

Abstract
Tuberous sclerosis (TSC) is a hamartoma syndrome attributable to mutations in either TSC1 or TSC2 in which brain involvement causes epilepsy, mental retardation, and autism. We have reported recently (Meikle et al., 2007) a mouse neuronal model of TSC in which Tsc1 is ablated in most neurons during cortical development. We have tested rapamycin and RAD001 [40-O-(2-hydroxyethyl)-rapamycin], both mammalian target of rapamycin mTORC1 inhibitors, as potential therapeutic agents in this model. Median survival is improved from 33 d to more than 100 d; behavior, phenotype, and weight gain are all also markedly improved. There is brain penetration of both drugs, with accumulation over time with repetitive treatment, and effective reduction of levels of phospho-S6, a downstream target of mTORC1. In addition, there is restoration of phospho-Akt and phospho-glycogen synthase kinase 3 levels in the treated mice, consistent with restoration of Akt function. Neurofilament abnormalities, myelination, and cell enlargement are all improved by the treatment. However, dysplastic neuronal features persist, and there are only modest changes in dendritic spine density and length. Strikingly, mice treated with rapamycin or RAD001 for 23 d only (postnatal days 7–30) displayed a persistent improvement in phenotype, with median survival of 78 d. In summary, rapamycin/RAD001 are highly effective therapies for this neuronal model of TSC, with benefit apparently attributable to effects on mTORC1 and Akt signaling and, consequently, cell size and myelination. Although caution is appropriate, the results suggest the possibility that rapamycin/RAD001 may have benefit in the treatment of TSC brain disease, including infantile spasms.
Keywords: tuberous sclerosis, TSC, TSC1, TSC2, rapamycin, RAD001
Introduction
Tuberous sclerosis complex (TSC) is a clinically devastating neurocutaneous syndrome in which benign tumors termed hamartomas develop in multiple organ systems (Gomez et al., 1999; Curatolo, 2003; Crino et al., 2006). Neurological manifestations are a predominant clinical feature and include early-onset epilepsy, mental retardation, developmental delay, and autism (Ess, 2006; Marcotte and Crino, 2006; Holmes and Stafstrom, 2007; Winterkorn et al., 2007). Most neurological symptoms are thought to be attributable to the occurrence of cortical tubers, which typically form at the gray–white matter junction. The laminar structure within these lesions is severely disrupted with occurrence of poorly differentiated giant cells, dysplastic neurons and astrocytes, and a variety of reactive cells (Huttenlocher and Wollmann, 1991; Ess, 2006). The number and location of cortical tubers, as well as more generalized cortical abnormalities, and the timing of onset and duration of infantile spasms all appear to have some relationship to the severity of the neurological manifestations that are seen in TSC patients (Doherty et al., 2005; Marcotte and Crino, 2006; Winterkorn et al., 2007).
TSC is attributable to inactivating mutations in either the TSC1 or the TSC2 gene and segregates in an autosomal dominant manner (Cheadle et al., 2000). TSC1 mutations account for 20–25% of all mutations identified, whereas TSC2 mutations account for the remainder. TSC1 disease is less severe than TSC2 disease in multiple respects (Jones et al., 1999; Dabora et al., 2001; Sancak et al., 2005; Au et al., 2007; Winterkorn et al., 2007), and this appears to be attributable to a reduced frequency of second-hit events in the TSC1 gene compared with the TSC2 gene (Dabora et al., 2001).
The TSC1 and TSC2 proteins form a relatively tight stoichiometric complex in cells, which functions in an ancestrally conserved signaling pathway that regulates the state of activation of mammalian target of rapamycin (mTOR) and thereby cell growth (Goncharova et al., 2002; Kwiatkowski et al., 2002; Inoki et al., 2005; Tee and Blenis, 2005; Avruch et al., 2006; Sabatini, 2006). Loss of either TSC1 or TSC2 leads to elevated rheb-GTP levels, a ras family GTPase, which interacts with the mTORC1 complex to cause its activation. mTORC1 activation leads to a downstream kinase signaling cascade, including activation of the S6 kinases and feedback inhibition of Akt activation (Hay, 2005; Manning et al., 2005), as well as translational activation of a select subset of mRNAs (Bilanges et al., 2007).
A conditional allele of Tsc1 has been developed and combined with different brain-specific cre recombinase alleles to generate models of TSC brain disease (Uhlmann et al., 2002; Meikle et al., 2007). We used a synapsin I promoter-driven cre allele to generate a neuronal model of TSC1, in which recombination and loss of the Tsc1 gene occurs in differentiating neurons (Meikle et al., 2007). These mice develop several pathologic features seen in TSC tubers, including enlarged and dysplastic neurons, which can occur ectopically in the cortex, persistently reduced myelination attributable to a neuronal inductive defect, and high expression of phospho-S6 (pS6), a protein downstream of mTORC1 (Meikle et al., 2007). Clinically, these mice are hyperactive and tremulous, with poor weight gain, development of seizures, and median survival of 33 d. Here we present a detailed study of the pharmacokinetics and clinical benefit of rapamycin and RAD001 [40-O-(2-hydroxyethyl)-rapamycin] [also called SDZ RAD, everolimus (Schuler et al., 1997)] in the treatment of the TSC1 neuronal model.
Materials and Methods
Mouse procedures.
As described previously (Meikle et al., 2007), mice with these alleles began in a mixed strain background (129S4/SvJae, C57BL/6, and CBA) but have been maintained as an inbred population in our colony for >3 years. Mice bearing the SynICre allele were the generous gift of Jamey Marth (University of San Diego, La Jolla, CA) (Zhu et al., 2001). Mice were generated through matings between parents of genotype Tsc1cc and Tsc1w−SynICre+ or Tsc1w−SynICre++ or Tsc1cwSynICre+. [We use c, w, and − to denote the conditional (floxed), wild-type, and null alleles of Tsc1, respectively; the formal name of the c allele is Tsc1tm1Djk.] We use the term Tsc1null-neuron mice to denote mice with the genotype Tsc1c− SynICre+ or Tsc1cc SynICre+. As reported previously, mice with these two genotypes have identical phenotypes and survival (Meikle et al., 2007). Controls usually had the genotype Tsc1c+ SynICre+, but some were Tsc1c− SynICre−, Tsc1c+ SynICre−, or Tsc1cc SynICre−. Littermate controls were used whenever possible, but all mutants and controls were closely related from this inbred colony.
All procedures were performed in accordance with the Guide for the Humane Use and Care of Laboratory Animals, and the study was approved by the Animal Care and Use Committee of Children's Hospital. Individual mice were killed when weight loss of 20%, greatly reduced movement, or other signs of morbidity were seen.
Neurologic assessment was performed by an observer blinded to genotype and treatment status of the mice. This included the following: assessment of hindleg clasping behavior when suspended by the tail, scored as absent (0) or present (1); whole-body tremor assessed by placing the palm of the hand on the back of the mouse, scored on a scale from absent (0) to severe and persistent (5); kyphosis, scored as absent (0) or present (1); and tail position observed during a 3 min interval during which the animal was allowed to walk freely in a confined space, scored as normal (0), held horizontal (1), held above horizontal (2), or Straub position (elevated highly, 3). Scores for these traits were compared using the Fisher's exact test (clasp, kyphosis) or the Mann–Whitney U test (tremor, tail position) in Prism (GraphPad Software).
Mice were anesthetized, and total body weight was determined before they were killed. The brains were rapidly removed, snap frozen in liquid nitrogen, and stored as half brains at −80°C until use. Half-brain weights were measured in milligrams accurate to ±0.1 mg. Brain weight to body weight ratios were calculated, and measurements were compared using the Mann–Whitney U test.
DNA analyses.
DNA was prepared from mouse toes/tails by standard procedures for genotyping. Genotyping at the Tsc1 gene was performed using a four-primer system that allows simultaneous analysis of the c, w, and − alleles, followed by agarose gel electrophoresis (Meikle et al., 2005). Primers that amplify a 300 bp portion of the cre recombinase gene were used to assess the presence of the SynICre allele (Meikle et al., 2005).
Antibodies.
Antibodies used were as follows: Tsc2 (C20), Akt (C20), extracellular signal-regulated kinase 2 [ERK2 (K23)], pCofilin(S3) from Santa Cruz Biotechnology; pS6(S240/244), pS6(S235/236), Tsc1, pAKT(S473), S6, Cofilin, glycogen synthase kinase (GSK) 3β, and pGSK3β(S9) from Cell Signaling Technology; neuronal-specific nuclear protein (NeuN) (MAB 377), neurofilament (MAB4245), MBP (AB980), high- and medium-molecular-weight neurofilament from Millipore Bioscience Research Reagents; nonphosphorylated neurofilament (SMI311), phosphorylated neurofilament (SMI312), and MBP (SMI99) from Sternberger Monoclonals.
Immunoblotting.
Mice were killed, and brains were harvested as above. Each frozen half brain was homogenized in 5× volume TBSV lysis buffer (in mm: 20 Tris-Cl, pH 7.5, 140 NaCl, 10 NaF, 1 Na3VO4, and 1 EDTA) with 0.1% Triton X-100, 10 mm β-glycerol phosphate, 1% phosphatase inhibitor cocktail 1 (Sigma), and 1% phosphatase inhibitor cocktail 2 (Sigma). Any solid insoluble matter was removed by centrifugation at 15,000 × g at 4°C for 10 min, and the supernatant was mixed with Laemli's SDS-sample buffer. Samples were put through a 22 gauge needle, boiled, and centrifuged before loading. Proteins were separated by electrophoresis on 4–12% Bis-Tris gels (Novex) and transferred onto trans-blot nitrocellulose membranes. Coomassie staining was performed to confirm that the samples were loaded equally. The membranes were blocked in 5% nonfat dry milk in PBS, pH 7.4, with 0.1% Tween 20 (PBS–Tween) for 1 h at room temperature. Primary antibodies were diluted in blocking solution, and membranes were incubated overnight at 4°C or 1 h at room temperature. The primary antibody was removed, and the blots were washed in PBS–Tween and then incubated for 1 h at room temperature in horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology). Reactive proteins were visualized using SuperSignal West Pico chemiluminescence reagent (Pierce) and exposure to x-ray film (BioMax MR; Eastman Kodak). All immunoblots shown in one row of a figure are from the same gel-blot exposure.
Histological preparation, immunohistological staining, and microscopy.
For immunohistochemistry, mice at ages postnatal day 21 (P21) to P100 were anesthetized, and transcardiac perfusion was performed using PBS, followed by 4% paraformaldehyde (PFA) in PBS (Boston BioProducts). Whole brains were then removed and postfixed overnight at 4°C in 4% PFA solution. Fixed brains were then cryoprotected in 30% sucrose in PBS for 1–14 d at 4°C, and coronal sections were cut at 50 μm using a Microm K400 Fast Freezing Unit and HM 450 Sliding Electronic Microtome and were then stored in PBS at 4°C until use (Meikle et al., 2007) Free-floating sections were incubated in blocking solution (PBS, 0.1% Triton X-100, and 5% normal goat serum) for 1 h at room temperature, followed by incubation with primary antibody overnight at 4°C. After three 20-min PBS washes, sections were incubated in secondary antibody solution containing Alexa Fluor 568 goat anti-rabbit/anti-mouse IgG/IgM or Oregon Green 488 goat anti-rabbit/anti-mouse IgG (Invitrogen), for 1 h at room temperature. After two washes in PBS, for some sections, 0.5 μg/ml Hoechst 33258 was added to each well for 5 min at room temperature, and the sections were then washed twice in PBS, mounted onto positively charged slides, and coverslipped with antifade medium (Fluoromount-G; Southern Biotechnology Associates). Control sections were done with omission of one or both primary antibodies, adding both secondary antibodies, to establish specificity. Slides were viewed on a Nikon TE2000-E inverted microscope. Illumination wavelength, intensity, and exposure times were selected using a dual filter-wheel apparatus containing an electronically controlled shutter and filters for the different fluorophore wavelengths (Lambda 10-2; Sutter Instrument). Digital images were captured using an Orca-II ER cooled CCD camera (Hamamatsu) and MetaMorph (Molecular Devices). Digital images were then processed and analyzed using Adobe Photoshop (Adobe Systems), including pseudocolorization.
Standard histology sections were prepared after Bouins fixation at room temperature. After hematoxylin and eosin staining, slides were viewed on a Nikon Eclipse E400 microscope, and images were captured using Spot software version 4.0.5.
For all histological and immunostaining observations, a minimum of three (range of 3–12) pairs of mutant and control mice were examined from stereotactically matched brain sections.
Confocal images were captured using a Zeiss LSM510 META two-photon confocal microscope using 20× and 63× objectives. For cell size determination and other images, a Z-stack of confocal images at 0.5 μm intervals were collected from the somatosensory cortex at layer V (−2 to −2.2 mm from bregma and 3.2–3.4 mm lateral to midline) for each of at least two matched mutant and control mice. Images were examined using NIH ImageJ software 1.31v to calculate cell size in square micrometers after manual drawing of cell margins. All SMI311-positive (SMI311+) cells in a 20× field were measured, independent of pS6 staining, for a minimum of eight cells per field, and the largest eight cells were included in the measurements. Measurements were compared using the Mann–Whitney U test.
To quantify dendritic orientation as a measure of neuronal dysplasia, the direction of the apical dendrite of each SMI311+ pyramidal neuron in layer V of a section of somatosensory cortex was assessed. Neurons with apical dendrites oriented within 15° of a vertical line to the pia from the cell center were considered normal. Neurons with apical dendrites oriented outside of this 15° range in either direction were considered to have aberrant orientation.
Drug handling, administration, and pharmacokinetic analyses.
Rapamycin was obtained from LC Laboratories, dissolved at 20 mg/ml in ethanol, and stored at − 20°C for up to 1 month. Before each administration, rapamycin was diluted in 5% Tween 80, 5% polyethylene glycol 400 (0.5–1.5 mg/ml). Rapamycin was given at 6 mg/kg intraperitoneally every 2 d. RAD001 was provided by Novartis in a proprietary vehicle at 20 mg/ml. Before each administration, RAD001 was diluted in water to 0.5–2 mg/ml and was given at either 3 or 6 mg/kg intraperitoneally every 2 d. Both drugs were given beginning at P7–P9, every other day, for up to 92 d (P100).
For pharmacokinetic analyses, each drug was given once or multiple times intraperitoneally to control, nonmutant mice from the Tsc1 synapsin–cre colony. Mice were killed at 1, 4, 12, 24, or 48 h after drug administration, blood was collected in acid–citrate–dextrose solution, and plasma was isolated by centrifugation at 5000 rpm for 5 min. Organs were then quickly removed and frozen at − 80°C. Organ extracts were prepared by homogenization in 5× (w/v) of PBS until a fine suspension was achieved. This solution was clarified by centrifugation at 12,000 × g for 15 min and then frozen at −80°C until ready for assay. Rapamycin levels were determined after solid-phase extraction using liquid chromatography/tandem mass spectrometry (LC/MS/MS) on an API-2000 (Applied Biosystems) instrument in the Clinical Laboratories, Children's Hospital, Boston. This is a Clinical Laboratory Improvement Amendments-certified laboratory that routinely determines rapamycin levels on pediatric samples. RAD001 levels were determined using LC/MS/MS by Ann Brown at Novartis Biomedical Research Institute (Cambridge, MA). Briefly, tissue lysates, separated plasma, and calibration standards were extracted using the acetonitrile (ACN) protein precipitation technique. Reverse-phase chromatography (column: ACE C18 30 × 2.1 mm, 3 μm) was used using a gradient elution with 0.1% HCOOH/water and 0.1% HCOOH/ACN as mobile phase A and B, respectively. The analyses were performed in electrospray-positive mode using multiple reaction monitoring conditions on a Sciex API 4000 instrument (Applied Biosystems).
DiI staining of neurons and dendritic spine measurements.
Spine tracing and analysis was performed as described previously (Murai et al., 2003). Briefly, anesthetized mice were transcardially perfused with PBS and fixed with 4% PFA at 2 ml/min for 5 min. Whole brains were removed and postfixed for 30 min in 4% PFA on ice. Brains were then embedded in 3% agarose and sectioned at 150 μm using a vibrating-blade microtome (Leica). Introduction of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) (Invitrogen)-coated gold particles was performed using the Helios Gene-Gun (Bio-Rad) at 80 psi. Coated particles were prepared by mixing 20 mg of gold particles with 5 mg of DiI. After gene gun particle injection, sections were placed in 4% PFA for 20 h to allow DiI diffusion along the cell membrane. Sections were then imaged by confocal microscopy. A Z-stack of confocal images at 0.5 μm intervals was collected from the somatosensory cortex in layer V. Spine analysis was performed using MetaMorph software (Molecular Devices). Spine lengths were measured from the tip to the intersection of the spine with the dendrite. Measurements of spine length (n = 856–1742 spines) and spine density (n = 10–19 dendrites) were compared using the Mann–Whitney U test in Prism (GraphPad Software).
Results
Pharmacokinetics of rapamycin and RAD001 in the brain
We explored the pharmacokinetics and pharmacodynamics of rapamycin and RAD001 to help define an effective dose of each compound for treatment of the previously described Tsc1null-neuron mice (Meikle et al., 2007). These drugs were chosen for study because they are both orally formulated for administration in humans, and both drugs are known to inhibit the kinase activity of mTOR by binding to FK506 binding protein 12 (FKBP12), which then binds to the mTORC1 complex (Brown et al., 1994). In addition, because the drugs are structurally different (Schuler et al., 1997), a direct comparison of both compounds was performed to determine whether they had different pharmacologic properties, particularly their relative penetration into the brain. Although both rapamycin and RAD001 can be given orally to older mice, we administered both drugs intraperitoneally because it is difficult to perform gavage on P7 mice, the age at which we chose to initiate treatment.
A single dose of either drug given at 6 mg/kg intraperitoneally to control mice aged P30–P45 led to substantial drug levels in plasma, liver, and brain (Fig. 1A,D). Brain levels remained markedly lower than systemic levels at all time points, consistent with an effect of the blood–brain barrier in reducing penetration into the CNS. Nonetheless, brain levels of each drug remained above the level required to inhibit mTORC1 (10 ng/ml for either drug when applied to cells in vitro) throughout the 48 h period after administration (rapamycin, 38.6 ng/g; RAD001, 40.5 ng/g; at 48 h after drug administration). Note also that therapeutic trough levels for each drug in humans are 3–20 ng/ml in whole blood. Both drugs displayed higher levels in nearly all cases when given every other day over a 3 week period (12 doses) compared with a single dose (Fig. 1B,E). Brain levels 48 h after the last of 12 doses were rapamycin at 88.4 ng/g and RAD001 at 48.9 ng/g. In an initial pharmacodynamic analysis, pS6(S235/236) and pS6(S240/244) levels were assessed by immunoblot analysis of whole-brain lysates from the Tsc1null-neuron mice, at 24 h and 48 h after the last treatment at 6 mg/kg intraperitoneally in a 12 dose every other day of treatment regimen. Phosphorylation of S6 at both sites returned to control (or lower) levels 24 h after the last dose (Fig. 1I) and remained low at 48 h (data not shown). However, this normalization of pS6 levels at 48 h after the last dose was not as consistent in mice similarly treated with 3 mg/kg (data not shown).
Figure 1.

Pharmacokinetics, survival benefit, and brain signaling effects of rapamycin and RAD001 in Tsc1null-neuron mice. A–F, Pharmacokinetic analyses of rapamycin (A–C) and RAD001 (D–F) in P30–P45 mice. Both drugs were given at 6 mg/kg intraperitoneally. SEMs are indicated. n = 3 for all rapamycin measurements; n = 3 for all but 5 RAD001 measurements. A, D, Drug levels in plasma, brain, and liver. Time after a single intraperitoneal injection is shown on the x-axis. Note that y-axis is exponential. B, E, Drug levels at 24 and 48 h after single and multiple doses. Multiple doses were given every other day. C, F, Drug levels in older (P30–P45) mice compared with young (P10) mice 24 h after a single intraperitoneal dose. G, H, Survival curves of Tsc1null-neuron mice treated with rapamycin (G) or RAD001 (H). Doses of each drug, periods of treatment, and numbers of treated mice are indicated. Treatment began at P7–P9 for all mice. Note that dashed lines reflect a cohort of mice in which treatment was discontinued after P30. Tsc1c− SynICre+ mice comprised 58, 78, and 61% of the rapamycin, RAD001, and untreated cohorts, respectively. The remainder were Tsc1cc SynICre+. Note that the survival of untreated Tsc1c− SynICre+ and Tsc1cc SynICre+ mice were not different, with medians of 32 and 34 d, respectively (not shown). I, Immunoblot analysis of P30 brain lysates collected 24 h after the last dose of drug shows reduction of Tsc1 protein levels (*) in all Tsc1null-neuron (mutant) brain lysates and reduction in pS6(240/4) and pS6(235/6) in lysates from treated mutant mice. AKT and ERK2 are loading controls. Mut, Mutant; Cont, controls. Both rapamycin (RAP) and RAD001 were given 6 mg/kg intraperitoneally every other day, beginning at P7–P9. Lanes 1, 2, 6, 8, and 9 were Tsc1c− SynICre+; lanes 5, 7, and 10 were Tsc1cc SynICre+; lanes 3 and 4 were Tsc1c+ SynICre−.
We also assessed whether the pharmacokinetics of these drugs was different in younger mice. Liver levels were increased 3.5-fold for rapamycin and 4.1-fold for RAD001 in P10 mice 24 h after a single intraperitoneal injection compared with similarly treated P30–P45 mice (Fig. 1C,F). These data indicate that overall clearance of each drug is reduced at this age. In addition, brain levels of each drug were similar to liver levels at P10 24 h after injection (Fig. 1C,F), indicating that the blood–brain barrier was not developed at P10.
These data indicated that penetration of rapamycin and RAD001 into the CNS was substantial, although it is clearly much higher in younger mice. Although levels were high at P10, we elected to use 6 mg/kg intraperitoneally every other day as our standard dose for several reasons. First, we wanted to ensure that we would have effective mTOR inhibition at the dose used throughout the period of treatment, to have maximal potential therapeutic effect. Second, although levels clearly rose with repeat dosing, we were concerned that these levels might be misleading in reflecting retention of drug in a lipid compartment in the brain or drug bound to protein, which would not be free to enter into a complex with FKBP12, required for mTORC1 inhibition. Finally, as noted above, mTORC1 inhibition in the brain, as assessed by pS6 immunoblotting, was more effective at this dose than at 3 mg/kg for either drug.
Therapeutic effects of rapamycin/RAD001 in Tsc1null-neuron mice
Both rapamycin and RAD001, when given intraperitoneally at 6 mg/kg every other day beginning at P7–P9, caused dramatic therapeutic benefit. Tsc1null-neuron mice on these regimens demonstrated 90–100% survival at 80 d of age, and this improvement continued until the experiment was terminated at P100 (Fig. 1G,H; note that median survival for the untreated mutants was 33 d). In addition, Tsc1null-neuron mice receiving either drug displayed dramatic clinical improvement with a marked reduction in clasping behavior when suspended by their tails, tremor, kyphosis, and aberrant tail position (Straub tail) (Fig. 2 A–C, Table 1). Using a blinded observer to assess these four phenotypic measures, all four were significantly improved at all follow-up times (P30, P60, and P100) in both rapamycin- and RAD001-treated mice (Table 1, p ≤ 0.01 for each comparison). Consistent with a marked improvement in phenotype and development, there was also an improvement in the brain/body weight ratio after rapamycin treatment, which was markedly elevated (2.4-fold) in untreated Tsc1null-neuron mice compared with controls (Fig. 2M). Despite this phenotypic improvement, the weight of rapamycin/RAD001-treated Tsc1null-neuron mice at P30 was similar, although slightly higher on average, to that of untreated mutants (Table 1). However, with longer follow up, significant weight gain was seen, with rapamycin- and RAD001-treated Tsc1null-neuron mice having average weights of 18.1 and 19.6 g at P100 (Table 1, compared with 23.2 g in untreated control P100 mice). Both drugs had a similar effect in suppressing the growth and weight gain of control mice at P30 (Table 1).
Figure 2.

Clinical and histological improvement in Tsc1null-neuron mice treated with rapamycin. A–D, Pictures of mice. Control and mutant (Tsc1null-neuron) mice with or without rapamycin treatment are shown. Rapamycin was given 6 mg/kg intraperitoneally every other day from P7 to P9 and was discontinued at P30 in the on/off mouse. A, C, D are all P45; B is P30. Note abnormal posture of mutant and mutant on/off treatment mice. E–I, Combined NeuN (red)–pS6(S235/236) (green) stains of base of cortex over the anterior hippocampus. P45 control, P30 mutant (Tsc1null-neuron), P45 rapamycin-treated mutant, P45 rapamycin-treated mutant until P30 then taken off drug (on/off), and P45 rapamycin-treated control mouse sections are shown. Scale bar, 50 μm. Images were collected using the confocal microscope on a single slice at 200× with 0.7 zoom. J–L, Hematoxylin and eosin stains of the cortex demonstrate mild cortical disorganization in the mutant mouse with global cell enlargement particularly at the base of cortex (*), with improvement with treatment. Ages and treatments are as in E–G. Genotypes for mice were as follows: A, E, I, J, Tsc1c+ SynICre+; F, L, Tsc1c− SynICre+; B, C, D, G, H, K, Tsc1cc SynICre+. M, Brain weight to body weight ratio in these same groups of mice, all at P30. Each point is a different mouse; the line indicates the mean. p < 0.01 for each group compared with untreated mutant. Numbers and genotypes of mice were as follows: Mutant, six Tsc1c− SynICre+; Control, three Tsc1c+ SynICre+; Mutant + Rap, three Tsc1cc SynICre+, three Tsc1c− SynICre+; Control + Rap, two Tsc1c+ SynICre+.
Table 1.
Neurologic features in Tsc1null-neuron mice according to age and treatment
| Genotype class | Treatment | Age | n | Weight (g) | Weight SD | Clasp | Tremor | Kyphosis | Tail | |
|---|---|---|---|---|---|---|---|---|---|---|
| % Tsc1c− SynICre+ | ||||||||||
| 46% | Mutant | None | P30 | 16 | 9.87 | 1.59 | 0.63 | 3.28 | 0.81 | 2.73 |
| 65% | Mutant | Rapamycin | P30 | 17 | 10.27 | 2.05 | 0 | 0.12 | 0 | 0.88 |
| 88% | Mutant | RAD001 | P30 | 17 | 11.14 | 1.14 | 0 | 0.26 | 0 | 1.24 |
| 92% | Mutant | Rapamycin | P60 | 12 | 14.39 | 2.68 | 0 | 0.42 | 0.17 | 0.83 |
| 86% | Mutant | rap-P30 | P60 | 9 | 16.13 | 2.48 | 0 | 1.94 | 0.67 | 1.14 |
| 71% | Mutant | Rapamycin | P100 | 14 | 18.13 | 1.84 | 0 | 0.64 | 0.14 | 0.79 |
| 82% | Mutant | RAD001 | P100 | 11 | 19.59 | 2.30 | 0.09 | 1.00 | 0.27 | 0.73 |
| % Tsc1c+ SynICre+ | ||||||||||
| 89% | Control | None | P30 | 10 | 20.32 | 2.85 | 0 | 0.35 | 0 | 0.30 |
| 100% | Control | Rapamycin | P30 | 4 | 11.50 | 0.27 | 0 | 0.25 | 0 | 0 |
| 86% | Control | RAD001 | P30 | 7 | 12.66 | 2.33 | 0.14 | 0.14 | 0 | 0.14 |
| 57% | Control | Rapamycin | P60 | 7 | 18.39 | 2.31 | 0 | 0.36 | 0 | 0.57 |
| 75% | Control | Rapamycin | P100 | 9 | 19.56 | 2.25 | 0 | 0.39 | 0 | 0.56 |
| 78% | Control | RAD001 | P100 | 9 | 20.76 | 2.91 | 0 | 0.22 | 0.22 | 0.44 |
Mutant refers to Tsc1null-neuron mice. Mutants had genotypes Tsc1c− SynICre+ or Tsc1cc SynICre+; % Tsc1c− SynICre+ is shown in the first column for mutants. Controls had genotypes Tsc1c+ SynICre+, Tsc1c+ SynICre−, Tsc1cc SynICre−, Tsc1c− SynICre−; % Tsc1c+ SynICre+ is shown in the first column for controls. All treatments were 6 mg/kg intraperitoneally every other day, beginning at P7–P9. Rapamycin on/off mice were treated until P30, and then drug was discontinued; all others were treated continuously. Average values for weight in grams and standard deviation (SD) are shown. Average values for clasping, tremor, kyphosis, and tail position scores are shown and were assessed as described in Materials and Methods. Tremor and tail position were scored on 0–5 and 0–3 scales, respectively. Clasping and kyphosis were assessed on a binary 0 (absent) or 1 (present) scale. Note that all neurologic scores as well as weight were significantly different (p < 0.003 for each) between untreated mutant and control mice at P30. All neurologic scores, but not weight, were also significantly different (p < 0.003 for each) between P30 mutant and rapamcyin- or RAD001-treated P30 mice. There was no significant difference between weight or any neurologic score between rapamycin- and RAD001- treated mice at either P30 or P100. There was a significant difference between rapamycin-treated P60 and rapamycin-treated 22 d only P60 (rap on/off) mice in both tremor (p = 0.0003) and kyphosis (p = 0.02).
Discontinuation of either drug treatment at P30 led to sustained clinical improvement for 1–2 weeks, followed by a gradual clinical deterioration (Fig. 2D, compare with C), which led to death at a median age of 79 d for rapamycin and 77 d for RAD001 (Fig. 1G,H). At P60, 30 d after stopping drug, rapamycin-treated mice had significant weight gain and significant phenotypic improvement in clasping behavior, tremor, and tail position (all p ≤ 0.03) compared with untreated P30 mutant mice (Table 1).
Effects of treatment on cortical organization, dysplasia, cell size enlargement, and myelination
We examined several aspects of cortical organization and neuronal morphology to provide insight into the mechanism of action of rapamycin/RAD001, following up previous observations in the Tsc1null-neuron mice (Meikle et al., 2007). Because these two compounds had equivalent therapeutic effects on survival (Fig. 1G,H) and in phenotypic improvement at both P30 and P100 (Table 1, all pairwise comparisons of phenotypic measures are nonsignificant), we focused these studies on mice treated with rapamycin.
Analysis of cortical sections from the rapamycin-treated Tsc1null-neuron mice showed several aspects of improvement (Fig. 2 E–L). First, treatment of the mutant mice with rapamycin led to a marked reduction in the extent of expression of pS6(S235/236) compared with untreated controls (Fig. 2 E–G). This was apparent throughout the cortex but was most marked in a subset of enlarged pS6+ cells seen at the base of the cortex (Fig. 2F, *) and in cortical layer V in the mutants. A considerable decrease in levels of pS6(S235/236) was also noted in the thalamus and CA3 region of the hippocampus in the treated mutant when compared with untreated mutants (see further below). Interestingly, these abnormal pS6-positive cells reappeared within 2 weeks of discontinuation of rapamycin (Fig. 2H, *). Treatment of control mice with rapamycin suppressed pS6(S235/236) levels even lower than in untreated controls (Fig. 2I). Standard histological sections also showed that these enlarged cells were markedly diminished in the rapamycin-treated mutants (Fig. 2 J–L, note * in K). However, the mild overall cortical disorganization noted in the untreated mutants was not affected by rapamycin treatment.
We also examined neuronal morphology in the treated mice, using the SMI311 antibody against nonphosphorylated neurofilament to identify a restricted neuronal population in lateral somatosensory cortex, as done previously (Meikle et al., 2007) (Fig. 3 A–J, see N for orientation). We observed that these cells were considerably enlarged in Tsc1null-neuron mice compared with SMI311+ cells from the identical region of control mice (Fig. 3A,B,F,G). Quantitative measurement showed that there was a considerable reduction in cell size after treatment (reduced from 1.81-fold to 1.18-fold when compared with untreated controls, p < 0.0001). However, this was a reversible phenomenon as the enlarged SMI311+ pyramidal cells reappeared when rapamycin treatment was discontinued for 2 weeks (Fig. 3D,I,M) (increased to 1.77-fold compared with untreated controls, p = 0.02).
Figure 3.
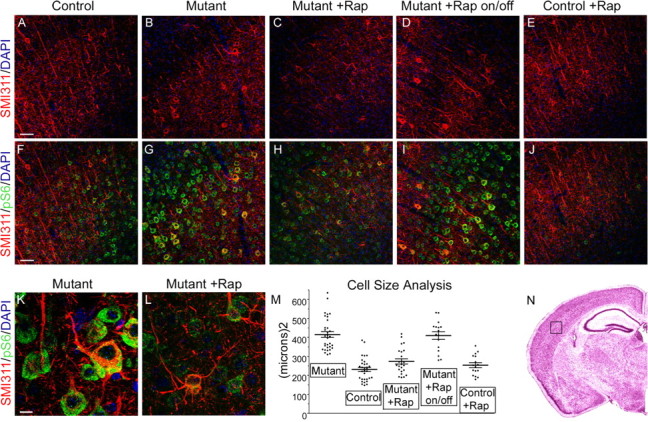
Effects on neuronal size and orientation in Tsc1null-neuron mice of treatment with rapamycin. A–J, Paired images are shown for P45 control, P30 mutant (Tsc1null-neuron), P45 rapamycin-treated mutant, and P45 rapamycin-treated until P30 then taken off drug (on/off) mutant, and P45 rapamycin-treated control, respectively. Scale bar, 50 μm. All images are Z-stacked 200× confocal images. Genotypes of mice were as follows: A, E, F, J, Tsc1c+ SynICre+; B, G, K, Tsc1c− SynICre+; C, D, H, I, L, Tsc1cc SynICre+. A–E, Nonphosphorylated neurofilament (SMI311) expression (red) and 4′,6′-diamidino-2-phenylindole (DAPI) (blue). F–J, Nonphosphorylated neurofilament (SMI311) expression (red) with pS6(S235/236) expression (green) and DAPI (blue) stains of lateral cortex. Note abnormal SMI311+ cells that are enlarged and dysplastic in the untreated mutant mice. These cells have reduced cell size but not degree of dysplasia in response to rapamycin. In mice taken off rapamycin, there is recurrence of cell enlargement. K, L, Higher-power view of cells from images G and H highlight the aberrant polarity of the apical dendrite of these neurons, despite the smaller size of the treated mutant neuron (L). Scale bar, 10 μm. Both images are Z-stacked 630×, 1.5× zoom confocal images. M, Cell size measurements in these same five categories of mice, from SMI311-stained lateral cortex. Cell size is indicated in square micrometers. Each point is the size of one SMI311-stained neuron, with eight neurons counted per mouse section. The average size and SEM are shown. Comparisons between each of the mutant and the on/off mutant and each of the control, treated mutant, and treated control are significant at p = 0.02 to <0.0001. All other comparisons have p > 0.05. Numbers and genotypes of mice were as follows: Mutant, four Tsc1c− SynICre+; Control, four Tsc1c+ SynICre+; Mutant + Rap, three Tsc1cc SynICre+; Mutant + Rap on/off, two Tsc1cc SynICre+; Control + Rap, two Tsc1c+ SynICre+. N, Cortical section indicates region of cortex and orientation shown in A–J.
Thus, rapamycin was very effective at reducing cell size in Tsc1null-neuron mice. However, despite this drastic reduction in cell size, rapamycin treatment appeared to have little effect on the dysplastic features of the neurons in this model (Fig. 3K,L). To examine this quantitatively, we assessed the orientation of the apical dendrite in SMI311+ layer V neurons in somatosensory cortex (Table 2). In control mice, nearly all (96%) neurons were polarized with a long apical dendrite that was oriented directly (±15°) toward the pial surface. In contrast, Tsc1null-neuron neurons often had major dendrites that extended tangentially and diagonally to the pia. In addition, rapamycin treatment initiated at P7 did not reduce the percentage of SMI311+ neurons with abnormally oriented dendrites in Tsc1null-neuron mice (Table 2, 38.8% without treatment, 43.5% with rapamycin treatment).
Table 2.
Apical dendrite orientation
| Genotype | Treatment | n sections/neurons | % abnormal orientation |
|---|---|---|---|
| Control | None | 6/304 | 4.2 ± 3.0 |
| Control | Rapamycin | 6/272 | 9.9 ± 2.5 |
| Mutant | None | 6/293 | 38.8 ± 3.5 |
| Mutant | Rapamycin | 4/199 | 43.5 ± 8.7 |
Mean ± SD are shown for the percentage of SMI311+ neurons in somatosensory cortex that have abnormal orientation (>15° angle of apical dendrite compared with a vertical line to the pia). Each control group is significantly different at p < 0.001 from each mutant group (one-way ANOVA with Tukey's correction). Neither the two control groups nor the two mutant groups are statistically significantly different from each other. Genotypes were all Tsc1c− SynICre+ for the untreated mutant group; all Tsc1cc SynICre+ for the rapamycin-treated mutants; and Tsc1c+ SynICre+ for all control mice.
Tsc1null-neuron mice have impaired myelination to the extent that the cerebral cortex of a P30 mouse had only a faint patchy myelin stain, consistent with reduced myelin synthesis by oligodendrocytes (Meikle et al., 2007) (Fig. 4B). Rapamycin treatment effectively restored myelination in the Tsc1null-neuron brain (Fig. 4 A–J; compare C with A, B; compare H with F, G). Although restoration of myelin was seen throughout the brain, the most dramatic improvement was seen in the cortex in which MBP+ myelin sheaths were evident coating radiating fibers extending out from the base of the cortex and in the pericallosal portion of the retrosplenial granular region (Fig. 4C). An improvement in myelination was also seen in the hippocampus (Fig. 4H). Double staining with pS6(S235/236) and MBP showed that there was a clear concordance between reduction in pS6 levels and restoration of myelin expression, as seen in the CA3 region of the hippocampus (Fig. 4 K–O). Despite reducing pS6 levels to a subnormal level, rapamycin appeared to have little effect on myelination in the treated controls (Fig. 2E,J,O).
Figure 4.

Improvement in myelination in Tsc1null-neuron mice treated with rapamycin. Each of the five columns of figures is taken from P45 control, P30 mutant (Tsc1null-neuron), P45 rapamycin-treated mutant, and P45 rapamycin-treated until P30 then taken off drug (on/off) mutant, and P45 rapamycin-treated control, respectively. A–E, Myelin stain of the retrosplenial granular region of cortex demonstrates that there is a marked reduction in myelination in untreated Tsc1null-neuron mice, which improves with treatment, and that myelination in the on/off mice also begins to look somewhat more patchy than in other groups. Scale bar, 100 μm. All images captured on Nikon fluorescence microscope at 60× and constant exposure. F–J, Myelin stain of the CA3 region of anterior hippocampus shows similar findings to those in cortex. Scale bar, 100 μm. All images captured on Nikon fluorescence microscope at constant exposure. K–O, Combined myelin (red)–pS6(S235/236) (green) stain of CA3 region of hippocampus demonstrates that there are enlarged pS6+ cells with reduced myelin in this region in untreated mutant mice, both of which improve with rapamycin treatment. pS6 expression and cell enlargement recur in the on/off treated mutant mice. Scale bar, 20 μm. All images are Z-stacked 630× confocal images. Genotypes of mice were as follows: A, E, F, J, K, O, Tsc1c+ SynICre+; B, G, L, Tsc1c− SynICre+; C, D, H, I, M, N, Tsc1cc SynICre+.
Biochemical and signaling effects of rapamycin treatment in the Tsc1null-neuron mice
Recent studies indicate that an important signaling effect in cells lacking Tsc1 or Tsc2 is a reduction in activation of Akt in response to normal stimuli (Zhang et al., 2003; Harrington et al., 2004; Shah et al., 2004). There has been speculation that this effect might have significant pathophysiological consequences in addition to that of mTORC1 activation in cells lacking Tsc1/Tsc2 (Manning et al., 2005). We assessed this possibility in brain extracts from the Tsc1null-neuron mice (Fig. 5A). Whereas pS6(S235/236) and pS6(S240/244) levels were significantly increased in the mutant mice, pAkt(S473) levels were reduced compared with controls. Moreover, rapamycin treatment led to restoration of pAkt(S473) levels, just as it decreased pS6 levels at both phosphorylation sites. Both of these effects were reversed when rapamycin treatment was discontinued. We also explored downstream signaling effects of this suppression of Akt signaling in the Tsc1null-neuron mice. pGSK3β(S9) levels were also reduced in the Tsc1null-neuron mice and were reversed by treatment with rapamycin (Fig. 5B), consistent with diminished signaling downstream of Akt.
Figure 5.
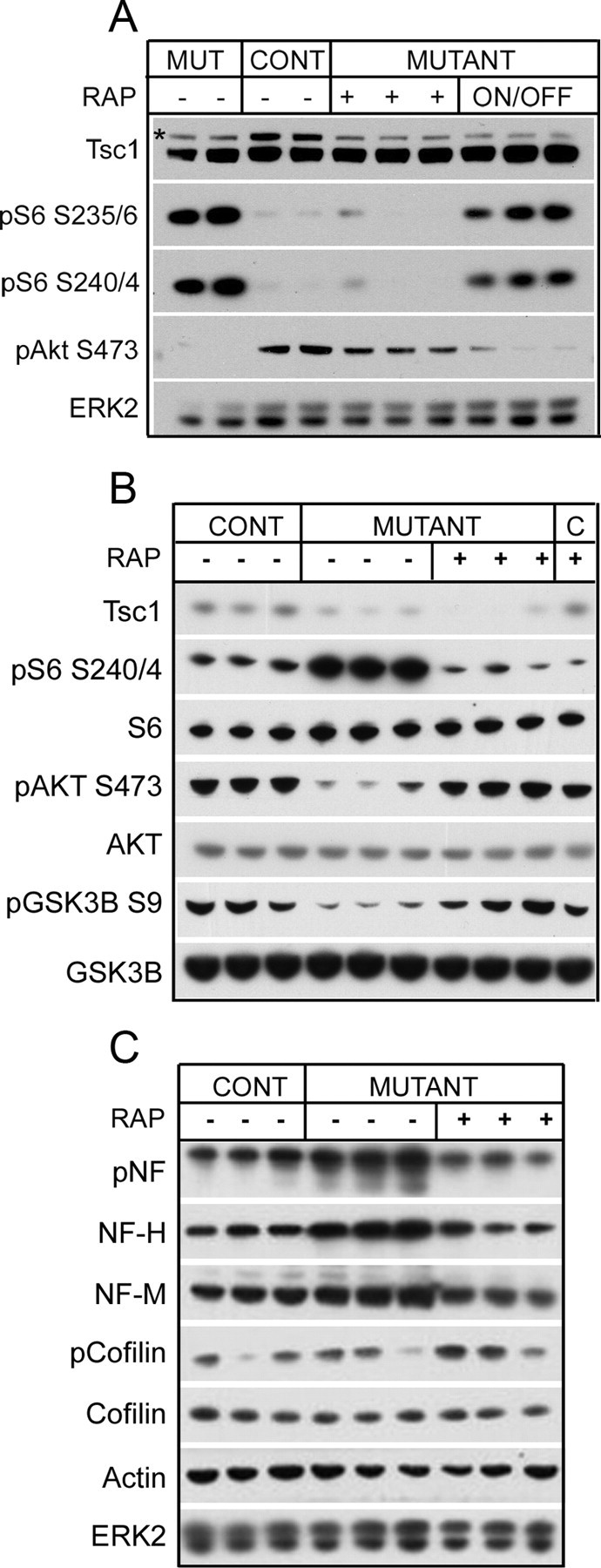
Signaling and cytoskeletal effects of rapamycin treatment in Tsc1null-neuron mice. A, Immunoblot analysis of brain lysates from P30 mutant (Tsc1null-neuron), P60 control, P60 rapamycin-treated mutant, and P60 rapamycin-treated until P30 then taken off drug (on/off). Note that treatment-induced changes in both pS6 levels and in pAkt S473 levels reverse when treatment is discontinued. * denotes Tsc1 protein band. All mutants were Tsc1c− SynICre+; both controls were Tsc1c+ SynICre+. B, C, Immunoblot analysis of brain lysates from P30 control, P30 mutant (Tsc1null-neuron), and P30 rapamycin-treated mutant and control (B) mice are shown. B, Note reduction in elevated pS6(S240/244) levels as well as an increase in reduced pS473–AKT and pS9–GSK3B levels of the mutant mice with treatment. C, In addition, abnormal increases in phosphorylated neurofilament (pNF) and neurofilament heavy (NF-H) and medium (NF-M) chains are reduced in response to rapamycin. Note that pCofilin(S3) levels are not consistently different among mutant and control mice. The untreated and treated controls were all Tsc1c+ SynICre+; untreated mutants were all Tsc1cc SynICre+; treated mutants were one Tsc1cc SynICre+ and two Tsc1c− SynICre+.
Cytoskeletal abnormalities have also been reported in cells and neurons lacking Tsc1/Tsc2 (Lamb et al., 2000; Goncharova et al., 2004). Levels of phosphorylated neurofilament, neurofilament heavy chain, and neurofilament medium chain were all increased in the Tsc1null-neuron mice compared with controls (Fig. 5C). Furthermore, these effects on the neuronal cytoskeleton were effectively reversed by rapamycin treatment. In contrast, we found no consistent evidence of significant alterations in pCofilin(S3) levels in the Tsc1null-neuron mice compared with controls (Fig. 5C).
Dendritic spine abnormalities in the Tsc1null-neuron mice
Because of a previous report of significant effects of loss of Tsc1 or Tsc2 on dendritic spine density, shape, and length in in vitro hippocampal slice cultures (Tavazoie et al., 2005), we examined dendritic spine morphology in the Tsc1null-neuron mice including in response to rapamycin treatment, using biolistics with DiI to label a small subset of cortical neurons. Confocal microscopy demonstrated that robust staining was achieved in select neurons (Fig. 6A). Quantitative analysis of spine density and length indicated that dendrites of cortical neurons from Tsc1null-neuron mice had a significant (p < 0.05), 22% reduction in spine density compared with neuronal dendrites from control mice (Fig. 6B,C, Table 3). However, there was no significant difference in spine length in neurons from these two types of mice. In response to rapamycin treatment of the Tsc1null-neuron mice, there was a small (10%) increase in spine density (p value was nonsignificant) toward a normal density. In addition, there was an 9% increase in spine length in the rapamycin-treated Tsc1null-neuron mice compared with both mutant and control mice (Table 3, p ≤ 0.002).
Figure 6.

Dendritic spines in Tsc1null-neuron brains and response to rapamycin. A–E, Confocal images of DiI-stained P23–P30 brain sections are shown. All are Z-stacked. A, C, and D were Tsc1cc SynICre+; B and E were Tsc1c+ SynICre+. A, Representative pyramidal neuron with apical dendrite. Scale bar, 10 μm. 200× confocal image with 1.5 zoom. B–E, Dendrite segments from control, mutant (Tsc1null-neuron), rapamycin-treated mutant, and rapamycin-treated control mice. Scale bar, 2 μm. 630× confocal images with 1.5 zoom.
Table 3.
Spine density and length measurements
| Genotype | Treatment | n cells/spines | Density (μm−1) | Length (μm) |
|---|---|---|---|---|
| Control | None | 16/1105 | 1.73 ± 0.14a | 1.38 ± 0.02c |
| Control | Rapamycin | 19/1742 | 1.73 ± 0.12b | 1.38 ± 0.02d |
| Mutant | None | 17/1524 | 1.35 ± 0.10a,b | 1.39 ± 0.02e |
| Mutant | Rapamycin | 10/856 | 1.49 ± 0.12 | 1.51 ± 0.03c,d,e |
a,bPairwise comparison p < 0.05 by both unpaired t and Mann–Whitney tests. All other comparisons in this column are not significant.
c,d,ePairwise comparison p < 0.002 by both unpaired t and Mann–Whitney tests. All other comparisons in this column are not significant.
Discussion
The Tsc1null-neuron mice studied here replicate several of the clinical and pathologic features seen in TSC patients (Huttenlocher and Wollmann, 1991; Crino, 2004; Zikou et al., 2005; Ess, 2006; Marcotte and Crino, 2006; Holmes and Stafstrom, 2007). There are enlarged and ectopic cells, with prominent dysplasia, and high level expression of pS6, as well as reduced myelination (Meikle et al., 2007). The mice show a progressive neurologic phenotype with tremor, hyperactivity, poor weight gain, seizure tendency, and limited survival. The current work demonstrates the marked therapeutic benefit of both rapamycin and RAD001 to effect both dramatic clinical and substantial histologic improvement in this TSC model. Mice treated at 6 mg/kg intraperitoneally every other day with either drug enjoyed survival out past 100 d in the vast majority of mice, with persistent improvement in clinical phenotype, weight gain, and behavior and complete absence of spontaneous clinical seizures. This study provides the first evidence that rapamycin/RAD001 can induce significant physiologic improvement in vivo through effects on postmitotic cells, in this case, neurons that are lacking Tsc1. Because loss of Tsc1 leads to reduced stability and functional loss of Tsc2 (Chong-Kopera et al., 2006; Meikle et al., 2007) and cells lacking either Tsc1 or Tsc2 have identical activation of mTORC1 (Zhang et al., 2003; Inoki et al., 2005), rapamycin would very likely have similar benefit in a Tsc2 neuronal model. It is notable that similar therapeutic benefit with reduction in cell size has been seen using CCI-779 [42(3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate)rapamycin], a rapamycin prodrug, in the treatment of a mouse brain model in which the phosphatase and tensin homolog PTEN is deleted (Kwon et al., 2003).
We explored several aspects of brain pathology in these mice to attempt to determine the cause of the clinical improvement that was seen. A reduction in cell size, improvement in biochemical and signaling profiles, reduction in neurofilament expression and phosphorylation, and markedly improved myelination were all seen. Strikingly, major clinical benefit persisted for several weeks when drug treatment was discontinued. Whereas cell enlargement and biochemical and signaling profiles reverted to their pretreatment patterns within 2 weeks (Figs. 2H, 3I, 4N, 5A), myelination remained intact. It is therefore likely that improved myelination played a major role in the reduction in tremor, ataxia, and spasticity seen in the treated mutant mice. As shown previously (Meikle et al., 2007), this defect in myelination is not attributable to abnormal oligodendrocyte number or distribution, but rather there is a neuronal inductive defect, which as shown here is responsive to rapamycin/RAD001 treatment. Although the precise mechanism requires additional study, it is likely attributable to overactive mTORC1.
In contrast to the many features that were improved in this model in response to treatment, neuronal dysplasia and neuronal migration were both unchanged (Fig. 3K,L, Table 2). This is consistent with completion of neuronal differentiation and cortical migration before institution of rapamycin/RAD001 therapy at P7–P9. It is possible that earlier treatment with either compound might reduce neuronal dysplasia, but any benefit might be offset by other growth and developmental consequences of mTORC1 blockade.
Although spine density was significantly reduced (22%) in the Tsc1null-neuron mice, there was no significant change in spine length or morphology (qualitative observations only) in these mice compared with controls. In response to rapamycin treatment, there was only a modest increase (10%) in spine density and a corresponding increase (9%) in spine length above normal, suggesting that these dendritic morphologic abnormalities had little direct importance for neuronal function in this model. Conversely, phosphorylated neurofilament, neurofilament heavy chain, and neurofilament medium chain levels were all increased in the Tsc1null-neuron mice and were reversed by rapamycin treatment (Fig. 5B). In contrast to a previous report from in vitro slice cultures (Tavazoie et al., 2005), we saw no significant change in pCofilin(S3) levels in brain extracts from the Tsc1null-neuron mice, suggesting that this actin regulatory protein had little to do with the in vivo phenotype produced by loss of Tsc1 in neurons.
Our pharmacokinetic studies demonstrate that rapamycin/RAD001 brain levels are approximately one-third that of systemic levels at 48 h after the last dose, in both acute and chronic treatment paradigms (Fig. 1A,B,D,E). These findings are consistent with the clear treatment benefit noted. The buildup of each drug that is seen over time in the brain may serve as a reservoir for slow release when treatment is discontinued. This phenomenon may help to explain the prolonged symptom-free interval and survival seen after drug withdrawal at P30 in the treated mice. Although rapamycin/RAD001 levels achieved in these mice were significantly higher than are typically sought in patients, it is notable that a lower dose of drug could have been used to achieve both low therapeutic range brain levels (∼5 ng/g) and concurrent high therapeutic range plasma levels (15–20 ng/ml). This is consistent with more limited studies we have performed, in which both rapamycin and RAD001 at 1 or 3 mg/kg given intraperitoneally three times per week led to clear therapeutic benefit in this model.
Loss of TSC1/TSC2 is now well known to lead to constitutive elevation of rheb-GTP levels and consequent constitutive activation of mTORC1, which causes transcriptional effects to influence cell size increase and growth by phosphorylation and activation of S6 kinase and phosphorylation and inactivation of eIF-4E binding protein 1. In addition to these direct or “downstream” effects, TSC1/TSC2 loss also leads to indirect (also termed “feedback” or “upstream”) effects that limit the phosphorylation and activation of AKT (Zhang et al., 2003; Harrington et al., 2004; Shah et al., 2004). This is the first work to demonstrate these complex effects of loss of Tsc1/Tsc2 in the brain, with strong AKT downregulation seen concurrent with activation of mTORC1. Rapamycin/RAD001 had prominent activity in blocking both direct and indirect effects of mTORC1 activation, restoring Akt phosphorylation. Reduced AKT expression has been engineered in mice, although it is difficult because of the existence of three different AKT isoforms with varying expression levels in various tissues. Mice with significant (50% or more) reduction in brain AKT expression have a significant phenotype with microcephaly and reduced numbers and size of neurons, although neurologic and behavioral abnormalities have not been investigated in detail (Easton et al., 2005; Tschopp et al., 2005; Dummler et al., 2006). Reduced AKT expression leads to a major (80–90%) reduction in pAKT(S473) levels in these brains, whereas pTsc2 and pGSK3β levels were near normal (Dummler et al., 2006). As we see the opposite clinical phenotype in the Tsc1null-neuron mice, with brain and neuronal enlargement, it is clear that these potential effects of AKT downregulation are overcome by the effects of mTORC1 activation in the Tsc1null-neuron mice. Nonetheless, as levels of pGSK3β were more reduced in the Tsc1null-neuron brains than in AKT-deficient brains (Dummler et al., 2006), it is possible that restoration of Akt function contributed significantly to the improvement in neurologic function seen in the Tsc1null-neuron mice in response to treatment. Significant concern has been raised by the possibility that elevation in pAKT(S473) might occur because of rapamycin/RAD001 treatment of malignancy, leading to a growth effect that could negate the potential benefits of mTORC1 blockade (O'Reilly et al., 2006). In this model, elevation of pAKT(S473) did occur in response to these drugs, concurrent with a marked phenotypic and histologic improvement, suggesting that it contributed to rather than impeded the clinical response.
Finally, given the similarities between the cellular and pathological abnormalities seen in this model and cortical tubers (Meikle et al., 2007), these findings suggest the possibility that rapamycin/RAD001 might have clinical benefit in the treatment of TSC patients. Indeed, rapamycin has been shown to have significant benefit, with shrinkage in size of TSC subependymal giant cell tumors (Franz et al., 2006). In addition, the brain penetration shown here in P10 mice suggests that rapamycin would also penetrate the CNS at high levels in infants. Therefore, these drugs may possibly have benefit in the treatment of TSC-associated infantile spasms, often a difficult clinical problem (Curatolo et al., 2006; Holmes and Stafstrom, 2007). Because similar although not identical histologic features, including evidence of mTORC1 activation and alteration of neurofilament expression, are seen in focal cortical dysplasias (FCD) (Taylor et al., 2001; Baybis et al., 2004; Miyata et al., 2004; Ljungberg et al., 2006), rapamycin may possibly be of benefit in the treatment of neurological manifestations associated with FCD as well. However, it is important to note that this model does not replicate the focal nature of cortical tubers/FCD, nor their full spectrum of abnormal cell types, including giant/balloon cells, so that translation of these findings to patients should be considered carefully. In addition, potential major side effects of rapamycin/RAD001 in infants and young children, including effects on growth as seen here in mice that began treatment at P7, also mandates a cautious approach to the investigation of the potential clinical translation of these findings.
Note added in proof.
We draw the reader's attention to concurrent work published recently by Zeng et al. (2008). These investigators report that rapamycin is very effective in the treatment of a mouse astrocyte model of TSC based on this same Tsc1 conditional allele (Uhlmann et al., 2002) with marked improvement in survival, epilepsy, and brain pathology.
Footnotes
This work was supported by the National Institutes of Health–National Institute of Neurological Disorders and Stroke Grants NS024279 (D.J.K.) and NS058956 (M.S.), the Tuberous Sclerosis Alliance (L.M., M.S., D.J.K.), and Novartis (D.J.K.). We thank Lihong Bu and the Children's Hospital Boston Mental Retardation and Developmental Disabilities Research Center for assistance with confocal microscopy (supported by National Institutes of Health Grant P01HD18655), Victoria Walker and Hongbing Zhang for technical assistance, Elena Pasquale (Burnham Institute for Medical Research, La Jolla, CA) for assistance with diolistic labeling, and Ann Brown for determination of RAD001 levels. RAD001 is a proprietary compound of Novartis AG.
References
- Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP, Delgado MR, Wheless JW, Baumgartner JE, Roa BB, Wilson CM, Smith-Knuppel TK, Cheung MY, Whittemore VH, King TM, Northrup H. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. 2007;9:88–100. doi: 10.1097/gim.0b013e31803068c7. [DOI] [PubMed] [Google Scholar]
- Avruch J, Hara K, Lin Y, Liu M, Long X, Ortiz-Vega S, Yonezawa K. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene. 2006;25:6361–6372. doi: 10.1038/sj.onc.1209882. [DOI] [PubMed] [Google Scholar]
- Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhann G, II, Aronica E, Crino PB. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56:478–487. doi: 10.1002/ana.20211. [DOI] [PubMed] [Google Scholar]
- Bilanges B, Argonza-Barrett R, Kolesnichenko M, Skinner C, Nair M, Chen M, Stokoe D. Tuberous sclerosis complex proteins 1 and 2 control serum-dependent translation in a TOP-dependent and -independent manner. Mol Cell Biol. 2007;27:5746–5764. doi: 10.1128/MCB.02136-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Cheadle JP, Reeve MP, Sampson JR, Kwiatkowski DJ. Molecular genetic advances in tuberous sclerosis. Hum Genet. 2000;107:97–114. doi: 10.1007/s004390000348. [DOI] [PubMed] [Google Scholar]
- Chong-Kopera H, Inoki K, Li Y, Zhu T, Garcia-Gonzalo FR, Rosa JL, Guan KL. TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J Biol Chem. 2006;281:8313–8316. doi: 10.1074/jbc.C500451200. [DOI] [PubMed] [Google Scholar]
- Crino PB. Molecular pathogenesis of tuber formation in tuberous sclerosis complex. J Child Neurol. 2004;19:716–725. doi: 10.1177/08830738040190091301. [DOI] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- Curatolo P. Cambridge, UK: Mac Keith; 2003. Tuberous sclerosis complex: from basic science to clinical phenotypes. [Google Scholar]
- Curatolo P, Bombardieri R, Cerminara C. Current management for epilepsy in tuberous sclerosis complex. Curr Opin Neurol. 2006;19:119–123. doi: 10.1097/01.wco.0000218225.50807.12. [DOI] [PubMed] [Google Scholar]
- Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, Choy YS, Reeve MP, Thiele E, Egelhoff JC, Kasprzyk-Obara J, Domanska-Pakiela D, Kwiatkowski DJ. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68:64–80. doi: 10.1086/316951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty C, Goh S, Young Poussaint T, Erdag N, Thiele EA. Prognostic significance of tuber count and location in tuberous sclerosis complex. J Child Neurol. 2005;20:837–841. doi: 10.1177/08830738050200101301. [DOI] [PubMed] [Google Scholar]
- Dummler B, Tschopp O, Hynx D, Yang ZZ, Dirnhofer S, Hemmings BA. Life with a single isoform of Akt: mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol. 2006;26:8042–8051. doi: 10.1128/MCB.00722-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ess KC. The neurobiology of tuberous sclerosis complex. Semin Pediatr Neurol. 2006;13:37–42. doi: 10.1016/j.spen.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopoulos A, Thomas G, Crone KR. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–498. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- Gomez M, Sampson J, Whittemore V, editors. Oxford: Oxford UP; 1999. The tuberous sclerosis complex, Ed 3. [Google Scholar]
- Goncharova E, Goncharov D, Noonan D, Krymskaya VP. TSC2 modulates actin cytoskeleton and focal adhesion through TSC1-binding domain and the Rac1 GTPase. J Cell Biol. 2004;167:1171–1182. doi: 10.1083/jcb.200405130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharova EA, Goncharov DA, Eszterhas A, Hunter DS, Glassberg MK, Yeung RS, Walker CL, Noonan D, Kwiatkowski DJ, Chou MM, Panettieri RA, Jr, Krymskaya VP. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation: a role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM) J Biol Chem. 2002;277:30958–30967. doi: 10.1074/jbc.M202678200. [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, Gout I, Downes CP, Lamb RF. The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Holmes GL, Stafstrom CE. Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsia. 2007;48:617–630. doi: 10.1111/j.1528-1167.2007.01035.x. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR, Wollmann RL. Cellular neuropathology of tuberous sclerosis. Ann NY Acad Sci. 1991;615:140–148. doi: 10.1111/j.1749-6632.1991.tb37756.x. [DOI] [PubMed] [Google Scholar]
- Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- Jones AC, Shyamsundar MM, Thomas MW, Maynard J, Idziaszczyk S, Tomkins S, Sampson JR, Cheadle JP. Comprehensive mutation analysis of TSC1 and TSC2 and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet. 1999;64:1305–1315. doi: 10.1086/302381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, Onda H. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11:525–534. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci USA. 2003;100:12923–12928. doi: 10.1073/pnas.2132711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb RF, Roy C, Diefenbach TJ, Vinters HV, Johnson MW, Jay DG, Hall A. The TSC1 tumour suppressor hamartin regulates cell adhesion through ERM proteins and the GTPase Rho. Nat Cell Biol. 2000;2:281–287. doi: 10.1038/35010550. [DOI] [PubMed] [Google Scholar]
- Ljungberg MC, Bhattacharjee MB, Lu Y, Armstrong DL, Yoshor D, Swann JW, Sheldon M, D'Arcangelo G. Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann Neurol. 2006;60:420–429. doi: 10.1002/ana.20949. [DOI] [PubMed] [Google Scholar]
- Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005;19:1773–1778. doi: 10.1101/gad.1314605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte L, Crino PB. The neurobiology of the tuberous sclerosis complex. Neuromolecular Med. 2006;8:531–546. doi: 10.1385/NMM:8:4:531. [DOI] [PubMed] [Google Scholar]
- Meikle L, McMullen JR, Sherwood MC, Lader AS, Walker V, Chan JA, Kwiatkowski DJ. A mouse model of cardiac rhabdomyoma generated by loss of Tsc1 in ventricular myocytes. Hum Mol Genet. 2005;14:429–435. doi: 10.1093/hmg/ddi039. [DOI] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata H, Chiang AC, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol. 2004;56:510–519. doi: 10.1002/ana.20234. [DOI] [PubMed] [Google Scholar]
- Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB. Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat Neurosci. 2003;6:153–160. doi: 10.1038/nn994. [DOI] [PubMed] [Google Scholar]
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C, Maat-Kievit A, Zonnenberg B, Verhoef S, Halley D, van den Ouweland A. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype-phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet. 2005;13:731–741. doi: 10.1038/sj.ejhg.5201402. [DOI] [PubMed] [Google Scholar]
- Schuler W, Sedrani R, Cottens S, Häberlin B, Schulz M, Schuurman HJ, Zenke G, Zerwes HG, Schreier MH. SDZ RAD, a new rapamycin derivative: pharmacological properties in vitro and in vivo. Transplantation. 1997;64:36–42. doi: 10.1097/00007890-199707150-00008. [DOI] [PubMed] [Google Scholar]
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Sater R, French J, Baltuch G, Crino PB. Transcription of intermediate filament genes is enhanced in focal cortical dysplasia. Acta Neuropathol. 2001;102:141–148. doi: 10.1007/s004010000348. [DOI] [PubMed] [Google Scholar]
- Tee AR, Blenis J. mTOR, translational control and human disease. Semin Cell Dev Biol. 2005;16:29–37. doi: 10.1016/j.semcdb.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Tschopp O, Yang ZZ, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, Michaelis T, Frahm J, Hemmings BA. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development. 2005;132:2943–2954. doi: 10.1242/dev.01864. [DOI] [PubMed] [Google Scholar]
- Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, Yamada K, Gutmann DH. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- Winterkorn EB, Pulsifer MB, Thiele EA. Cognitive prognosis of patients with tuberous sclerosis complex. Neurology. 2007;68:62–64. doi: 10.1212/01.wnl.0000250330.44291.54. [DOI] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, Kwiatkowski DJ. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003;112:1223–1233. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikou A, Ioannidou MC, Tzoufi M, Astrakas L, Argyropoulou MI. Magnetization transfer ratio measurements of the brain in children with tuberous sclerosis complex. Pediatr Radiol. 2005;35:1071–1074. doi: 10.1007/s00247-005-1539-y. [DOI] [PubMed] [Google Scholar]