

Abstract
The presence of an impermeable surface barrier is an essential homeostatic mechanism in almost all living organisms. We have recently described a novel gene that is critical for the developmental instruction and repair of the integument in mammals. This gene, Grainy head-like 3 (Grhl3) is a member of a large family of transcription factors that are homologs of the Drosophila developmental gene grainy head (grh). Mice lacking Grhl3 fail to form an adequate skin barrier, and die at birth due to dehydration. These animals are also unable to repair the epidermis, exhibiting failed wound healing in both fetal and adult stages of development. These defects are due, in part, to diminished expression of a Grhl3 target gene, Transglutaminase 1 (TGase 1), which encodes a key enzyme involved in cross-linking of epidermal structural proteins and lipids into the cornified envelope (CE). Remarkably, the Drosophila grh gene plays an analogous role, regulating enzymes involved in the generation of quinones, which are essential for cross-linking structural components of the fly epidermis. In an extension of our initial analyses, we focus this report on additional defects observed in the Grhl3-null epidermis, namely defective extra-cellular lipid processing, altered lamellar lipid architecture and cellular hyperproliferation. These abnormalities suggest that Grhl3 plays diverse mechanistic roles in maintaining homeostasis in the skin.
Key Words: grainy head, Grhl3, skin barrier, lipids, proliferation
Evolutionary Parallels in the Function of Grh-Like Genes in Maintenance of the Surface Barrier
The formation of an impermeable surface barrier is a homeostatic mechanism common to most species. Mammals, terrestrial reptiles, amphibians, insects, and even xeric-adapted plants have developed divergent, lipid-based strategies to maintain internal fluid homeostasis. In Drosophila development, the epidermal epithelium deposits the embryonic cuticle, a rigid waterproof layer that is also essential for maintaining the structural integrity and microbial resistance of the organism. In mammalian skin development, the epidermis becomes multi-layered with the outermost stratum corneum (SC) layer providing the equivalent barrier functions. Despite the significant structural differences between the impermeable epidermal barriers of invertebrates and vertebrates, the mechanisms that establish and maintain these barriers have been evolutionarily conserved.1 This is evident in the studies of the Drosophila grh gene and a mammalian homolog, Grhl3, which both regulate enzymes essential for the cross-linking of proteins to lipids within the respective protective barrier layers. In the Drosophila cuticle, grh regulates Dopa decarboxylase (Ddc) and tyrosine hydroxylase (ple), enzymes involved in the generation of quinones, which are essential for the cross-linking process.2 In the outer epidermis of terrestrial mammals, Grhl3 regulates the cross-linking enzyme, TGase1.3 Remarkably, the enzymes regulated by the grh-like factors in flies and mammals are completely unrelated structurally, but still play the same functional role in fortifying the surface barrier. Thus, in evolutionary terms, the players are diverse, but the mechanism is highly conserved.
Grhl3-Null Stratum Corneum Reveals Defective Extracellular Lipid Organization
In addition to altered expression of TGase 1, the Grhl3-null epidermis also displays abnormalities of lipid processing. Ultrastructural analysis of Grhl3-/- epidermis using electron microscopy (EM) displayed a normal number of compressed, tightly interdigitated corneocyte layers, consistent with depletion of the intercorneocyte lipid mortar (Fig. 1A).3 Higher resolution examination of multiple EM epidermal sections revealed no obvious differences in either the number or structure of corneodesmosomes between wild type and mutant skin (data not shown). In contrast, morphometric analyses on corneocytes from wild type and Grhl3-/- embryos revealed that the CE thickness was significantly reduced (11.5%) in the mutant SC, consistent with loss of external lipids from the surface of this structure (Fig. 1B).3 Examination of E18.5 skin post-fixation, utilizing ruthenium tetroxide to visualize extracellular membrane domains in the wild type SC, revealed several alternating layers of electron-dense and electronlucent lipid lamellae between adjacent corneocytes (Fig. 1C).3 In contrast, the Grhl3-/- SC displayed a near absence of lamellar membranes, with only occasional fragments of replete lamellar arrays visualized (Fig. 1D).3 These marked differences in lamellar membrane structure could be, in turn, attributable to abnormalities in the internal contents of lamellar bodies (LBs). In wild-type epidermis, LBs showed characteristic multi-layered stacks (see inset of Fig. 1E).3 Although the quantity of LBs were essentially normal in Grhl3-/- epidermis, qualitatively the majority of these LBs lacked internal lamellar contents (Fig. 1E).3 These findings, coupled with the abnormality in CE thickness, suggest that the absence of Grhl3 results in gross defects in the processing of LB contents, required for assembly of the cornified lipid envelope and the inter-corneocyte lamellae of the SC.
Figure 1.
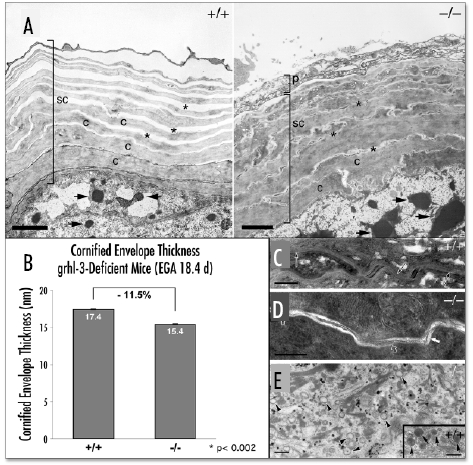
Grhl3-null epidermis displays abnormal lamellar body contents, and decreased stratum corneum membrane structures. (A) Ultrastructural analysis by electron microscopy of skin sections from E18.5 embryos. In the wild type skin (left panel, +/+), the stratum corneum (SC) has normal multiple layers of corneocytes (C) with normal EM artefact-induced, well-spaced intercellular layers (asterisk). In comparison, although the number of corneocyte layers is similar in mutant skin (right panel, -/-), the intercellular layers are essentially non-existent (asterisk). Keratohyaline granules (arrows) seem somewhat larger in the mutant skin. Scale bar = 2 µm (B) Morphometric analysis of cornified envelope thickness from multiple E18.5 wild type (+/+) and mutant (-/-) epidermis sections shows a significant decrease (11.5%) in the overall CE thickness in the mutant epidermis. (C) Focusing on the lipid structures shows that the E18.5 wild type (+/+) stratum corneum displays a full complement of extracellular lamellar membrane structures (arrows) and that the lamellar bodies show multiple layers of lipid containing lamellae (insert of panel E, arrows). Whereas, the extracellular spaces of the (D) mutant (-/-) stratum corneum displays a paucity of lamellar bilayers (open arrows), although occasional fragments of replete lamellar arrays are seen (solid arrow). (E) This is as a result of almost all the lamellar organelles lacking internal contents (arrowheads) in the mutant epidermis, even though there are a normal complement of lamellar bodies. Panels (A & E), osmium tetroxide post-fixation; Panels (C & D), ruthenium tetroxide post-fixation. Previously published in Science 2005; 308:411–3.
Cellular Hyperproliferation in the Grhl3-Null Epidermis
In comparison to normal murine epidermal development at E15.5 and E18.5 (Fig. 2, E15.5+/+ and E18.5+/+),3 the epidermal histology of Grhl3-/- embryos at both these time points was abnormal. At E15.5, the Grhl3-/- epidermis exhibited loss of ridging and a flattened appearance, but displayed no obvious disorganization of the individual cell layers (Fig. 2, E15.5-/-).3 In contrast, the E18.5 Grhl3-/- epidermis displayed compact hyperkeratosis of the SC, hyperproliferative spinous and granular layers, and a disorganized basal layer (Fig. 2, E18.5-/-).3 Immunofluorescence studies on Grhl3-/- epidermis with the basal cell-specific markers, keratin 5 and keratin 14 showed expression extending supra-basally into the spinous and granular layers. This was accompanied by a marked increase in the expression of the proliferative marker, Ki67 (data not shown). The epidermal hyperproliferation persisted when Grhl3-/- skin was grafted onto nude mice, suggesting that this defect was cell autonomous, rather than a secondary homeostatic response to loss of barrier function (data not shown).
Figure 2.
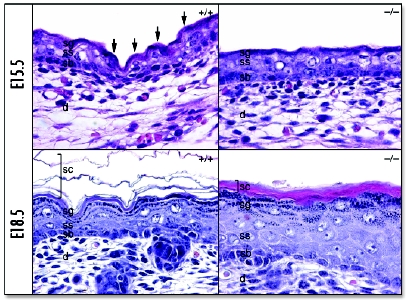
Abnormalities in differentiation and CE formation in Grhl3-null epidermis. Skin from wild type (+/+) and mutant (-/-) embryos taken at E15.5 and E18.5 as indicated. Epithelial ridges (arrows) in wild type skin at E15.5 (top left panel) are absent in the E15.5 mutant skin (top right panel). Basal disorganisation and suprabasal hyperproliferation and a compacted SC are evident in the E18.5 mutant skin (bottom right) compared to the wild type (bottom left). d, dermis; sb, stratum basale; ss, stratum spinosum; sg, stratum granulosum; sc, stratum corneum. Previously published in Science 2005; 308:411–3.
Discussion
The coordinated assembly of the structural proteins and lipids that comprise the CE and the intercellular lipid lamellae is essential for effective physical and water barrier function of the epidermis. The importance of lipid synthesis and post-secretory processing for formation of the skin barrier is evident from studies of human patients with genetic abnormalities of lipid synthetic or processing enzymes. For example, the most severe forms of Gaucher's disease also exhibit skin barrier abnormalities. This occurs due to loss of β-glucocerebrosidase, the enzyme that catalyses the hydrolysis of glucosylceramides to ceramides, which results in abnormal, incompletely processed lamellar body-derived sheets throughout the SC interstices.4 Linoleic acid is an important component of epidermal ceramides, and dietary insufficiency of this fatty acid in patients receiving intravenous alimentation can lead to ichthyosiform dermatitis.5 Several other acquired and inherited icthyoses are due to abnormal intercellular deposition of lipids, but the pathogenesis in many of these disorders remains unknown. One exception to this is lamellar ichthyosis, which is caused by mutations in TGase 1,6,7 which we have shown to be regulated by Grhl3.3
Several mouse mutants also have abnormal barrier formation in conjunction with defects in lipid processing. For example, skin-specific disruption of the Pig-a gene, which is responsible for GPI-anchor synthesis, leads to narrowing of the intercellular space due to reduced GPI-anchored protein-mediated ceramide transport.8 Affected mice died early in the neonatal period, presumably due to a barrier defect deriving from defects in the lipid (LBs) secretory system, although diminished filaggrin processing was also noted.9 In addition, wrinkle-free (wrfr), a spontaneous mouse mutant characterized by absent skin wrinkles and a restrictive dermopathy, is due to a mutation in the fatty acid transport protein gene, Fatp4.10 Targeted disruption of the Fatp4 gene revealed an equivalent, abnormal skin phenotype, due to production of ceramides with shortened fatty acid chains with an increased content of unsaturated fatty acids.11 Finally, mice deficient in Elovl3, a gene encoding a fatty acid elongation enzyme, also have defective epidermal permeability barrier function,12 further demonstrating the importance of lipid composition for effective barrier formation.
In this report, we demonstrate that altered processing of key lipids, with subsequent disruption of lamellar body structure is a likely contributing mechanism to the surface barrier defect in Grhl3-null mice. As a consequence, the intercellular lipid lamellae are largely absent in the mutant epidermis and the CEs display reduced thickness and an abnormal appearance, likely reflecting loss of cross-linked and externally bound lipid. Although the mechanisms underlying these defects are as yet unknown, our findings are consistent with a defect in lipid biosynthesis, transport and/or processing. We are currently examining a range of genes involved in these processes to determine whether they are direct target genes of Grhl3.
Another area of future investigation centres on the hyperproliferative defect observed in the Grhl3-null epidermis. A similar cell autonomous hyperproliferative response has been noted in another barrier defective mouse model, the Klf4-nullizygous mice.13 Accumulating evidence suggests that loss of this transcription factor leads to abnormal cellular proliferation, consistent with a tumor suppressor role for Klf4.14–18 We are currently exploring a similar hypothesis for Grhl3.
In conclusion, Grhl3 is indispensable for a functional mammalian epithelial barrier. Integral to this is the regulation of TGase 1 expression,3 although other potential Grhl3-dependent mechanisms, such as lipid processing and regulation of cellular proliferation are emerging. The integration of these mechanisms, coupled with the elucidation of the signalling cascades that control them,19,20 remain substantial challenges for the immediate future.
Acknowledgements
We thank members of the Jane lab for helpful discussions, and S. Vasudevan for technical assistance, and D. Crumrine for expert elecron microscopy work. Animal support was provided by staff from the Walter and Eliza Hall Institute. S.M.J. is a Principal Research Fellow of the Australian National Health and Medical Research Council (NHMRC). S.B.T. is a scholar of the Cancer Council of Victoria. J.M.C. is supported by the Cancer Center Support CORE Grant P30 CA 21765, the American Lebanese Syrian Associated Charities (ALSAC), and the Assisi Foundation of Memphis. This work was also supported by Project Grant 282400 from the NHMRC, and the National Institutes of Health (USA) through grants: PO1 HL53749-03 (J.M.C.) and PO1-AR39448 (W.M.H. and P.M.E.), and the Department of Veterans Affairs (P.M.E.).
Footnotes
Previously published online as an Organogenesis E-publication: http://www.landesbioscience.com/journals/organogenesis/abstract.php?id=2167 Once the issue is complete and page numbers have been assigned, the citation will change accordingly.
References
- 1.Jane SM, Ting SB, Cunningham JM. Epidermal impermeable barriers in mouse and fly. Curr Opin Genet Dev. 2005;15:447–453. doi: 10.1016/j.gde.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Mace KA, Pearson JC, McGinnis W. An epidermal barrier wound repair pathway in Drosophila is mediated by grainy head. Science. 2005;308:381–385. doi: 10.1126/science.1107573. [DOI] [PubMed] [Google Scholar]
- 3.Ting SB, Caddy J, Hislop N, Wilanowski T, Auden A, Zhao LL, Ellis S, Kaur P, Uchida Y, Holleran WM, Elias PM, Cunningham JC, Jane SM. A homolog of Drosophila grainy head is essential for epidermal integrity in mice. Science. 2005;308:411–413. doi: 10.1126/science.1107511. [DOI] [PubMed] [Google Scholar]
- 4.Holleran WM, Ginns EI, Menon GK, Grundmann JU, Fartasch M, McKinney CE, Elias PM, Sidransky E. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. J Clin Invest. 1994;93:1756–1764. doi: 10.1172/JCI117160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman Z. Essential fatty acid consideration at birth in the premature neonate and the specific requirement for preformed prostaglandin precursors in the infant. Prog Lipid Res. 1986;25:355–364. doi: 10.1016/0163-7827(86)90073-1. [DOI] [PubMed] [Google Scholar]
- 6.Huber M, Rettler I, Bernasconi K, Frenk E, Lavrijsen SP, Ponec M, Bon A, Lautenschlager S, Schorderet DF, Hohl D. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science. 1995;267:525–528. doi: 10.1126/science.7824952. [DOI] [PubMed] [Google Scholar]
- 7.Russell LJ, DiGiovanna JJ, Rogers GR, Steinert PM, Hashem N, Compton JG, Bale SJ. Mutations in the gene for transglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat Genet. 1995;9:279–283. doi: 10.1038/ng0395-279. [DOI] [PubMed] [Google Scholar]
- 8.Hara-Chikuma M, Takeda J, Tarutani M, Uchida Y, Holleran WM, Endo Y, Elias PM, Inoue S. Epidermal-specific defect of GPI anchor in Pig-a null mice results in Harlequin ichthyosis-like features. J Invest Dermatol. 2004;123:464–469. doi: 10.1111/j.0022-202X.2004.23227.x. [DOI] [PubMed] [Google Scholar]
- 9.Tarutani M, Itami S, Okabe M, Ikawa M, Tezuka T, Yoshikawa K, Kinoshita T, Takeda J. Tissue-specific knockout of the mouse Pig-a gene reveals important roles for GPI-anchored proteins in skin development. Proc Natl Acad Sci USA. 1997;94:7400–7405. doi: 10.1073/pnas.94.14.7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moulson CL, Martin DR, Lugus JJ, Schaffer JE, Lind AC, Miner JH. Cloning of wrinkle-free, a previously uncharacterized mouse mutation, reveals crucial roles for fatty acid transport protein 4 in skin and hair development. Proc Natl Acad Sci USA. 2003;100:5274–5279. doi: 10.1073/pnas.0431186100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herrmann T, van der Hoeven F, Grone HJ, Stewart AF, Langbein L, Kaiser I, Liebisch G, Gosch I, Buchkremer F, Drobnik W, Schmitz G, Stremmel W. Mice with targeted disruption of the fatty acid transport protein 4 (Fatp 4, Slc27a4) gene show features of lethal restrictive dermopathy. J Cell Biol. 2003;161:1105–1115. doi: 10.1083/jcb.200207080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westerberg R, Tvrdik P, Unden AB, Mansson JE, Norlen L, Jakobsson A, Holleran WH, Elias PM, Asadi A, Flodby P, Toftgard R, Capecchi MR, Jacobsson A. Role for ELOVL3 and fatty acid chain length in development of hair and skin function. J Biol Chem. 2004;279:5621–5629. doi: 10.1074/jbc.M310529200. [DOI] [PubMed] [Google Scholar]
- 13.Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999;22:356–360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 14.Katz JP, Perreault N, Goldstein BG, Actman L, McNally SR, Silberg DG, Furth EE, Kaestner K. H. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005;128:935–945. doi: 10.1053/j.gastro.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 15.Ohnishi S, Ohnami S, Laub F, Aoki K, Suzuki K, Kanai Y, Haga K, Asaka M, Ramirez F, Yoshida T. Downregulation and growth inhibitory effect of epithelial-type Kruppel-like transcription factor KLF4, but not KLF5, in bladder cancer. Biochem Biophys Res Commun. 2003;308:251–256. doi: 10.1016/s0006-291x(03)01356-1. [DOI] [PubMed] [Google Scholar]
- 16.Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of Kruppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene. 2004;23:395–402. doi: 10.1038/sj.onc.1207067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shie JL, Chen ZY, Fu M, Pestell RG, Tseng CC. Gut-enriched Kruppel-like factor represses cyclin D1 promoter activity through Sp1 motif. Nucleic Acids Res. 2000;28:2969–2976. doi: 10.1093/nar/28.15.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Johns DC, Geiman DE, Marban E, Dang DT, Hamlin G, Sun R, Yang VW. Kruppel-like factor 4 (gut-enriched Kruppel-like factor) inhibits cell proliferation by blocking G1/S progression of the cell cycle. J Biol Chem. 2001;276:30423–30428. doi: 10.1074/jbc.M101194200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harden N. Of grainy heads and broken skins. Science. 2005;308:364–365. doi: 10.1126/science.1112050. [DOI] [PubMed] [Google Scholar]
- 20.Stramer B, Martin P. Cell biology:master regulators of sealing and healing. Curr Biol. 2005;15:R425–R427. doi: 10.1016/j.cub.2005.05.034. [DOI] [PubMed] [Google Scholar]