

Abstract
Organ specific metastasis might be based on the specific interactions between chemokines expressed in premetastatic sites and their receptors on tumor cells. The ligand/receptor system in host defense mechanism pertinent to immune cells like macrophages is supposed to be hijacked by tumor cells. Ectopic expression of receptors in tumor cells enables bidirectional signaling between primary tumors and distant metastatic organs. VEGF and TNFα secreted from primary tumors signal through circulatory system to stimulate lung endothelial cells and macrophages to enhance production of S100A8 and A9 as well as MIP-1α, which in turn stimulate primary tumor cells as well as macrophages in bone marrow to migrate over to the lungs presumably via local chemokine gradient. Although it is beyond discussion to determine which came first, tumor cells or macrophages, the bidirectional signals could amplify the migration of both cells to accomplish metastasis.
Key Words: premetastasis, S100A8, S100A9, chemokine, VEGF, LLC
Metastasis has a multifactorial nature involving physical factors such as circulatory pathways and factors derived from biological properties of both tumor cells and host cells. It consists of at least four steps: (1) loss of adhesion in primary tumors and invasion to surrounding normal tissues, (2) intravasation, (3) extravasation into metastatic sites, and (4) regrowth. Growth factor receptors for EGF, HGF, and TGFβ and VEGF induce epithelial mesenchymal transition promoting the steps 1 and 2. Selective pressure from the tumor microenvironment such as hypoxia or contacts with extracellular matrix is supposed to promote dominance of metastatic subclones with activated signaling through those receptors.1
Historically, much attention has been paid to metastasis gene(s) in tumor cells. For example, differences in expression levels revealed NDP kinase Nm23, RhoC and chemokine receptors. Given the complicated biological process in metastasis, experimental dissections would provide better understanding of each step. One idea is to utilize subclones of tumor cells with differential biological abilities2,3 as represented by the discovery of the transcription factor Twist.2 Another idea is a procedure-based dissection that focuses on host cells instead of tumor cells.4 We subcutaneously transplant nonmetastasizing tumor cells LLC or B16 in mice, which grow up as primary tumors in two weeks. Then, by skipping the above-mentioned steps 1 and 2, the tumor cells are directly injected via tail veins into circulation to promote lung metastasis. We could monitor transcriptional changes in premetastatic lungs and eventually found that S100A8 and S100A9 are upregulated at metastatic sites.4
Injection into tail veins forces tumor cells to encounter lungs as the first organ. However, injection into the portal vein failed to enhance metastasis in liver although S100A8 and A9 are transcriptionally activated. (Hiratsuka S, Maru Y, unpublished results). Given that subcutaneously transplanted LLC and 3LL, a highly metastatic subclone of LLC, spontaneously metastasize to lungs in a long and short period of time, respectively, it can safely be said that LLC has organ specificity in metastasis. Then what determines tumor and metastatic site pairing? The most understandable idea is paired expression of growth factor in metastatic sites and its receptor in tumor cells, as evidenced in CXCR4-expressing breast cancer cells vs. high CXCL12 (SDF-1) in lungs.5 The chemokines promote lymphocyte homing to specific sites in the body. In our metastasis assays, LLC and 3LL but not benign tumor cells induced mRNA upregulation of chemokines S100A8, A9, and MIP-1α but not SDF-1 or MCP-1. However, anti-chemokine antibody therapy gave roughly 20% reduction in metastasis against MIP-1α vs. 80% against S100A8 in five hours. In some diabetic conditions, upregulated S100A8 causes enhanced adhesion of macrophages to fibronectin.6 This is consistent with what Kaplan et al. proposed in the initiation of premetastatic niche.7 Another example of strict pairing is observed in RANK-expressing melanoma vs. RANKL production in osteoblasts.8 RANK is physiologically expressed in osteoclast progenitor cells and RANKL is an osteoclast differentiation factor essential in bone physiology. Given the high specificity of those ligand/receptor interations, metastatic preference to certain organs is not a matter of chance but tumor cells might be destined to be recruited to metastatic sites if they pass through the steps 1 and 2.
To overcome the steps 3 and 4, tumor cells homing to metastatic sites need to invade and restart growing without angiogenesis at the beginning. CXCL12 can activate Pyk2 and MAP kinase to induce cell proliferation.9 S100A8 and A9 belong to the divalent cation-binding EF-hand family of proteins and appear to function both intracellularly and extracellularly as proinflammatory chemokines.10 One major activity inside the cells would be superoxide production as a regulator of Nox2.11 Once released outside the cells, they bind endothelial cells via glycans,12 induce activation of NFκB, p38, and MAP kinases in some tumor cells,13 or could be mitogenic in some fibroblasts,14 although no specific receptor has been found so far.
We repeatedly observed that both S100A8 and A9 induce migration of macrophages as well as tumor cells by signaling through p38 kinase. Whatever their receptors are, noteworthy is the activation of macrophages, which is highly augmented in S100A8- or A9-incubated conditioned medium of organ culture of lung slices. S100A8 appears to induce other chemokine(s) in lungs more potent to confer invasive and migrating abilities on tumor cells. This suggests that both S100A8 and A9 could potentially amplify macrophage-migrating activity within the premetastatic pulmonary microenvironment. Since anti-VEGF neutralizing antibody could partially block S100A8 and A9 induction in lungs, at least VEGF produced in primary tumors could induce their expression. S100A8 and A9 might then induce another potent chemokine(s) in lungs, mobilizing macrophages from bone marrow. Since VEGF but not chemokines appears to play an essential role in endothelial progenitor cell migration from bone marrow15 and increment in the number of macrophages but not endothelial cells in lungs is evident in our assays, it is likely that at least the steps 3 and 4 in metastasis are dictated by chemokines and the late step 4 by VEGF.
Based on the “seed and soil” hypothesis in metastasis, our finding suggests that lungs are cultivated by tumor challenges to express seed-guiding and -rearing chemokines S100A8 and S100A9 in endothelial cells and macrophages. This raises a fundamental question of whether or not primary tumors really make a remote operation for metastasis. Given that macrophages are mobilized from bone marrow to pulmonary microenvironment by the insult of development of primary tumors, one idea could be that host defense mechanisms are hijacked by tumor cells. Macrophages are supposed to express receptors, if any, for S100A8, S100A9, or other unknown chemokines secreted in a paracrine fashion in lungs. If tumor cells express the same receptor(s), tumor cells and defending macrophages should be simultaneously recruited to the chemokine-producing microenvironment. Lungs may be the most sensitive organ for attacks from exogenous pathogens that might be inhaled on each respiration or endogenous tumor cells as it is the first organ for circulating tumor cells to encounter in the body. This could partly explain the high expression levels of S100A8 in lungs but not in liver or kidney where metastasis failed to take place. The trigger of lung metastasis might be a very small number of tumor cells that migrate to lungs. That could be beyond detection by any experimental ways but induce chemokine production in that microenvironment for macrophage mobilization from bone marrow. Recruited macrophages in lungs also secrete chemokines and an amplification mechanism begins (Fig. 1). Alternatively, defending macrophages are recruited to lungs before arrival of any single tumor cell, similar to the question of which came first, the chicken or the egg. For prevention of metastasis, it is important to elucidate circulating chemokines and their receptors on tumor cells in each cancer.
Figure 1.
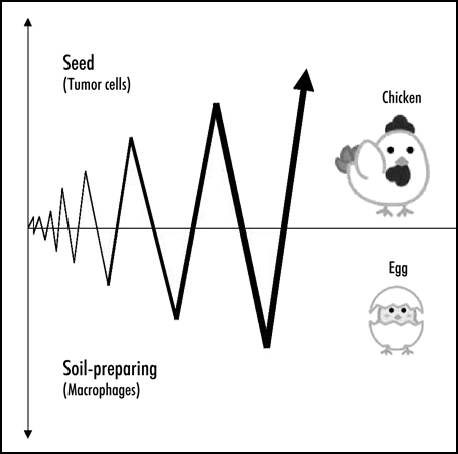
Both tumor cells and macrophages express receptors for chemokines produced in premetastatic organs. Migration could be amplified by bidirectional signaling between the two. Macrophages could be soil-carrying seeds.
Abbreviations
- VEGF
vascular endothelial growth factor
- EGF
epidermal growth factor
- HGF
hepatocyte growth factor
- TNF
tumor necrosis factor
- TGF
transforming growth factor
- LLC
Lewis lung carcinoma
- MIP
macrophage inflammatory protein
- SDF
stromal cell derived factor
- RANKL
receptor activator of NFκB ligand
- RAGE
receptor for advanced glycation end products
- MCP
monocyte chemoattractant protein
Footnotes
Previously published online as a Cell Adhesion & Migration E-publication: http://www.landesbioscience.com/journals/celladhesion/article/4489
References
- 1.Anderson ARA, Weaver AM, Cummings PT, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell. 2006;127:905–915. doi: 10.1016/j.cell.2006.09.042. [DOI] [PubMed] [Google Scholar]
- 2.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massagua J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8:1369–1375. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 5.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 6.Bouma G, Lam-Tse WK, Wierenga-Wolf AF, Drexhage HA, Versnel MA. Increased serum levels of MRP-8/14 in type 1 diabetes induce an increased expression of CD11b and an enhanced adhesion of circulating monocytes to fibronectin. Diabetes. 2004;53:1979–1986. doi: 10.2337/diabetes.53.8.1979. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the premetastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, Morony S, Rubin E, Sarao R, Hojilla CV, Komnenovic V, Kong YY, Schreiber M, Dixon SJ, Sims SM, Khokha R, Wada T, Penninger JM. Regulation of cancer cell migration and bone metastasis by RANKL. Nature. 2006;440:692–696. doi: 10.1038/nature04524. [DOI] [PubMed] [Google Scholar]
- 9.Florio T, Casagrande S, Diana F, Bajetto A, Porcile C, Zona G, Thellung S, Arena S, Pattarozzi A, Corsaro A, Spaziante R, Robello M, Schettini G. Chemokine Stromal Cell-derived factor 1 induces proliferation and growth hormone release in GH4C1 rat pituitary adenoma cell line through multiple intracellular signals. Mol Pharmacol. 2006;69:539–546. doi: 10.1124/mol.105.015255. [DOI] [PubMed] [Google Scholar]
- 10.Ryckman C, Vandal K, Rouleau P, Talbot M, Philippe A. Tessier. Proinflammatory activities of S100: Proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol. 2003;170:3233–3242. doi: 10.4049/jimmunol.170.6.3233. [DOI] [PubMed] [Google Scholar]
- 11.Kerkhoff C, Nacken W, Benedyk M, Dagher MC, Sopalla C, Doussiere J. The arachidonic acid-binding protein S100A8/A9 promotes NADPH oxidase activation by interaction with p67phox and Rac-2. FASEB J. 2005;19:467–469. doi: 10.1096/fj.04-2377fje. [DOI] [PubMed] [Google Scholar]
- 12.Robinson MJ, Tessier P, Poulsom R, Hogg N. The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J Biol Chem. 2002;277:3658–3665. doi: 10.1074/jbc.M102950200. [DOI] [PubMed] [Google Scholar]
- 13.Hermani A, Servi BD, Medunjanin S, Tessier PA, Mayer D. S100A8 and S100A9 activate MAP kinase and NF-κB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res. 2006;312:184–197. doi: 10.1016/j.yexcr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 14.Shibata F, Miyama K, Shinoda F, Mizumoto J, Takano K, Nakagawa H. Fibroblast growth-stimulating activity of S100A9 (MRP-14) Eur J Biochem. 2004;271:2137–2143. doi: 10.1111/j.1432-1033.2004.04129.x. [DOI] [PubMed] [Google Scholar]
- 15.Spring H, Schüler T, Arnold B, Hämmerling GJ, Ganss R. Chemokines direct endothelial progenitors into tumor neovessels. Proc Natl Acad Sci USA. 2005;102:18111–18116. doi: 10.1073/pnas.0507158102. [DOI] [PMC free article] [PubMed] [Google Scholar]