. [DOI] [PubMed] [Google Scholar]
- [33].Jabrane-Ferrat N, Nekrep N, Tosi G, Esserman L, Peterlin BM. MHC class II enhanceosome: how is the class II transactivator recruited to DNA-bound activators? Int Immunol. 2003;15(4):467–75. doi: 10.1093/intimm/dxg048. [DOI] [PubMed] [Google Scholar]
- [34].Jabrane-Ferrat N, Fontes JD, Boss JM, Peterlin BM. Complex architecture of major histocompatibility complex class II promoters: reiterated motifs and conserved protein-protein interactions. Mol Cell Biol. 1996;16(9):4683–90. doi: 10.1128/mcb.16.9.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Peretti M, Villard J, Barras E, Zufferey M, Reith W. Expression of the three human major histocompatibility complex class II isotypes exhibits a differential dependence on the transcription factor RFXAP. Mol Cell Biol. 2001;21(17):5699–709. doi: 10.1128/MCB.21.17.5699-5709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ivashkiv LB, Liou HC, Kara CJ, Lamph WW, Verma IM, Glimcher LH. mXBP/CRE-BP2 and c-Jun form a complex which binds to the cyclic AMP, but not to the 12-O-tetradecanoylphorbol-13-acetate, response element. Mol Cell Biol. 1990;10(4):1609–21. doi: 10.1128/mcb.10.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liou HC, Boothby MR, Finn PW, Davidon R, Nabavi N, Zeleznik-Le NJ, et al. A new member of the leucine zipper class of proteins that binds to the HLA DR alpha promoter. Science. 1990;247(4950):1581–4. doi: 10.1126/science.2321018. [DOI] [PubMed] [Google Scholar]
- [38].Andersson G, Peterlin BM. NF-X2 that binds to the DRA X2-box is activator protein 1. Expression cloning of c-Jun. J Immunol. 1990;145(10):3456–62. [PubMed] [Google Scholar]
- [39].Wright KL, Vilen BJ, Itoh-Lindstrom Y, Moore TL, Li G, Criscitiello M, et al. CCAAT box binding protein NF-Y facilitates in vivo recruitment of upstream DNA binding transcription factors. EMBO J. 1994;13(17):4042–53. doi: 10.1002/j.1460-2075.1994.tb06721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Muhlethaler-Mottet A, Otten LA, Steimle V, Mach B. Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO J. 1997;16(10):2851–60. doi: 10.1093/emboj/16.10.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Landmann S, Muhlethaler-Mottet A, Bernasconi L, Suter T, Waldburger JM, Masternak K, et al. Maturation of dendritic cells is accompanied by rapid transcriptional silencing of class II transactivator (CIITA) expression. J Exp Med. 2001;194(4):379–91. doi: 10.1084/jem.194.4.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ghosh N, Piskurich JF, Wright G, Hassani K, Ting JP, Wright KL. A novel element and a TEF-2-like element activate the major histocompatibility complex class II transactivator in B-lymphocytes. J Biol Chem. 1999;274(45):32342–50. doi: 10.1074/jbc.274.45.32342. [DOI] [PubMed] [Google Scholar]
- [43].Waldburger JM, Suter T, Fontana A, Acha-Orbea H, Reith W. Selective abrogation of major histocompatibility complex class II expression on extrahematopoietic cells in mice lacking promoter IV of the class II transactivator gene. J Exp Med. 2001;194(4):393–406. doi: 10.1084/jem.194.4.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Li G, Harton JA, Zhu X, Ting JP. Downregulation of CIITA function by protein kinase a (PKA)-mediated phosphorylation: mechanism of prostaglandin E, cyclic AMP, and PKA inhibition of class II major histocompatibility complex expression in monocytic lines. Mol Cell Biol. 2001;21(14):4626–35. doi: 10.1128/MCB.21.14.4626-4635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shuai K, Ziemiecki A, Wilks AF, Harpur AG, Sadowski HB, Gilman MZ, et al. Polypeptide signalling to the nucleus through tyrosine phosphorylation of Jak and Stat proteins. Nature. 1993;366(6455):580–3. doi: 10.1038/366580a0. [DOI] [PubMed] [Google Scholar]
- [46].Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, et al. Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci USA. 1998;95(18):10626–31. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, et al. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387(6636):924–9. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- [48].Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387(6636):921–4. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- [49].Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387(6636):917–21. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- [50].David M, Chen HE, Goelz S, Larner AC, Neel BG. Differential regulation of the alpha/beta interferon-stimulated Jak/Stat pathway by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol Cell Biol. 1995;15(12):7050–8. doi: 10.1128/mcb.15.12.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chang Y, Ceacareanu B, Dixit M, Sreejayan N, Hassid A. Nitric oxide-induced motility in aortic smooth muscle cells: role of protein tyrosine phosphatase SHP-2 and GTP-binding protein Rho. Circ Res. 2002;91(5):390–7. doi: 10.1161/01.res.0000033524.92083.64. [DOI] [PubMed] [Google Scholar]
- [52].Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23(2):96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- [53].Song JH, So EY, Lee CE. Increased serine phosphorylation and activation of STAT1 by oncogenic Ras transfection. Mol Cells. 2002;13(2):322–6. [PubMed] [Google Scholar]
- [54].Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275(6 Pt 1):C1640–52. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- [55].Cunnick JM, Dorsey JF, Mei L, Wu J. Reversible regulation of SHP-1 tyrosine phosphatase activity by oxidation. Biochem Mol Biol Int. 1998;45(5):887–94. doi: 10.1002/iub.7510450506. [DOI] [PubMed] [Google Scholar]
- [56].Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20(10):2175–83. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- [57].Abate C, Patel L, Rauscher FJ, 3rd, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science. 1990;249(4973):1157–61. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- [58].Tabuchi A, Sano K, Oh E, Tsuchiya T, Tsuda M. Modulation of AP-1 activity by nitric oxide (NO) in vitro: NO-mediated modulation of AP-1. FEBS Lett. 1994;351(1):123–7. doi: 10.1016/0014-5793(94)00839-6. [DOI] [PubMed] [Google Scholar]
- [59].Duh JL, Zhu H, Shertzer HG, Nebert DW, Puga A. The Y-box motif mediates redox-dependent transcriptional activation in mouse cells. J Biol Chem. 1995;270(51):30499–507. doi: 10.1074/jbc.270.51.30499. [DOI] [PubMed] [Google Scholar]
- [60].Nenoi M, Ichimura S, Mita K, Yukawa O, Cartwright IL. Regulation of the catalase gene promoter by Sp1, CCAAT-recognizing factors, and a WT1/Egr-related factor in hydrogen peroxide-resistant HP100 cells. Cancer Res. 2001;61(15):5885–94. [PubMed] [Google Scholar]
- [61].Luo D, Rando TA. The regulation of catalase gene expression in mouse muscle cells is dependent on the CCAAT-binding factor NF-Y. Biochem Biophys Res Commun. 2003;303(2):609–18. doi: 10.1016/s0006-291x(03)00397-8. [DOI] [PubMed] [Google Scholar]
- [62].Alfano M, Poli G. Cytokine and chemokine based control of HIV infection and replication. Curr Pharm Design. 2001;7(11):993–1013. doi: 10.2174/1381612013397591. [DOI] [PubMed] [Google Scholar]
- [63].Gomariz RP, Martinez C, Abad C, Leceta J, Delgado M. Immunology of VIP: a review and therapeutical perspectives. Curr Pharm Design. 2001;7(2):89–111. doi: 10.2174/1381612013398374. [DOI] [PubMed] [Google Scholar]
- [64].Kurowska EM. Nitric oxide therapies in vascular diseases. Curr Pharm Design. 2002;8(3):155–66. doi: 10.2174/1381612023396429. [DOI] [PubMed] [Google Scholar]