

As defined in this chapter by Drs. Fried and Arbiser, angiogenesis is “the process by which normal and pathologic tissue derives a blood supply”. Increasingly, angiogenesis has been shown to play critical roles in inflammatory as well as neoplastic processes. Understanding the molecular basis of angiogenesis, including the homing of bone marrow endothelial cell precursors to newly established sites of angiogenesis, has given us many potential therapeutic targets related to formation or maintenance of blood vessels. The recent approval of an anti-VEGF antibody, bevacizumab, for cancer therapy is one such example and many more appear to be on their way. Dr. Jack Arbiser, the senior author of this review, has been a leader in elucidating the molecular basis of angiogenesis (e.g., the role of reactive oxygen species) as well as pioneer in using new and established drugs (e.g., gentian violet) to block angiogenesis in a variety of settings, including the treatment of hemangiomas, that have relevance in dermatology.
Sam Hwang
Angiogenesis is the process through which normal and pathologic tissue derives a blood supply, which is necessary for the perfusion of these tissues with oxygen and removal of waste. Impairment of angiogenesis to normal tissues or abnormal tissues has immediate consequences, including central necrosis and apoptosis. When this occurs in normal tissue, this is called infarction, which can have life-ending consequences (myocardial infarction, stroke). When this occurs in pathologic tissues, ie tumors or inflammatory processes, central necrosis and apoptosis may occur, and this may be therapeutically beneficial. Excess angiogenesis underlies many disease processes in dermatology, from those that seem obvious (neoplastic disease), but also ulceration, in which excess angiogenesis prevents reepithelialization, to inflammatory processes (psoriasis, atopic dermatitis, lupus), and even chronic infections like leprosy. In this review, we will discuss common disorders which are angiogenic and discuss how novel and existing treatments may be utilized to treat these disorders.
Angiogenesis is the process in which tissue recruits blood vessels to form a neovasculature to vascularize the tissue. In most cases, the tissue experiences physiologic hypoxia, in which the tissue generates angiogenic growth factors such as vascular endothelial growth factors (VEGF A, B,C, D), basic fibroblast growth factor (bFGF, FGF-2), placenta growth factor (PLGF), stromal derived factor (SDF-1), corticotropin releasing hormone (CRH), angiopoietins 1 and 2, leptin, monocyte chemotactic factor (MCP-1) and others (1-8). These factors encourage both local angiogenesis, through mobilizing small vessel endothelial cells from pre-existing capillaries, as well as recruitment of bone marrow endothelial precursors. The ability of processes to recruit endothelial cells from these processes differs between processes and in different individuals, and may be different between males and females due to estrogenic stimuli. Tumors injected into mice are highly dependent on recruitment of bone marrow precursors, while tumors that are induced in mice through chemical carcinogenesis are dependent on local recruitment. Evidence of this is provided by the Id-1/Id-3 knockout mice, which are highly resistant to implanted tumors, but are not resistant to carcinogen induced tumors (9, 10).
Once endothelial cells are attracted to the source of blood vessels, they must form a functional lumen, allowing for the transport of red blood cells and metabolic wastes. Initial lumen formation is a state in which blood vessels are not invested with pericytes, and is thus unstable. This vasculature, called a neovasculature, is leaky and unstable, and is prone to constant remodeling. Interestingly, tumors which have a high degree of vascular remodeling have a poor prognosis (11-13). If the neovasculature is not actively remodeling, it becomes invested with pericytes and becomes stabilized and has a decreased ability to remodel. Pericytes are cells with smooth muscle characteristics that invest small blood vessels. It is harder to regress blood vessels that have been stabilized with pericytes, but these vessels may have less capability to vascularize a rapidly proliferating tumor (14, 15). There is thus a tradeoff between vessel growth and stability.
Targeting the neovasculature has been a focus of pharmacologic efforts to inhibit angiogenesis. Most neovasculatures are characterized by high levels of VEGF, but also angiopoeitin-2 (ang-2), which has the dual effect of destablizing vessels and attracting endothelial precursor cells. Tumors with high levels of ang-2 have a poor prognosis compared with tumors with low levels of ang-2. Both ang-2 and ang-1 bind to the same receptor, and stimulate similar signaling cascades, but ang-2 causes leaky vessels and vascular leak, while ang-1 decreases vascular leak and promotes vessel stabilization (16, 17). These findings have been thought to be couterintuitive, but more recent studies show that ang-1 and ang-2 are both survival factors for endothelial cells under different combinations of growth factors, and that ang-2 promotes endothelial cell survival under conditions of severe stress, such as the acidotic and hypoxic environment of a solid tumor. For the clinician, the important thing to remember is that redness is often a sign not just of angiogenesis, but vascular leak, and that these conditions are often accompanied by high level expression of ang-2.
Vascular leak as a target for therapy
Vascular leak is a common component of multiple pathologic processes, which may be systemic or local. Dramatic evidence of systemic vascular leak is seen in sepsis, which is often accompanied by peptides that enhance vascular leak, such as tumor necrosis factor alpha (TNF-α). The edema of a glioblastoma multiforme or toxoplasmosis causing the enhancing ring phenomenon on radiography is an example of vascular leak in an area that is compressed. In areas that are not compressed, such as the peritoneum or pleura, vascular leak manifests itself as ascites. In the skin, vascular leak if rapid, manifests itself as bullae, which may account for the blisters seen in pemphigus or pemphigoid. Lower level vascular leak manifests as microvesicles, such as seen in allergic contact dermatitis, dermatitis herpetiformis, psoriasis, and atopic dermatitis(18-22). Vascular leak is associated in most cases with a limited genetic repertoire, consisting of an inflammatory cytokine (Often TNF-α), NFkB activation, endothelial expression of surface ICAM-1, VCAM-1 and E-selectin, which promotes lymphocytic, neutrophil, and platelet adherence to endothelial cells, and high levels of VEGF and ang-2 (23, 24). These processes can be seen in a variety of infection processes as well, ranging from soft tissue cellulitis to meningococcal meningitis to cerebral malaria(25). Currently the major treatment of vascular leak is systemic glucocorticoids which decrease vascular leak, and have lytic effects on neutrophils and lymphocytes which perpetuate the inflammatory effects. Unfortunately, steroids are highly immunosuppressive and have other effects, such as promotion of insulin resistance. This may be due to increased production of reactive oxygen as a downstream effect of glucocorticoid action (26), and may explain the flare of psoriasis seen when systemic steroids are used to treat psoriasis and tapered off.
Targeting reactive oxygen to treat skin diseases
Oxygen is the major component of cellular respiration. When fully metabolized, oxygen is converted to water. However, oxygen has additional metabolic fates, such as conversion to superoxide by enzymes such as the NADPH oxidase complex (Nox)(27). This is a family of enzymes that converts molecular oxygen to superoxide, a highly reactive and potentially destructive molecule. Superoxide has many physiologic functions, such as the destruction of pathogens by neutrophils, as neutrophils can use superoxide directly or metabolites of superoxide, such as hypochloric acid (bleach) to kill organisms. Deficiency of the ability to produce superoxide is manifested in chronic granulomatous disorder (CGD), the most common form is X-linked CGD, in which nox2 is deficient (28, 29).
The nox enzyme family in humans consists of 5 nox genes and 2 dual oxygenase (Duox genes). Nox2, which is the primary gene of CGD, resides at the cell membrane in a complex with other genes, such as gp22phox, gp47phox, p67phox and the small GTPases rac1 and rac2. Deficiency in any of these genes can result in a CGD like phenotype, in which neutrophils can engulf bacteria, but not destroy them, leading to granulomas that are inflammatory. Rac is required for the activity of Nox 1,2, and 3, while gp22phox is required for the activity of nox4 (30).
Generation of superoxide has several important physiologic consequences for cells, whether or not the cells are specialized to kill pathogens. Superoxide oxidizes IkB, resulting in activation of nuclear factor kappa beta (NFkB) and downstream target genes, such as ICAM-1, VCAM-1, E-selectin, ang-2 and VEGF(31-33). Superoxide can also promote tumors through epigenetic phenomena including hypermethylation of tumor suppressor genes and oxidation of other tumor suppressor genes, such as PTEN(34). Hyperproliferative activity of reactive oxygen may be halted by activation of the tumor suppressor gene p53(35).
Reactive oxygen has been shown to control angiogenesis in many tumors, especially those deficient in the tumor suppressor gene p16ink4a(36). Prominent among them are melanomas, the third most common form of skin cancer and the cancer responsible for most death due to a skin condition. Most melanomas exhibit a phenomenon called the “reactive-oxygen driven tumor” in which angiogenesis and resistance to apoptosis are mediated by a reactive oxygen (superoxide)-NFkB mechanism (37, 38). Blockade of reactive oxygen may sensitize these tumors to chemotherapy and radiation, in part by reducing NFkB mediated angiogenesis. Recently, we have found that triphenylmethane dyes, such as gentian violet, block the activity of nox2 and nox4 and eliminate the growth of experimental hemangiomas in mice (39). Gentian violet has been shown to be clinically effective against oral hairy leukoplakia, a condition caused by epithelial infection with Epstein Barr virus (EBV) which causes reactive oxygen generation(40, 41). Currently, novel derivatives of gentian violet are being synthesized for systemic use against solid and hematopoietic tumors that use reactive oxygen for signaling.
Angiogenesis and Psoriasis
Psoriasis is a common papulosquamous disorder that is present in 1-2% of the US population, and can range in severity from a few asymptomatic plaques to total body involvement (3, 42). Psoriasis can overlap with other rheumatologic disorders, including arthritis and inflammatory bowel disease. Psoriasis is seen exclusively in humans, and results from a complex interaction between lymphocytes and keratinocytes. The earliest evidence of increased angiogenesis in psoriasis was elucidated by Braverman, who demonstrated increased vascularity in the psoriatic lesion and determined that the epidermis was the major source of the angiogenic activity (43-45) (Figure 1). A major component of that activity was found to be VEGF, and transgenic overexpression of VEGF-A in the epidermis gives rise to some features of the psoriatic phenotype(46, 47). The dependence of the psoriatic process on lymphocytes was elegantly demonstrated by Nickoloff, who transplanted uninvolved psoriatic skin onto SCID mice, infused stimulated lymphocytes into the mice, and demonstrated regeneration of the psoriatic lesion along with a vigorous neovasculature (48).
Figure 1. Angiogenesis in Psoriasis.
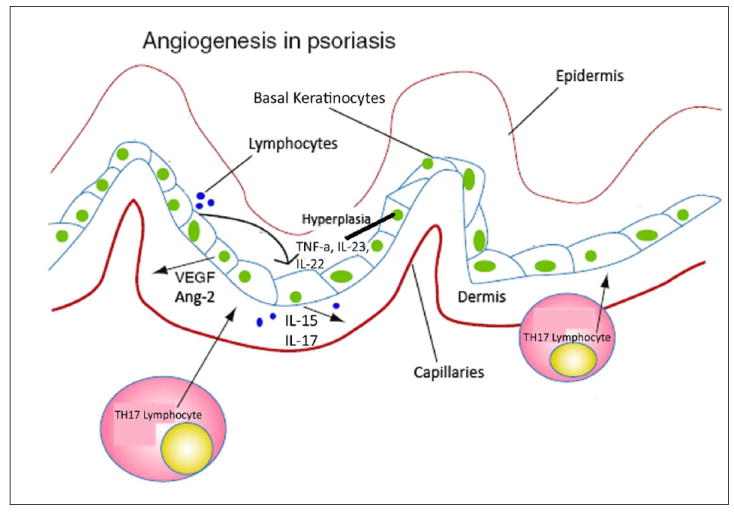
Lymphocytes act to release TNF-a, IL-23 and IL-22 which induced hyperplasia in basal keratinocytes. This causes the release of VEGF, Ang2, IL-15 and IL-17 into the dermis which causes inflammation.
Psoriasis was once thought to be due to a Th1 lymphocytic process, but more recently has been demonstrated to be mediated by a Th17 lymphocytic process, mediated by T cells that have some similarity to NK cells(49-55). This finding might explain why psoriasis often worsens in patients with advanced HIV infection, in which Th1 function is severely dysfunctional. Infiltrating T lymphocytes in psoriasis produce IL-17, which stimulates NFkB targets in keratinocytes, including ICAM-1 (53, 56, 57). Synergistic activity is seen with interferon gamma, and other cytokines potentiate the inflammatory and angiogenic response, including interleukins 15,22, and 23, as well as TNF-α. IL-23 shares the p40 subunit of its receptor with the IL-12 receptor, and recent clinical trial has shown efficacy of targeted therapy in patients with psoriasis. IL-22 has shown to cause epidermal hyperplasia, and possibly increased epidermal angiogenesis. These cytokines are controlled by TNF-α, and blockade of TNF-α has been shown to be efficacious in reducing levels of IL-22 and IL-23. Inhibitors of TNF alpha, including traps and blocking antibodies, are in widespread use in psoriasis, although complicated by expense and side effects (58-60). The side effects include reactivation of chronic infections, Ie tuberculosis), worsening of autoimmune diseases that may be controlled by TNF-α (lupus, multiple sclerosis), or worsening of congestive heart failure. In virtually all cases, psoriasis returns after cessation of TNF inhibitor therapy, necessitating long time therapy for efficacy.
Other drugs used in psoriasis may have antiangiogenic properties. Methotrexate was once the most popular systemic drug against psoriasis, and has been demonstrated to have antiangiogenic properties. Other compounds used against psoriasis, including cyclosporine, retinoids, and vitamin D analogs have also been shown to have antiangiogenic properties. Coal tar, one of the oldest remedies for psoriasis, has been shown to induce long term remissions in psoriasis. Recently, we fractionated coal tar and found an angiogenesis inhibitor, carbazole, which blocks rac-stat3 function and may account for the therapeutic activity of coal tar in psoriasis, as well as block production of interleukin-15 (61). Antimicrobial peptides which are overexpressed in psoriasis stimulate generation of reactive oxygen. Thus, it is likely that psoriasis represents a reactive oxygen driven process, and that inhibitors of this process may be beneficial in the treatment of angiogenesis associated with psoriasis (62, 63).
Angiogenesis and Vascular Lesions
Vascular lesions comprise a large and heterogenous population of lesions that are not fully understood. While the molecular events of only a few of these lesions are understood, the clinical course of these lesions fits into one of three possibilities: Rapid growth and regression (hemangioma), growth proportional to growth of patient without regression, and growth with local invasion and metastasis without regression (malignant endothelial tumor). Of these categories, hemangioma is the most common, found in 1% of all births and up to 10% of premature infants. Risk factors for hemangioma include female sex (4 fold increase), premature birth, and chorionic villus biopsy (64). The endothelial cells that comprise hemangiomas express placental markers, so it was proposed that hemangiomas arise from placental rests. Clonality has been assigned to hemangiomas, but their true neoplastic nature has not been elucidated. Currently, the most likely hypothesis is that hemangiomas represent population of embryonic bone marrow endothelium that homes to skin (or other organs). Factors which may play a role in this phenomenon are ang-2, which is highly expressed in hemangiomas of infancy, as well as a mouse model, bend3 cells, derived from polyoma middle T transformation of embryonic endothelial cells. Bend3 cells form tumors exclusively by recruiting host endothelial cells, and of interest, polyoma middle T only transforms embryonic or neonatal endothelial cells, and not from older mice, suggesting that like in humans (65). Intriguingly, verruga peruana, caused by infection of endothelial cells by Bartonella baciliformis, is associated with rac elevation and increased production of ang-2, and is histologically indistinguishable from hemangiomas (66, 67).
Hemangiomas exhibit a distinct “life cycle”, which has been divided into three phases (64). The proliferative state, is characterized by rapid growth of the lesion. The second state, is spontaneous involution, in which endothelial apoptosis is noted. The final stage is the involuted state, in which the hemangiomas are replaced by a connective tissue scar. Unlike other angiogenic lesions, hemangiomas do not use vascular endothelial growth factor (VEGF) as their major angiogenic factor, but use angiopoietin-2 (ang-2), which recruits endothelial precursor cells (68). Finally, we demonstrated that polyoma transformed endothelial cells produce high levels of ang-2 compared to VEGF, and the growth of these cells in vivo could be inhibited by a trap against ang-2. Therefore, strategies that block the production of ang-2 might be potentially useful in the treatment of hemangiomas(39). To this end, we demonstrated that triphenylmethane dyes, which block the production of ang-2 in endothelial cells, block the growth of polyoma transformed murine hemangiomas in vivo. In order to determine whether these findings could be translated into human disease, we treated ulcerated hemangiomas in infants with the triphenylmethane eosin (manuscript submitted), and showed significant efficacy.
Vascular malformations are the second most common class of endothelial lesions (69) (Figure 2). Unlike hemangiomas, vascular malformations do not regress with time, and often have large lumens with diminished smooth muscle investment. This is commonly seen in Sturge-Weber syndrome. Recently, we demonstrated that overexpression of Akt in endothelial cells could reproduce the large vessel phenotype. Interestingly, while there is evidence that hemangiomas also have high levels of Akt, they also express reactive oxygen markers such as ICAM-1 and Wilms tumor 1 (WT-1), while hemangiomas do not (70-72). Thus, large vessel vascular malformations use Akt, while hemangiomas use both Akt and reactive oxygen. Another class of small vessel vascular hemangiomas includes the acquired vascular disorder, rosacea. We hypothesize that while large vessel malformations use Akt alone, and hemangiomas use Akt/reactive oxygen, rosacea may use reactive oxygen alone. Rosacea can be treated with tetracycline class antibiotics, which inhibit matrix metalloproteinases, or by drugs such as metronidazole, which inhibits reactive oxygen. Finally, sulfacetamide is also a reducing agent which may inhibit reactive oxygen.
Figure 2. Pathways of different Vascular Diseases.
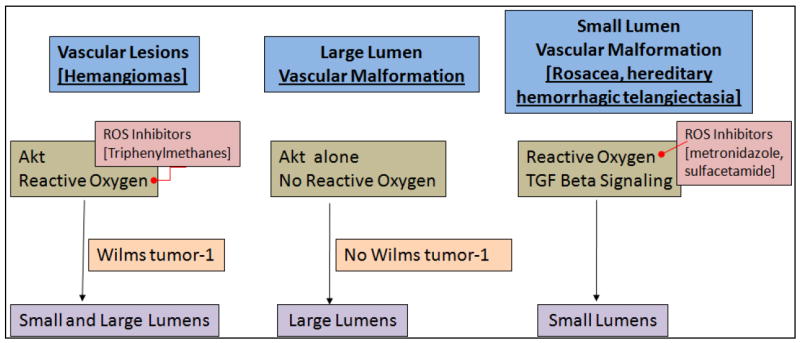
Vascular diseases such as Malformations and lesions have differing pathways which lead to the creation of lumens of varying size.
Angiogenesis and melanoma
Melanoma is the third most common skin cancer, but the leading cause of death due to a dermatologic condition. The factors that make melanoma such a lethal cancer is its propensity to metastasize and the resistance of metastatic tumors to chemotherapy and radiation. Both of these adverse prognostic factors are directly related to angiogenesis. Most melanomas arise as a result of mutations in B-raf or N-ras in the setting of loss of the tumor suppressor p16ink4a, and this combination of oncogenes and tumor suppressor genes may account for up to 90% of cutaneous melanomas (36). We have previously demonstrated an association between loss of p16ink4a and activation of p42/44 MAP kinase and reactive oxygen/akt activation. This association holds true in cutaneous melanoma. Over 90% of early melanomas are positive for MAP kinase activation and expression of VEGF(73, 74). Introduction of MAP kinase into immortalized melanocytes results in induction of VEGF. In deeper melanomas, VEGF is similarly highly expressed, but MAP kinase activation is replaced by activation of Akt/reactive oxygen. These discoveries lead to potential therapies for melanoma (38).
Sorafenib is a small molecule inhibitor of B-raf and VEGFR2 tyrosine kinases. The impetus for the development of sorafenib is the high rate of B-raf mutations in melanoma. However, sorafenib did not have appreciable activity in melanoma as monotherapy (75, 76). The likeliest reason for this result is that in deeper melanomas, additional signaling pathways can substitute for MAP kinase activation. Blockade of additional signaling pathways will be required to treat melanoma. Interferon alpha is currently the treatment of choice in high risk melanoma, but only yields a 10% survival advantage. Resistance to interferon may be due to induction of Akt by both endothelial and tumor cells. Avastin (bevacizumab), an anti-VEGF antibody, has been tried in combination with interferon alpha (77). Avastin alone had modest effects, and the addition of interferon did not increase efficacy. It is becoming clear that treatment of melanoma will require multiple drug cocktails, similar to our treatment of HIV infection.
Angiogenesis and nonmelanoma skin cancer
Basal and squamous cell carcinomas represent the most common skin cancers, affecting more than one million patients yearly in the US. While most of these tumors are curable through local excision and destructive modalities, a minority of these tumors exhibit aggressive and metastatic properties. These are most commonly seen in tumors which arise in transplant patients or in the setting of chronic inflammation, ie carcinomas arising in scars, ulcers, and recessive dystrophic epidermolysis bullosa (RDEB). We have demonstrated that a significant number of squamous cell carcinomas arising in RDEB exhibit loss of p16ink4a, in contrast to the mutations in p53 most commonly seen in cutaneous nonmelanoma skin cancers (5, 78). We have previously demonstrated that p16ink4a deficient tumors signal differently than mutant p53 tumors, with trends towards increased aggressiveness.
The major angiogenic factor in basal cell carcinoma is bFGF, while in squamous cell carcinoma is VEGF (79). The reason for this difference remains unclear, but may explain why BCC is usually less aggressive than SCC. Both tumors require an angiogenic switch between local inhibitors of angiogenesis (interferon alpha, interleukin-12) and local stimulators of angiogenesis (bFGF, VEGF) (80, 81). Angiogenesis inhibitors are already in use for the treatment of nonmelanoma skin cancers. These include imiquimod, which is FDA approved for nonmelanoma skin cancers, as well as warts, which are also angiogenic. Imiquimod is a TLR7 agonist which induces local production of interferon. Other topical antiangiogenic agents include topical vitamin D, while systemic agents that have antiangiogenic activity include retinoids and tyrosine kinase inhibitors that inhibit VEGF, EGFR, and her2/neu (geftinib, herceptin, erlotinib, sunitinib, etc). However, these agents are probably likely to be ineffective as monotherapy because activation of oncogenic ras genes leads to resistance to these targeted therapies(82). (Table 1)
Table 1.
| Angiogenesis Stimulators: | Description. |
|---|---|
| Vascular Endothelial Growth Factor (VEGF) | VEGF is a primary contributor to the growth of capillaries in a given network. In the presence of VEGF endothelial cells will undergo angiogenesis. VEGF causes a large tyrosine kinase signaling cascade in endothelial cells. This cascade causes other factors to be produced and these factors stimulate proliferation (survival) via bFGF, migration via MMP and differentiation into mature blood vessels.
The upregulation of VEGF is the main operator in the physiological response during exercise. Muscle contraction increases the blood flow to the affected areas. This increased flow causes an increase in the mRNA production of VEGF receptors 1 and 2. This increase in receptors increases the signaling cascades related to angiogenesis. |
| Basic Fibroblast Growth Factor (bFGF) | Basic fibroblast growth factor is a member of the fibroblast growth factor family. In normal tissue, basic fibroblast growth factor is present in basement membranes and in the subendothelial extracellular matrix of blood vessels. bFGF stays membrane-bound as long as there is no signal peptide. During both wound healing of normal tissues and tumor development, the action of heparan sulphate-degrading enzymes activates bFGF, thus mediating the formation of new blood vessels (angiogenesis). |
| Matrix Metalloproteinase (MMP) | MMPs help degrade the proteins that keep the vessel walls solid. This proteolysis allows the endothelial cells to escape into the interstitial matrix as seen in sprouting angiogenesis. These enzymes are highly regulated during the vessel formation process because this destruction of the extracellular matrix would destroy the integrity of the microvasculature. MMP2 and MMP9 are the two proteinases linked to angiogenesis. |
| COX2-PGE2 | COX-2 inhibitors are a class of nonsteroidal anti-inflammatory drugs (NSAIDs) that selectively block the COX-2 enzyme. This action impedes the production of the chemical messengers (prostaglandins) that cause the pain and swelling of arthritis inflammation. Being that COX-2 inhibitors selectively block the COX-2 enzyme and not the COX-1 enzyme, these drugs are uniquely different from traditional NSAIDs. |
| Platelet Derived Growth Factor (PDGF) | PDGF is a dimer that activates its signaling pathway by a ligand-induced receptor dimerization and autophosphorylation. PDGF has provided a market for protein receptor antagonists to treat disease. These antagonists include specific antibodies that target the molecule of interest. |
Kaposi’s sarcoma (KS) has been a prime focus for angiogenesis inhibition. KS is thought to originate from HHV8 infection of lymphatic endothelial cells. Drugs that have demonstrated efficacy against KS include thalidomide, rapamycin, liposomal doxorubicin, and docetaxol, all of which have antiangiogenic activities (83-88). Additive therapies that induce lytic replication of HHV8 in KS cells in combination with antiangiogenic agents, such as valproic acid and other HDAC inhibitors, may provide additional benefit in KS (89, 90).
Angiogenesis and infection
The host response to both acute and chronic infection requires angiogenesis. While it is important not to inhibit the host response to acute infection, chronic infection may require angiogenesis for maintenance. Most chronic infections result from intracellular colonization of bacteria, or adherence of bacteria to a foreign object, impairing bacterial clearance.
Bacterial infections associated with intracellular colonization include mycobacterial infections (leprosy, tuberculosis), syphilis (endothelial colonization), and bartonella (endothelial colonization). Each of these colonizations activates signaling abnormalities in the host cell to allow both persistence and maintenance. For example, bartonella infection induces rac activation and ang-2 production to mimic a hemangioma (66, 67). Increased angiogenesis is seen in lepromatous leprosy, in which an ineffective immune response, due in part to loss of interleukin-12 and other angiogenesis inhibitors, allows engorgement of cells into tumor-like masses(91). We have recently shown that lepromatous leprosy is highly angiogenic, and that administration of angiogenesis inhibitors may shorten the duration of therapy needed to treat mycobacterial infections, such as leprosy and tuberculosis.
Chronic viral infections often induce angiogenesis through oncogenes. Human papillomavirus (HPV) contains two oncogenes which can impact angiogenesis. E6 binds p53 and E7 binds Rb. The action of either or both of these genes is sufficient to increase angiogenesis. Epstein-Barr Virus, which causes Burkitt’s lymphoma, oral hairy leukoplakia, and leiomyosarcoma, expresses oncogenes such as LMP-1 which activate angiogenesis (40). HHV8 also expresses an oncogenic G protein that induces angiogenesis.
Chronic infections resist immune clearance by several mechanisms. These include VEGF, which impairs dendritic cell maturation, secretion of immunosuppressive cytokines, such as interleukin-10, and decreased expression of antigen presenting molecules. These may be controlled by similar pathways, such as phosphoinositol-3 kinase/Akt and reactive oxygen signaling. Blockade of these pathways may enhance clearance of chronic infection through angiogenesis inhibition and enhanced immune response.
Angiogenesis inhibition - an overview
Most conditions in dermatology are characterized by excess angiogenesis. These include inflammatory processes such as psoriasis, atopic dermatitis, rosacea, acne, venous ulcers, neoplastic conditions, such as melanoma, nonmelanoma skin cancer, and benign neoplasia, and chronic infections. As part of routine dermatologic practice, we use angiogenesis inhibition often without recognizing it. Drugs already in use are tetracycline antibiotics, which inhibit the activity of matrix metalloproteinases, retinoids which inhibit the synthesis of matrix metalloproteinases, interferon and interferon inducers (imiquimod), gentian violet, which inhibits NADPH oxidases, topical glucocorticoids, and TNF-α inhibitors/blockers. Drugs that are used in other specialties also impact on cutaneous angiogenesis. These include statins, tyrosine kinase inhibitors, Avastin, and PPAR agonists. Finally dietary supplements such as curcumin and tea polyphenols may systemically affect the angiogenic state of the patient. Knowledge of these basic mechanisms will enhance dermatologic therapy.
Acknowledgments
JLA was supported by the grant RO1 AR47901and P30 AR42687 Emory Skin Disease Research Core Center Grant from the National Institutes of Health, a Veterans Administration Hospital Merit Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Folkman J, Merler E, Abernathy C, Williams G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med. 1971;133:275–288. doi: 10.1084/jem.133.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M, De MM, Autiero M, Wyns S, Plaisance S, Moons L, van RN, Giacca M, Stassen JM, Dewerchin M, Collen D, Carmeliet P. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–475. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 3.Arbiser JL. Angiogenesis and the skin: a primer. J Am Acad Dermatol. 1996;34:486–497. doi: 10.1016/s0190-9622(96)90444-2. [DOI] [PubMed] [Google Scholar]
- 4.Arbiser JL, Karalis K, Viswanathan A, Koike C, nand-Apte B, Flynn E, Zetter B, Majzoub JA. Corticotropin-releasing hormone stimulates angiogenesis and epithelial tumor growth in the skin. J Invest Dermatol. 1999;113:838–842. doi: 10.1046/j.1523-1747.1999.00760.x. [DOI] [PubMed] [Google Scholar]
- 5.Arbiser JL, Fine JD, Murrell D, Paller A, Connors S, Keough K, Marsh E, Folkman J. Basic fibroblast growth factor: a missing link between collagen VII, increased collagenase, and squamous cell carcinoma in recessive dystrophic epidermolysis bullosa. Mol Med. 1998;4:191–195. [PMC free article] [PubMed] [Google Scholar]
- 6.Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, Hooper AT, Amano H, Avecilla ST, Heissig B, Hattori K, Zhang F, Hicklin DJ, Wu Y, Zhu Z, Dunn A, Salari H, Werb Z, Hackett NR, Crystal RG, Lyden D, Rafii S. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12:557–567. doi: 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol. 2007;28:299–307. doi: 10.1016/j.it.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li S, Takeuchi F, Wang JA, Fuller C, Pacheco-Rodriguez G, Moss J, Darling TN. MCP-1 overexpressed in tuberous sclerosis lesions acts as a paracrine factor for tumor development. J Exp Med. 2005;202:617–624. doi: 10.1084/jem.20042469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O’Reilly R, Bader BL, Hynes RO, Zhuang Y, Manova K, Benezra R. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–677. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- 10.Sikder H, Huso DL, Zhang H, Wang B, Ryu B, Hwang ST, Powell JD, Alani RM. Disruption of Id1 reveals major differences in angiogenesis between transplanted and autochthonous tumors. Cancer Cell. 2003;4:291–299. doi: 10.1016/s1535-6108(03)00245-9. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka S, Mori M, Sakamoto Y, Makuuchi M, Sugimachi K, Wands JR. Biologic significance of angiopoietin-2 expression in human hepatocellular carcinoma. J Clin Invest. 1999;103:341–345. doi: 10.1172/JCI4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalomenidis I, Kollintza A, Sigala I, Papapetropoulos A, Papiris S, Light RW, Roussos C. Angiopoietin-2 levels are elevated in exudative pleural effusions. Chest. 2006;129:1259–1266. doi: 10.1378/chest.129.5.1259. [DOI] [PubMed] [Google Scholar]
- 13.Hu B, Jarzynka MJ, Guo P, Imanishi Y, Schlaepfer DD, Cheng SY. Angiopoietin 2 induces glioma cell invasion by stimulating matrix metalloprotease 2 expression through the alphavbeta1 integrin and focal adhesion kinase signaling pathway. Cancer Res. 2006;66:775–783. doi: 10.1158/0008-5472.CAN-05-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger M, Bergers G, Arnold B, Hammerling GJ, Ganss R. Regulator of G-protein signaling-5 induction in pericytes coincides with active vessel remodeling during neovascularization. Blood. 2005;105:1094–1101. doi: 10.1182/blood-2004-06-2315. [DOI] [PubMed] [Google Scholar]
- 15.Hirschi KK, Rohovsky SA, D’Amore PA. PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141:805–814. doi: 10.1083/jcb.141.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daly C, Pasnikowski E, Burova E, Wong V, Aldrich TH, Griffiths J, Ioffe E, Daly TJ, Fandl JP, Papadopoulos N, McDonald DM, Thurston G, Yancopoulos GD, Rudge JS. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proc Natl Acad Sci USA. 2006;103:15491–15496. doi: 10.1073/pnas.0607538103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, Gale NW, Witzenrath M, Rosseau S, Suttorp N, Sobke A, Herrmann M, Preissner KT, Vajkoczy P, Augustin HG. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med. 2006;12:235–239. doi: 10.1038/nm1351. [DOI] [PubMed] [Google Scholar]
- 18.Voskas D, Jones N, Van SP, Sturk C, Chang W, Haninec A, Babichev YO, Tran J, Master Z, Chen S, Ward N, Cruz M, Jones J, Kerbel RS, Jothy S, Dagnino L, Arbiser J, Klement G, Dumont DJ. A cyclosporine-sensitive psoriasis-like disease produced in Tie2 transgenic mice. Am J Pathol. 2005;166:843–855. doi: 10.1016/S0002-9440(10)62305-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lind AJ, Wikstrom P, Granfors T, Egevad L, Stattin P, Bergh A. Angiopoietin 2 expression is related to histological grade, vascular density, metastases, and outcome in prostate cancer. Prostate. 2005;62:394–399. doi: 10.1002/pros.20163. [DOI] [PubMed] [Google Scholar]
- 20.Thurston G, Wang Q, Baffert F, Rudge J, Papadopoulos N, Jean-Guillaume D, Wiegand S, Yancopoulos GD, McDonald DM. Angiopoietin 1 causes vessel enlargement, without angiogenic sprouting, during a critical developmental period. Development. 2005;132:3317–3326. doi: 10.1242/dev.01888. [DOI] [PubMed] [Google Scholar]
- 21.Kim I, Kim JH, Moon SO, Kwak HJ, Kim NG, Koh GY. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Oncogene. 2000;19:4549–4552. doi: 10.1038/sj.onc.1203800. [DOI] [PubMed] [Google Scholar]
- 22.Hammes HP, Lin J, Wagner P, Feng Y, vom HF, Krzizok T, Renner O, Breier G, Brownlee M, Deutsch U. Angiopoietin-2 causes pericyte dropout in the normal retina: evidence for involvement in diabetic retinopathy. Diabetes. 2004;53:1104–1110. doi: 10.2337/diabetes.53.4.1104. [DOI] [PubMed] [Google Scholar]
- 23.Lee KH, Lawley TJ, Xu YL, Swerlick RA. VCAM-1-, ELAM-1-, and ICAM-1-independent adhesion of melanoma cells to cultured human dermal microvascular endothelial cells. J Invest Dermatol. 1992;98:79–85. doi: 10.1111/1523-1747.ep12495643. [DOI] [PubMed] [Google Scholar]
- 24.Mattila P, Majuri ML, Mattila PS, Renkonen R. TNF alpha-induced expression of endothelial adhesion molecules, ICAM-1 and VCAM-1, is linked to protein kinase C activation. Scand J Immunol. 1992;36:159–165. doi: 10.1111/j.1365-3083.1992.tb03087.x. [DOI] [PubMed] [Google Scholar]
- 25.Ockenhouse CF, Tegoshi T, Maeno Y, Benjamin C, Ho M, Kan KE, Thway Y, Win K, Aikawa M, Lobb RR. Human vascular endothelial cell adhesion receptors for Plasmodium falciparum-infected erythrocytes: roles for endothelial leukocyte adhesion molecule 1 and vascular cell adhesion molecule 1. J Exp Med. 1992;176:1183–1189. doi: 10.1084/jem.176.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 27.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 28.Babior BM, Curnutte JT. Chronic granulomatous disease--pieces of a cellular and molecular puzzle. Blood Rev. 1987;1:215–218. doi: 10.1016/0268-960x(87)90022-1. [DOI] [PubMed] [Google Scholar]
- 29.Dinauer MC, Orkin SH, Brown R, Jesaitis AJ, Parkos CA. The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome b complex. Nature. 1987;327:717–720. doi: 10.1038/327717a0. [DOI] [PubMed] [Google Scholar]
- 30.Bokoch GM, Zhao T. Regulation of the phagocyte NADPH oxidase by Rac GTPase. Antioxid Redox Signal. 2006;8:1533–1548. doi: 10.1089/ars.2006.8.1533. [DOI] [PubMed] [Google Scholar]
- 31.Lopes NH, Vasudevan SS, Gregg D, Selvakumar B, Pagano PJ, Kovacic H, Goldschmidt-Clermont PJ. Rac-dependent monocyte chemoattractant protein-1 production is induced by nutrient deprivation. Circ Res. 2002;91:798–805. doi: 10.1161/01.res.0000040421.54108.81. [DOI] [PubMed] [Google Scholar]
- 32.Chen XL, Zhang Q, Zhao R, Medford RM. Superoxide, H2O2, and iron are required for TNF-alpha-induced MCP-1 gene expression in endothelial cells: role of Rac1 and NADPH oxidase. Am J Physiol Heart Circ Physiol. 2004;286:H1001–H1007. doi: 10.1152/ajpheart.00716.2003. [DOI] [PubMed] [Google Scholar]
- 33.Yu L, Zhen L, Dinauer MC. Biosynthesis of the phagocyte NADPH oxidase cytochrome b558. Role of heme incorporation and heterodimer formation in maturation and stability of gp91phox and p22phox subunits. J Biol Chem. 1997;272:27288–27294. doi: 10.1074/jbc.272.43.27288. [DOI] [PubMed] [Google Scholar]
- 34.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101:16419–16424. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Govindarajan B, Klafter R, Miller MS, Mansur C, Mizesko M, Bai X, LaMontagne K, Jr, Arbiser JL. Reactive oxygen-induced carcinogenesis causes hypermethylation of p16(Ink4a) and activation of MAP kinase. Mol Med. 2002;8:1–8. [PMC free article] [PubMed] [Google Scholar]
- 36.Shields JM, Thomas NE, Cregger M, Berger AJ, Leslie M, Torrice C, Hao H, Penland S, Arbiser J, Scott G, Zhou T, Bar-Eli M, Bear JE, Der CJ, Kaufmann WK, Rimm DL, Sharpless NE. Lack of extracellular signal-regulated kinase mitogen-activated protein kinase signaling shows a new type of melanoma. Cancer Res. 2007;67:1502–1512. doi: 10.1158/0008-5472.CAN-06-3311. [DOI] [PubMed] [Google Scholar]
- 37.Fried L, Arbiser JL. The reactive oxygen-driven tumor: relevance to melanoma. Pigment Cell Melanoma Res. 2008;21:117–122. doi: 10.1111/j.1755-148X.2008.00451.x. [DOI] [PubMed] [Google Scholar]
- 38.Govindarajan B, Sligh JE, Vincent BJ, Li M, Canter JA, Nickoloff BJ, Rodenburg RJ, Smeitink JA, Oberley L, Zhang Y, Slingerland J, Arnold RS, Lambeth JD, Cohen C, Hilenski L, Griendling K, Martinez-Diez M, Cuezva JM, Arbiser JL. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest. 2007;117:719–729. doi: 10.1172/JCI30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perry BN, Govindarajan B, Bhandarkar SS, Knaus UG, Valo M, Sturk C, Carrillo CO, Sohn A, Cerimele F, Dumont D, Losken A, Williams J, Brown LF, Tan X, Ioffe E, Yancopoulos GD, Arbiser JL. Pharmacologic Blockade of Angiopoietin-2 Is Efficacious against Model Hemangiomas in Mice. J Invest Dermatol. 2006 doi: 10.1038/sj.jid.5700413. [DOI] [PubMed] [Google Scholar]
- 40.Cerimele F, Battle T, Lynch R, Frank DA, Murad E, Cohen C, Macaron N, Sixbey J, Smith K, Watnick RS, Eliopoulos A, Shehata B, Arbiser JL. Reactive oxygen signaling and MAPK activation distinguish Epstein-Barr Virus (EBV)-positive versus EBV-negative Burkitt’s lymphoma. Proc Natl Acad Sci U S A. 2005;102:175–179. doi: 10.1073/pnas.0408381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhandarkar SS, Mackelfresh J, Fried L, Arbiser JL. Targeted therapy of oral hairy leukoplakia with gentian violet. J Am Acad Dermatol. 2008;58:711–712. doi: 10.1016/j.jaad.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 42.Arbiser JL, Grossman K, Kaye E, Arndt KA. Use of short-course class 1 topical glucocorticoid under occlusion for the rapid control of erythrodermic psoriasis. Arch Dermatol. 1994;130:704–706. [PubMed] [Google Scholar]
- 43.Braverman IM, Sibley J. Role of the microcirculation in the treatment and pathogenesis of psoriasis. J Invest Dermatol. 1982;78:12–17. doi: 10.1111/1523-1747.ep12497850. [DOI] [PubMed] [Google Scholar]
- 44.Malhotra R, Stenn KS, Fernandez LA, Braverman IM. Angiogenic properties of normal and psoriatic skin associate with epidermis, not dermis. Lab Invest. 1989;61:162–165. [PubMed] [Google Scholar]
- 45.Braverman IM, Sibley J. The response of psoriatic epidermis and microvessels to treatment with topical steroids and oral methotrexate. J Invest Dermatol. 1985;85:584–586. doi: 10.1111/1523-1747.ep12283604. [DOI] [PubMed] [Google Scholar]
- 46.Detmar M, Brown LF, Claffey KP, Yeo KT, Kocher O, Jackman RW, Berse B, Dvorak HF. Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J Exp Med. 1994;180:1141–1146. doi: 10.1084/jem.180.3.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kunstfeld R, Hirakawa S, Hong YK, Schacht V, Lange-Asschenfeldt B, Velasco P, Lin C, Fiebiger E, Wei X, Wu Y, Hicklin D, Bohlen P, Detmar M. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood. 2004;104:1048–1057. doi: 10.1182/blood-2003-08-2964. [DOI] [PubMed] [Google Scholar]
- 48.Wrone-Smith T, Nickoloff BJ. Dermal injection of immunocytes induces psoriasis. J Clin Invest. 1996;98:1878–1887. doi: 10.1172/JCI118989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nickoloff BJ, Xin H, Nestle FO, Qin JZ. The cytokine and chemokine network in psoriasis. Clin Dermatol. 2007;25:568–573. doi: 10.1016/j.clindermatol.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 50.Nickoloff BJ, Bonish BK, Marble DJ, Schriedel KA, DiPietro LA, Gordon KB, Lingen MW. Lessons learned from psoriatic plaques concerning mechanisms of tissue repair, remodeling, and inflammation. J Investig Dermatol Symp Proc. 2006;11:16–29. doi: 10.1038/sj.jidsymp.5650010. [DOI] [PubMed] [Google Scholar]
- 51.Zenz R, Eferl R, Kenner L, Florin L, Hummerich L, Mehic D, Scheuch H, Angel P, Tschachler E, Wagner EF. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–375. doi: 10.1038/nature03963. [DOI] [PubMed] [Google Scholar]
- 52.Ma HL, Liang S, Li J, Napierata L, Brown T, Benoit S, Senices M, Gill D, Dunussi-Joannopoulos K, Collins M, Nickerson-Nutter C, Fouser LA, Young DA. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008;118:597–607. doi: 10.1172/JCI33263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fitch E, Harper E, Skorcheva I, Kurtz SE, Blauvelt A. Pathophysiology of psoriasis: recent advances on IL-23 and Th17 cytokines. Curr Rheumatol Rep. 2007;9:461–467. doi: 10.1007/s11926-007-0075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elder JT. IL-15 and psoriasis: another genetic link to Th17? J Invest Dermatol. 2007;127:2495–2497. doi: 10.1038/sj.jid.5700855. [DOI] [PubMed] [Google Scholar]
- 55.Li J, Chen X, Liu Z, Yue Q, Liu H. Expression of Th17 cytokines in skin lesions of patients with psoriasis. J Huazhong Univ Sci Technolog Med Sci. 2007;27:330–332. doi: 10.1007/s11596-007-0329-1. [DOI] [PubMed] [Google Scholar]
- 56.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 57.Zaba LC, Cardinale I, Gilleaudeau P, Sullivan-Whalen M, Suarez FM, Fuentes-Duculan J, Novitskaya I, Khatcherian A, Bluth MJ, Lowes MA, Krueger JG. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. 2007;204:3183–3194. doi: 10.1084/jem.20071094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kimball AB, Gordon KB, Langley RG, Menter A, Chartash EK, Valdes J. Safety and efficacy of ABT-874, a fully human interleukin 12/23 monoclonal antibody, in the treatment of moderate to severe chronic plaque psoriasis: results of a randomized, placebo-controlled, phase 2 trial. Arch Dermatol. 2008;144:200–207. doi: 10.1001/archdermatol.2007.63. [DOI] [PubMed] [Google Scholar]
- 59.Gottlieb AB, Matheson RT, Lowe N, Krueger GG, Kang S, Goffe BS, Gaspari AA, Ling M, Weinstein GD, Nayak A, Gordon KB, Zitnik R. A randomized trial of etanercept as monotherapy for psoriasis. Arch Dermatol. 2003;139:1627–1632. doi: 10.1001/archderm.139.12.1627. [DOI] [PubMed] [Google Scholar]
- 60.Tyring S, Gordon KB, Poulin Y, Langley RG, Gottlieb AB, Dunn M, Jahreis A. Long-term safety and efficacy of 50 mg of etanercept twice weekly in patients with psoriasis. Arch Dermatol. 2007;143:719–726. doi: 10.1001/archderm.143.6.719. [DOI] [PubMed] [Google Scholar]
- 61.Arbiser JL, Govindarajan B, Battle TE, Lynch R, Frank DA, Ushio-Fukai M, Perry BN, Stern DF, Bowden GT, Liu A, Klein E, Kolodziejski PJ, Eissa NT, Hossain CF, Nagle DG. Carbazole is a naturally occurring inhibitor of angiogenesis and inflammation isolated from antipsoriatic coal tar. J Invest Dermatol. 2006;126:1396–1402. doi: 10.1038/sj.jid.5700276. [DOI] [PubMed] [Google Scholar]
- 62.Zheng Y, Niyonsaba F, Ushio H, Nagaoka I, Ikeda S, Okumura K, Ogawa H. Cathelicidin LL-37 induces the generation of reactive oxygen species and release of human alpha-defensins from neutrophils. Br J Dermatol. 2007;157:1124–1131. doi: 10.1111/j.1365-2133.2007.08196.x. [DOI] [PubMed] [Google Scholar]
- 63.Buchau AS, Gallo RL. Innate immunity and antimicrobial defense systems in psoriasis. Clin Dermatol. 2007;25:616–624. doi: 10.1016/j.clindermatol.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takahashi K, Mulliken JB, Kozakewich HP, Rogers RA, Folkman J, Ezekowitz RA. Cellular markers that distinguish the phases of hemangioma during infancy and childhood. J Clin Invest. 1994;93:2357–2364. doi: 10.1172/JCI117241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams RL, Risau W, Zerwes HG, Drexler H, Aguzzi A, Wagner EF. Endothelioma cells expressing the polyoma middle T oncogene induce hemangiomas by host cell recruitment. Cell. 1989;57:1053–1063. doi: 10.1016/0092-8674(89)90343-7. [DOI] [PubMed] [Google Scholar]
- 66.Cerimele F, Brown LF, Bravo F, Ihler GM, Kouadio P, Arbiser JL. Infectious angiogenesis: Bartonella bacilliformis infection results in endothelial production of angiopoetin-2 and epidermal production of vascular endothelial growth factor. Am J Pathol. 2003;163:1321–1327. doi: 10.1016/S0002-9440(10)63491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Verma A, Ihler GM. Activation of Rac, Cdc42 and other downstream signalling molecules by Bartonella bacilliformis during entry into human endothelial cells. Cell Microbiol. 2002;4:557–569. doi: 10.1046/j.1462-5822.2002.00217.x. [DOI] [PubMed] [Google Scholar]
- 68.Yu Y, Varughese J, Brown LF, Mulliken JB, Bischoff J. Increased Tie2 expression, enhanced response to angiopoietin-1, and dysregulated angiopoietin-2 expression in hemangioma-derived endothelial cells. Am J Pathol. 2001;159:2271–2280. doi: 10.1016/S0002-9440(10)63077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chiller KG, Frieden IJ, Arbiser JL. Molecular pathogenesis of vascular anomalies: classification into three categories based upon clinical and biochemical characteristics. Lymphat Res Biol. 2003;1:267–281. doi: 10.1089/153968503322758076. [DOI] [PubMed] [Google Scholar]
- 70.Perry B, Banyard J, McLaughlin ER, Watnick R, Sohn A, Brindley DN, Obata T, Cantley LC, Cohen C, Arbiser JL. AKT1 overexpression in endothelial cells leads to the development of cutaneous vascular malformations in vivo. Arch Dermatol. 2007;143:504–506. doi: 10.1001/archderm.143.4.504. [DOI] [PubMed] [Google Scholar]
- 71.Shirazi F, Cohen C, Fried L, Arbiser JL. Mammalian target of rapamycin (mTOR) is activated in cutaneous vascular malformations in vivo. Lymphat Res Biol. 2007;5:233–236. doi: 10.1089/lrb.2007.1012. [DOI] [PubMed] [Google Scholar]
- 72.Lawley LP, Cerimele F, Weiss SW, North P, Cohen C, Kozakewich HP, Mulliken JB, Arbiser JL. Expression of Wilms tumor 1 gene distinguishes vascular malformations from proliferative endothelial lesions. Arch Dermatol. 2005;141:1297–1300. doi: 10.1001/archderm.141.10.1297. [DOI] [PubMed] [Google Scholar]
- 73.Cohen C, Zavala-Pompa A, Sequeira JH, Shoji M, Sexton DG, Cotsonis G, Cerimele F, Govindarajan B, Macaron N, Arbiser JL. Mitogen-actived protein kinase activation is an early event in melanoma progression. Clin Cancer Res. 2002;8:3728–3733. [PubMed] [Google Scholar]
- 74.Govindarajan B, Bai X, Cohen C, Zhong H, Kilroy S, Louis G, Moses M, Arbiser JL. Malignant transformation of melanocytes to melanoma by constitutive activation of mitogen-activated protein kinase kinase (MAPKK) signaling. J Biol Chem. 2003;278:9790–9795. doi: 10.1074/jbc.M212929200. [DOI] [PubMed] [Google Scholar]
- 75.Eisen T, Ahmad T, Flaherty KT, Gore M, Kaye S, Marais R, Gibbens I, Hackett S, James M, Schuchter LM, Nathanson KL, Xia C, Simantov R, Schwartz B, Poulin-Costello M, O’dwyer PJ, Ratain MJ. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer. 2006 doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ratain MJ, Eisen T, Stadler WM, Flaherty KT, Kaye SB, Rosner GL, Gore M, Desai AA, Patnaik A, Xiong HQ, Rowinsky E, Abbruzzese JL, Xia C, Simantov R, Schwartz B, O’dwyer PJ. Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24:2505–2512. doi: 10.1200/JCO.2005.03.6723. [DOI] [PubMed] [Google Scholar]
- 77.Varker KA, Biber JE, Kefauver C, Jensen R, Lehman A, Young D, Wu H, Lesinski GB, Kendra K, Chen HX, Walker MJ, Carson WE. III A randomized phase 2 trial of bevacizumab with or without daily low-dose interferon alfa-2b in metastatic malignant melanoma. Ann Surg Oncol. 2007;14:2367–2376. doi: 10.1245/s10434-007-9389-5. [DOI] [PubMed] [Google Scholar]
- 78.Arbiser JL, Fan CY, Su X, Van Emburgh BO, Cerimele F, Miller MS, Harvell J, Marinkovich MP. Involvement of p53 and p16 tumor suppressor genes in recessive dystrophic epidermolysis bullosa-associated squamous cell carcinoma. J Invest Dermatol. 2004;123:788–790. doi: 10.1111/j.0022-202X.2004.23418.x. [DOI] [PubMed] [Google Scholar]
- 79.Arbiser JL, Byers HR, Cohen C, Arbeit J. Altered basic fibroblast growth factor expression in common epidermal neoplasms: examination with in situ hybridization and immunohistochemistry. J Am Acad Dermatol. 2000;42:973–977. [PubMed] [Google Scholar]
- 80.Meeran SM, Katiyar S, Elmets CA, Katiyar SK. Interleukin-12 deficiency is permissive for angiogenesis in UV radiation-induced skin tumors. Cancer Res. 2007;67:3785–3793. doi: 10.1158/0008-5472.CAN-06-3134. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 81.Yusuf N, Nasti TH, Long JA, Naseemuddin M, Lucas AP, Xu H, Elmets CA. Protective role of Toll-like receptor 4 during the initiation stage of cutaneous chemical carcinogenesis. Cancer Res. 2008;68:615–622. doi: 10.1158/0008-5472.CAN-07-5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arbiser JL. Why targeted therapy hasn’t worked in advanced cancer. J Clin Invest. 2007;117:2762–2765. doi: 10.1172/JCI33190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kolhe N, Mamode N, Van der WJ, Pattison J. Regression of post-transplant Kaposi’s sarcoma using sirolimus. Int J Clin Pract. 2006;60:1509–1512. doi: 10.1111/j.1742-1241.2006.00815.x. [DOI] [PubMed] [Google Scholar]
- 84.Campistol JM, Gutierrez-Dalmau A, Torregrosa JV. Conversion to sirolimus: a successful treatment for posttransplantation Kaposi’s sarcoma. Transplantation. 2004;77:760–762. doi: 10.1097/01.tp.0000115344.18025.0b. [DOI] [PubMed] [Google Scholar]
- 85.Campistol JM, Schena FP. Kaposi’s sarcoma in renal transplant recipients--the impact of proliferation signal inhibitors. Nephrol Dial Transplant. 2007;22 Suppl 1:i17–i22. doi: 10.1093/ndt/gfm089. [DOI] [PubMed] [Google Scholar]
- 86.Fife K, Howard MR, Gracie F, Phillips RH, Bower M. Activity of thalidomide in AIDS-related Kaposi’s sarcoma and correlation with HHV8 titre. Int J STD AIDS. 1998;9:751–755. doi: 10.1258/0956462981921512. [DOI] [PubMed] [Google Scholar]
- 87.Cooley T, Henry D, Tonda M, Sun S, O’Connell M, Rackoff W. A randomized, double-blind study of pegylated liposomal doxorubicin for the treatment of AIDS-related Kaposi’s sarcoma. Oncologist. 2007;12:114–123. doi: 10.1634/theoncologist.12-1-114. [DOI] [PubMed] [Google Scholar]
- 88.Krown SE. Therapy of AIDS-associated Kaposi’s sarcoma: targeting pathogenetic mechanisms. Hematol Oncol Clin North Am. 2003;17:763–783. doi: 10.1016/s0889-8588(03)00042-x. [DOI] [PubMed] [Google Scholar]
- 89.Shaw RN, Arbiser JL, Offermann MK. Valproic acid induces human herpesvirus 8 lytic gene expression in BCBL-1 cells. AIDS. 2000;14:899–902. doi: 10.1097/00002030-200005050-00021. [DOI] [PubMed] [Google Scholar]
- 90.Klass CM, Krug LT, Pozharskaya VP, Offermann MK. The targeting of primary effusion lymphoma cells for apoptosis by inducing lytic replication of human herpesvirus 8 while blocking virus production. Blood. 2005;105:4028–4034. doi: 10.1182/blood-2004-09-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bhandarkar SS, Cohen C, Kuruvila M, Rea TH, Mackelfresh JB, Lee DJ, Modlin RL, Arbiser JL. Angiogenesis in cutaneous lesions of leprosy: implications for treatment. Arch Dermatol. 2007;143:1527–1529. doi: 10.1001/archderm.143.12.1527. [DOI] [PubMed] [Google Scholar]