

Abstract
Pharmacotherapy of CNS disorders, e.g., neurodegenerative diseases, epilepsy, brain cancer, and neuro-AIDS, is limited by the blood-brain barrier. P-glycoprotein, an ATP-driven, drug efflux transporter, is a critical element of that barrier. High level of expression, luminal membrane location, specificity and high transport potency make P-glycoprotein a selective gate-keeper of the blood-brain barrier and thus a primary obstacle to drug delivery into the brain. As such, P-glycoprotein limits entry into the CNS for a large number of prescribed drugs, contributes to the poor success rate of CNS drug candidates and likely contributes to patient-to-patient variability in response to CNS pharmacotherapy. Modulating P-glycoprotein could therefore improve drug delivery into the brain. Here we review the current understanding of signaling mechanisms responsible for the modulation of P-glycoprotein activity/expression at the blood-brain barrier with an emphasis on recent studies from our laboratories. Using intact brain capillaries from rats and mice, we have identified multiple extracellular and intracellular signals that regulate this transporter; several signaling pathways have been mapped. Three pathways are triggered by elements of the brain's innate immune response, one by glutamate, one by xenobiotic-nuclear receptor (PXR) interactions and one by elevated β-amyloid levels. Signaling is complex, with several pathways sharing common signaling elements (TNF-R1, ETB receptor, PKC, NOS), suggesting a regulatory network. Several pathways utilize autocrine/paracrine elements, involving release of the proinflammatory cytokine, TNF-α, and the polypeptide hormone, ET-1. Finally, several steps in signaling are potential therapeutic targets that could be used to modulate P-glycoprotein activity in the clinic.
I. Introduction
More than 98% of drug candidates for CNS disorders never make it to the clinic (Pardridge, 2007a). For most of these drugs, the major confounding issue is their inability to cross the blood-brain barrier at sufficient levels to have a therapeutic effect. This barrier resides within the brain's capillary endothelium and it has been an object of study for over 100 years. Research on the blood-brain barrier has occurred in several stages. Initial work focused on the barrier's physiological properties, i.e., the ability to prevent movement of solutes between blood and CNS. The morphological basis of the barrier was determined to be primarily the tight junctions that connect the endothelial cells. The molecular basis for the barrier's properties was explored as well as the involvement of specific transporters that increased or decreased solute permeability. Over the past several years, research on all of these aspects has continued within the context of the barrier as a dynamic tissue responding to changes in its environment and as part of a more complex neurovascular unit in which endothelial cells, astrocytes, pericytes and neurons interact.
It is in this context that the present review was written. It is focused on P-glycoprotein, the one blood-brain barrier transporter that is considered as the major obstacle to CNS entry of therapeutic drugs and is thus seen as the molecular basis for preclinical and clinical drug failure. Our emphasis in the present review is on the underlying mechanisms that modulate P-glycoprotein at the blood-brain barrier. We posit that an understanding of these mechanisms is important to provide new strategies for improving CNS pharmacotherapy and to appreciate how barrier properties change in disease.
II. The Blood-Brain Barrier
Although the vascular system penetrates every tissue of the body, blood vessels display a remarkable range of phenotypes with regard to structure, gene expression, function, cellular ultrastructure and blood-tissue exchange properties (Aird, 2007a; b). Indeed, even within a single organ the range of endothelial heterogeneity can be quite wide. This is certainly seen with regard to barrier properties of vessels within the central nervous system (CNS) where pial (surface) vessels present at most a moderate barrier, but cerebral microvessels (3-8 μm diameter) present a formidable barrier to macromolecules, small organic drugs and ions. These small vessels within the brain parenchyma constitute the blood-brain barrier. In man, their total length is estimated to be more than 600 km with a surface area of 10-30 m2 (Pardridge, 2003). This makes the blood-brain barrier the third largest discrete surface area for solute and water exchange after intestine and lung. However, as the name indicates, compared to capillaries in peripheral tissues, solute exchange between blood and brain is severely restricted and thus this barrier is a major impediment to CNS pharmacotherapy (Pardridge, 2007a). The mechanistic basis for restricted access of drugs to the CNS lies within the special properties of the cells that make up the brain capillary endothelium.
A. The Structural/Physical Barrier
The blood-brain barrier reflects the properties of two components (Begley, 2004; Hawkins and Davis, 2005; Loscher and Potschka, 2005). One forms a structural/physical barrier, comprised of the endothelial cells themselves and the extremely tight, intercellular junctional complexes that connect one cell to another. The structural barrier limits diffusion of solutes between blood and brain. For many solutes permeability is inversely related to size (most macromolecules have extremely low permeability) and directly related to lipophilicity. Indeed, for many small, uncharged molecules, in vivo blood-brain barrier passive permeability increases with octanol/water partition coefficient, likely indicating diffusion through the lipid-like core of cellular surface membranes (Smith, 2003).
Current research is focused on several strategies designed to circumvent the physical barrier: hyperosmotic blood-brain barrier opening, barrier opening using alkyl glycerols and drug delivery using surface-modified liposomes and nanoparticles, which hijack endothelial cell transcytotic mechanisms (Tiwari and Amiji, 2006). All have been shown to work in proof-of-principle experiments and some have increased the efficacy of therapeutics that normally only poorly enter the brain (see, e.g., (Kumar et al., 2007; Pardridge, 2007b) Tiwari and Amiji, 2006. However, in general, two factors have limited use of these promising approaches: toxicity and an inability to deliver enough drug to the CNS. At present, none of these is in routine clinical use for treating CNS disorders.
B. The Selective/Biochemical Barrier
Although many solutes follow the consensus relationship between blood-brain barrier permeability and octanol/water partition coefficient, there are a substantial number of outliers, indicating that one cannot think of the blood-brain barrier as being solely physical in nature. These outliers emphasize the higher than expected blood to brain permeability of certain highly hydrophilic solutes, e.g., glucose and amino acids, and the lower than expected permeability of certain hydrophobic foreign chemicals (xenobiotics), including, therapeutic drugs and CNS toxicants. This phenomenon is explained by the second component of the barrier, which is both biochemical and selective. Its molecular basis is a group of specific transport proteins expressed on the luminal (blood-facing) and abluminal (brain-facing) plasma membranes of the endothelial cells. Some of these transporters selectively increase barrier permeability to essential nutrients, while others selectively effectively prevent entry of xenobiotics (Dallas et al., 2006; Loscher and Potschka, 2005).
III. P-glycoprotein, A Critical Element of the Selective/Biochemical Barrier
Multiple drug transporters have been localized to brain capillary endothelial cells and several efflux transporters are positioned to play a gatekeeper role in the active, selective blood-brain barrier (Fig. 1). However, based on three critical defining criteria, specificity, energetics and location, it is clear why P-glycoprotein is such an important element of the blood-brain barrier. P-glycoprotein was originally discovered more than 30 years ago as an overexpressed gene (MDR1/ABCB1) in multidrug resistant human tumor cells (Juliano and Ling, 1976). that was subsequently shown to code for a plasma membrane protein that functions as an ATP-driven efflux pump (Gottesman and Pastan, 1988; Hennessy and Spiers, 2007). This pump handles a remarkably wide range of substrates, from about 300 to 4000 Da in mass. The list of substrates includes commonly prescribed drugs from many classes, such as, calcium channel blockers, statins, opioids, chemotherapeuitcs, HIV protease inhibitors, antibiotics, immunosuppressives and β-adrenergic antagonists (Ford and Hait, 1993). This broad substrate spectrum explains the ability of P-glycoprotein to provide cross-resistance to multiple classes of chemotherapeutics and to underlie drug-drug interactions involving structurally dissimilar classes of drugs, e.g., cyclosporines and cardiac glycosides (Marchetti et al., 2007).
Figure 1.
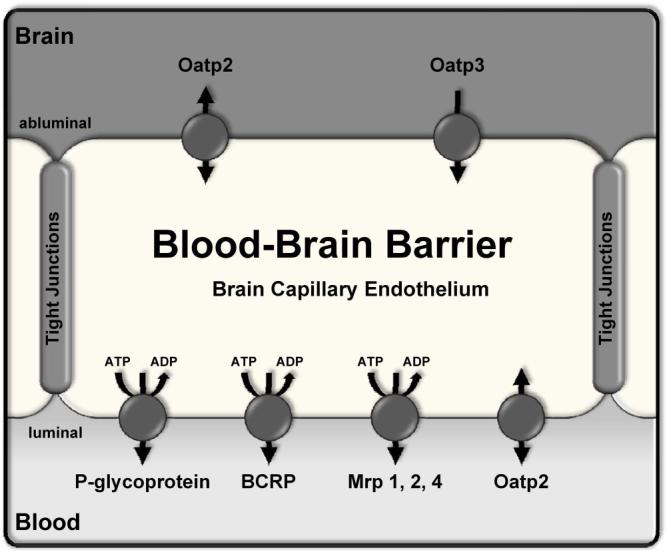
Localization of efflux transporters at the blood-brain barrier. Transporters shown have been demonstrated to be expressed in the brain capillary endothelium at the protein level. Arrows indicate direction of substrate transport; ABC transporters are marked with ATP/ADP hydrolysis. P-glycoprotein, multidrug resistance protein (Mrp) isoforms 1, 2, and 4, and breast cancer resistance protein (BCRP) are expressed in the luminal membrane. However, localization of Mrps within the brain capillary endothelium is still controversial (see, e.g., (Dallas et al., 2006; Loscher and Potschka, 2005)). We have localized Mrp2 and Mrp4 to the luminal membrane of rat brain capillaries (Bauer et al., 2008) and thus placed them there in the diagram. The organic anion transporting polypeptide 2, Oatp2, is localized to both luminal and abluminal membranes (Gao et al., 1999); Oatp3 has been detected in the abluminal membrane (Ohtsuki et al., 2004).
P-glycoprotein is not only highly expressed in multidrug resistant tumor cells, but is also physiologically present in a number of non-tumor cells, with highest levels of expression in barrier and excretory tissues (Fojo et al., 1987; Thiebaut et al., 1987). Of these normal tissues, brain capillaries express particularly high levels of P-glycoprotein protein (Fig. 2A (Cordon-Cardo et al., 1989)). In brain capillaries in situ, in isolated capillaries ex vivo and in brain capillary endothelial cell cultures, the transporter has been localized to the luminal (blood-side) plasma membrane (Fig. 2B). This is the ideal location to most efficiently limit diffusion of drugs from blood to endothelial cell cytoplasm and thus to the brain parenchyma. Recent studies using electron microscopy have also detected P-glycoprotein expression in intracellular membranes within brain capillary endothelial cells, e.g., ER, vesicles and nuclear envelope (Babakhanian et al., 2007; Bendayan et al., 2006). P-glycoprotein can be associated with elements of the cell's trafficking machinery, e.g. lipid rafts and caveolae (Barakat et al., 2007; Belanger et al., 2004; Orlowski et al., 2006), and at least in tumor cells internalized transporter appears to play some role in multidrug resistance (Molinari et al., 2002). At this time it is not clear what role this pool of internalized P-glycoprotein plays in endothelial cell function, although one could envision it providing transporter for rapid insertion into the plasma membrane, protecting cytoplasmic function by sequestering xenobiotics in vesicles and possibly limiting nuclear exposure to xenobiotics.
Figure 2.
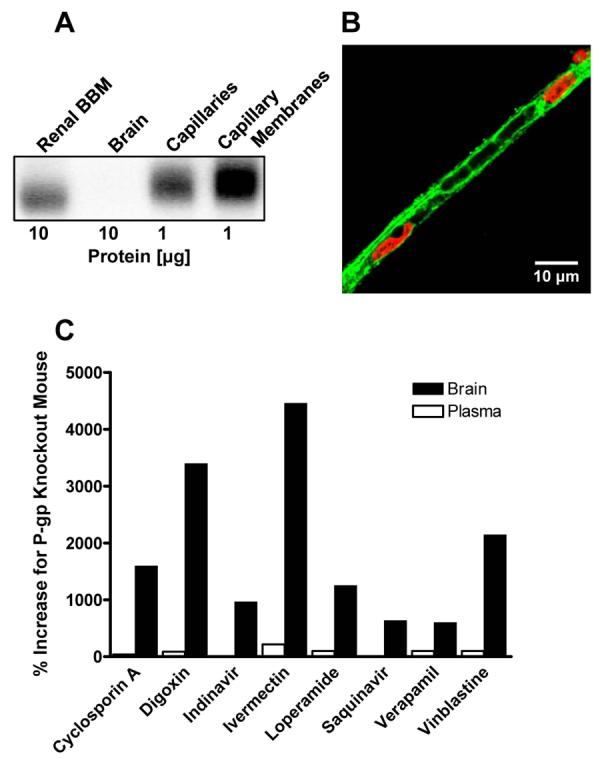
Expression of P-glycoprotein at the blood-brain barrier. (A) Western blot showing enrichment of P-glycoprotein from whole rat brain homogenate to capillary lysate to isolated capillary membranes. Note that P-glycoprotein expression levels are highest in capillary membranes (1 μg total protein loading) compared to brain and renal brush border membranes (10 μg total protein loading). (B) Immunostaining of an isolated rat brain capillary for P-glycoprotein (green). Nuclei are stained with propidium iodide (red). Note the “rail road track”-like P-glycoprotein staining of the luminal membrane that defines the capillary lumen and outlines trapped red blood cells. (C) Genetically knocking out P-glycoprotein increases drug levels in the brain. Plasma levels are not changed substantially, indicating a key role for P-glycoprotein at the blood-brain barrier. Data shown for each drug is the per cent increase in the two compartments calculated from data for wild-type and P-glycoprotein-null mice (graphed from data generated by Schinkel and coworkers and compiled in (Mizuno et al., 2003)).
Evidence for P-glycoprotein's importance comes from its remarkable ability to restrict drug access to the CNS. This is best seen in comparisons of drug distribution in wild-type and P-glycoprotein-null mice (genetic knockout) and in studies where transporter function is abolished by P-glycoprotein-specific and potent inhibitors (chemical knockout). These show10-50-fold increases in brain levels of a number of drugs (many highly lipophilic) when P-glycoprotein activity is abolished (Fig. 2C). Moreover, in spite of the fact that P-glycoprotein is expressed in liver, gut and kidney, for many drugs P-glycoprotein knockout mice show only modest increases in plasma levels. This likely reflects redundancy of transport function and the relative importance of other excretory transporters in liver and kidney. These results further highlight the important role P-glycoprotein plays as a specific gatekeeper of the CNS. Importantly, they also provide a scientific basis to modulate P-glycoprotein transport activity as a way to potentially improve CNS pharmacotherapy.
This potential has been investigated using animal models for specific diseases. Consider, a recent study with the chemotherapeutic, taxol (Paclitaxel®), a highly lipophilic antimitotic drug used to treat solid tumors (Fellner et al., 2002). Although taxol is very potent in vitro against a variety of tumors, including glioblastoma, it is ineffective against brain tumors in vivo, because it does not cross the blood-brain barrier. In nude mice, specific inhibition of P-glycoprotein by the second generation inhibitor, PSC833, increased taxol accumulation in brain by an order of magnitude; these increased brain taxol levels were sustained for at least 24 h. When given to nude mice with a cerebrally implanted human glioblastoma, taxol was ineffective. However, pretreating mice with PSC833 before taxol therapy, substantially reduced tumor volume by 90% (drugs given twice over a 5 week period; (Fellner et al., 2002)). These in vivo experiments using an animal model suggest one strategy for overcoming P-glycoprotein-based CNS pharmacoresistance: direct inhibition of transporter activity.
Similar results showing increased drug penetration into the brain have since been obtained with several other P-glycoprotein inhibitors, suggesting one general strategy to overcome CNS multidrug resistance based on P-glycoprotein (Kemper et al., 2003; Kemper et al., 2004). These studies dramatically demonstrate how P-glycoprotein can be an overriding impediment to CNS chemotherapy. In addition to the long list of failed CNS-drugs that are P-glycoprotein substrates and therefore, do not cross the blood-brain barrier, these studies further argue that the ability to control the activity/expression of this transporter in brain capillaries could be of substantial benefit in the treatment of a number of CNS diseases. Ideally, one would want to specifically and transiently reduce P-glycoprotein activity (as in the mouse study) before administering a normally impermeant therapeutic drug, which then could access the CNS during the window in time when efflux pump activity is reduced.
IV. Modulation of P-glycoprotein Transport Activity
A. In the Periphery
Since P-glycoprotein greatly influences CNS pharmacotherapy, it is important to know the extent to which transporter activity is pathophysiologically modulated. Currently, there is convincing evidence that disease processes, e.g., inflammation, can influence the expression of multiple hepatic ABC transporters, including P-glycoprotein (Ho and Piquette-Miller, 2006; McRae et al., 2003). At the blood-brain barrier emerging evidence shows that P-glycoprotein expression is also changed during disease, e.g. in epilepsy and stroke (see below). Since the brain capillary endothelium is the interface between the periphery and the CNS, it is exposed to normal and pathological signals from both the blood and the CNS. The extent to which these signals change P-glycoprotein-mediated transport is likely to be a significant factor in devising treatment regimens. Thus, it is also important to know how signals derived from both CNS and periphery influence transporter expression and overall blood-brain barrier function. Because of its role in multidrug resistance in cancer, there is a substantial body of literature on signals that modulate P-glycoprotein expression in tumor cells. Transcriptional regulation of MDR1 gene expression through the binding of several trans-acting proteins to consensus cis-elements in the promoter region is complex and not yet fully understood. For the hMDR1 gene, several promoter elements have been identified, including, a GC-box, a Y-box, a p53 element, a PXR element, an inverse MED1 element, an AP-1 element, a NF-κB element and a heat shock protein element. A detailed description can be found in (Labialle et al., 2004; Labialle et al., 2002; Scotto, 2003). Among these are binding sites for transcription factors that respond to environmental cues, such as, oxidative stress, inflammation, hypoxia, xenobiotics including drugs and toxicants, heavy metal salts, etc.
hMDR1 gene expression can also be substantially affected by epigenetic mechanisms, including, DNA methylation and histone acetylation (Baker and El-Osta, 2003; 2004; Baker et al., 2005). Finally, a number of intracellular signals were found to affect transporter activity without changing transporter protein expression, suggesting multiple post-translational mechanisms of modulation, including, transporter trafficking (Kipp and Arias, 2002), degradation (Zhang et al., 2004), protein phosphorylation/dephosphorylation (Chambers et al., 1990; Sachs et al., 1999), and specific association with other membrane proteins (e.g., caveolin, (Schlachetzki and Pardridge, 2003)).
B. At the Blood-Brain Barrier
Until a few years ago, it was uncertain whether P-glycoprotein activity in brain capillaries could be influenced by any of these signals. Now it is clear that both transporter activity and expression can be modulated by a number of physiological and pathological signals to either increase or decrease P-glycoprotein transport activity. The remainder of this review is focused primarily on recent studies in intact brain capillaries and in vivo animal models demonstrating how signaling alters P-glycoprotein expression and activity at the blood-brain barrier. Use of freshly isolated brain capillaries provides a means of investigating mechanisms of transporter function and regulation in an intact, integrated tissue without the uncertainties inherent in use of cells in culture (primary cells in culture and cell lines). These capillaries retain the ability to support for many hours specific, ATP-driven transport mediated by P-glycoprotein, Mrp2 and BCRP, providing a platform to study both rapid and transcriptional regulation at the functional, protein and mRNA levels (Bauer et al., 2004; Bauer et al., 2008; Hartz et al., 2004). Changes in tight junction function and specific junctional protein expression can also be assessed in these isolated capillaries (Hartz et al., 2004). Although adherent pericytes are retained by the capillaries, connections with astrocytes and neurons are broken during isolation; thus, potentially important interactions within the neurovascular unit are not present. As described below, we have used these capillaries along with pharmacological tools and knockout and transgenic animals to examine multiple signaling pathways in detail in vitro and to verify several pathways in vivo.
What follows is organized into eight sections, seven dealing with specific signals and diseases followed by a perspectives section that describes challenges and certain emerging areas of research.
1. Ligand-Activated Nuclear Receptors
Geick et al. (Geick et al., 2001) discovered a complex regulatory cluster of several binding sites for the ligand activated nuclear receptor, pregnane X receptor (PXR, NR1I2) in the 5′-upstream region of hMDR1. Three DR4 motifs (direct repeats of a AG(G/T)TCA motif with a spacer of 4 nucleotides in between the binding motif), one DR3 motif and one ER6 motif (everted repeat) were identified at about -8 kilobase pairs. Electrophoretic mobility shift assays further revealed that PXR binds as a heterodimer with the retinoid X receptor α (RXRα) to all DR4 motifs (Geick et al., 2001). In addition, reporter gene assays confirmed that this cluster of response elements is responsible for PXR-mediated hMDR1 induction.
PXR is a member of a superfamily of ligand-activated transcription factors, the so-called orphan nuclear receptors. It is activated by naturally occurring steroids such as pregnenolone and progesterone, and synthetic glucocorticoids and anti-glucocorticoids, but also by a wide range of xenobiotics including dietary compounds, toxicants and a large number of commonly prescribed drugs, e.g., steroids, chemotherapeutics, HIV protease inhibitors, glucocorticoids and anticonvulsants. Studies in liver and gut have shown that PXR targets genes that are responsible for xenobiotic metabolism (phase I and phase II) and efflux. Therefore PXR is considered to be a ‘master regulator’ of defenses against xenobiotics at the cellular and molecular levels (Dussault and Forman, 2002). Efflux transporters known to be regulated by PXR include organic anion transporting polypeptide isoform 2 (SLCO1A4), bile salt export pump (ABCB11), multidrug resistance-associated proteins isoforms 2 and 3, Mrp2 and Mrp3 (ABCC2, ABCC3) and P-glycoprotein (ABCB1, MDR1) (Geick et al., 2001; Kliewer et al., 1998; Lehmann et al., 1998; Teng et al., 2003). Certainly in hepatocytes, PXR, other nuclear receptors, e.g., CAR, xenobiotic metabolizing enzymes and efflux transporters appear to comprise a regulated network of core defense mechanisms (Hartley et al., 2004; Rosenfeld et al., 2003).
Initial studies with whole brain homogenates found no evidence of PXR expression (Jones et al., 2000; Kliewer et al., 1998; Zhang et al., 1999). However, we reasoned that capillaries comprise less than 1% of brain volume and if PXR expression were restricted to those structures, mRNA levels in whole brain might be too low to detect (Bauer et al., 2004). Using RT-PCR, we detected PXR mRNA in freshly isolated rat brain capillaries; immunostaining confirmed receptor expression within the capillary endothelial cells (Bauer et al., 2004). Consistent with this, two PXR ligands, pregnenolone 16α-carbonitrile (PCN; specific for rodent PXR) and dexamethasone (ligand for PXR and glucocorticoid receptor),both increased levels of P-glycoprotein protein expression (Western blots and quantitative immunostaining) as well as increased P-glycoprotein-mediated transport of a fluorescent cyclosporine A derivative into isolated rat brain capillary lumens (Bauer et al., 2004). Importantly, dosing rats with PCN and dexamethasone increased P-glycoprotein expression in plasma membranes from liver and brain capillaries and upregulated P-glycoprotein-specific transport in the capillaries. Expression of Mrp2, another drug efflux pump expressed at the blood-brain barrier and glutathione transferase π, both PXR target genes, were similarly upregulated by PCN and dexamethasone in vivo and in vitro (Bauer et al., 2004; Bauer et al., 2008). These in vitro and in vivo dosing experiments provided the first evidence for PXR expression in brain and for regulation by nuclear receptors of xenobiotic efflux pumps at the blood-brain barrier. As noted by Bauer et al, one cannot rule out the possibility that at least a part of dexamethasone's effects in these experiments were mediated by the glucocorticoid receptor.
Experiments with transgenic mice expressing hPXR further emphasized the clinical implications of these findings (Bauer et al., 2006). Although the DNA binding domain of PXR is highly conserved across species, the ligand binding domain is not. Because of this there are substantial species differences in ligand affinities for rodent vs. human PXR. For example, PCN is a ligand for rodent PXR but not hPXR; and rifampin, an antibiotic, and hyperforin, a component of the herbal remedy St. John's wort, are high affinity ligands for hPXR but not rodent PXR. Consistent with these differences in receptor specificity, rifampin and hyperforin increased P-glycoprotein expression in brain capillaries from hPXR transgenic mice, but PCN did not (Bauer et al., 2006). Conversely, PCN increased P-glycoprotein expression in brain capillaries from wild-type mice, but rifampin and hyperforin did not. In additional experiments, hPXR transgenic mice were given rifampin at a dose adjusted to result in free plasma drug levels equivalent to those seen in patients undergoing a course of rifampin treatment. In rifampin-pretreated hPXR mice, P-glycoprotein expression in liver, intestine and brain capillaries was substantially increased. Moreover, the antinociceptive effects of injected methadone, a CNS-acting analgesic and weak P-glycoprotein substrate, was reduced by 70% when compared to hPXR mice not pretreated with rifampin (Bauer et al., 2006). Plasma methadone levels were not affected by rifampin pretreatment, indicating that P-glycoprotein is not a major determinant of plasma methadone levels; it is likely that other hepatic and renal transporters are more important for drug excretion. However, methadone antinociception was shown to be a simple function of brain drug levels (Bauer et al., 2006). These studies with an animal model indicate that clinically relevant decreases in the efficacy of CNS-acting drugs that are P-glycoprotein substrates would be one consequence of PXR activation at the blood-brain barrier.
These findings have several important implications for the clinic. First, because of the large number of xenobiotics that are PXR ligands, theyse studies suggest that one basis for patient-to-patient variation in the effectiveness of CNS pharmacotherapy could be variable levels of blood-brain barrier efflux transporter induction by therapeutic drugs (polypharmacy) and dietary constituents. Second, blood-brain barrier P-glycoprotein expression has been shown to be elevated in epilepsy and stroke (see below), but it is not clear to what extent increases in transporter expression reflect activation of PXR or other nuclear receptors by prescribed drugs and endogenous ligands. Third, a recent report shows that PXR activation provides increased neuroprotection in a mouse model of Niemann-Pick type C1 disease, a fatal lipid storage disorder (Langmade et al., 2006). This finding suggests that disease-induced, progressive neurodegeneration could be delayed by therapy designed to activate PXR. Choosing to target PXR in a disease with substantial neurological complications could have unintended consequences, especially if other CNS-acting drugs are prescribed.
Finally, PXR is only one member of the family of ligand activated nuclear receptors that regulates expression of drug metabolizing enzymes and transporters (Francis et al., 2003). Our preliminary studies with rat brain capillaries show mRNA expression of CAR, FXR, LXR, VDR as well as additional target drug efflux transporters and phase I and phase II drug metabolizing enzymes (Bauer et al., 2008). Each of these receptors recognizes a somewhat different set of ligands and activates a different pattern of genes coding for enzymes and transporters. However, in other tissues, these nuclear receptors form an interacting, regulatory network in which crosstalk among receptors is the rule rather than the exception (Pascussi et al., 2008). Thus, both the identity of the receptor ligands that enter brain capillary endothelial cells and the structure of the regulatory network will importantly determine the range of chemicals that alter the BBB, the range of drugs for which barrier function is modified, and the nature and extent of the change in the barrier seen by each specific CNS-acting drug.
2. Inflammation
The acute inflammatory response is a complex immunological reaction that is triggered by a wide variety of stimuli, including infection, trauma, and cell stress. Inflammation is a major factor in a number of diseases, including diseases of the brain, many of which have a major inflammatory component that aggravates therapy. Inflammation results in the release of a number of proinflammatory cytokines which can have systemic effects and signal changes in multiple tissues. The complexity of the acute response is reflected in differences in the extent and time courses of cytokine release, immune cell infiltration and individual tissue responses to the various triggering events. In experimental studies, this complexity can make generalizations difficult since patterns of responses can be dose-, time-, context-, and model-dependent. With these caveats in mind, it is interesting to note that expression of P-glycoprotein appears to be tied in with the innate immune response. First, P-glycoprotein expression is upregulated in inflamed bowel (Farrell et al., 2000) and mild colitis is one of the few phenotypes other than altered sensitivity to xenobiotics that is associated with the P-glycoprotein-null mouse (Wilk et al., 2005). Second, a number of studies in liver and intestine show that inflammation, usually induced experimentally by injection of lipopolysaccharide (LPS, bacterial endotoxin), reduces expression (mRNA and protein) of certain drug metabolizing enzymes and drug efflux transporters, including P-glycoprotein (Ho and Piquette-Miller, 2006; McRae et al., 2003). In the LPS model, changes in the disposition of a number of xenobiotics are consistent with the effects on enzyme and transporter expression.
A similar hepatic response is seen with treatments that induce cholestasis, for example, experimental bile duct ligation (Ho and Piquette-Miller, 2006; McRae et al., 2003). The proinflammatory mediators responsible likely include IL-6, IL-β and TNF-α. These cytokines downregulate transporter expression, possibly by suppressing nuclear receptors, e.g., PXR, HNF-1α and CAR, from transcriptional activation of genes coding for luminal drug efflux transporters. However, signals generated by bile salts, proinflammatory cytokines, oxidative stress, retinoids, drugs, and hormones are involved and the mechanisms responsible for the profound changes in hepatic enzyme and transporter expression in cholestasis are expected to be complex.
Severe inflammation has profound effects on the blood-brain barrier (Huber et al., 2001). Inflammatory mediators (e.g., TNF-α, IL-1β and INF-γ) can increase junctional permeability and recent evidence shows that inflammation alters P-glycoprotein expression in the brain and in brain capillary endothelial cells (Seelbach et al., 2007; Tan et al., 2002; Theron et al., 2003). However, both increases and decreases in transporter expression have been observed and it is not clear from these studies what mechanisms are responsible and whether the changes in tight junction structure and function are somehow linked to altered P-glycoprotein expression. Consider two examples which show opposite effects of inflammation on P-glycoprotein expression. First, Goralski et al. (Goralski et al., 2003) showed that intracranial ventricle injection of LPS in rats both reduced expression of P-glycoprotein in the brain and increased accumulation of the P-glycoprotein substrate, digoxin, in brain tissue. Similar effects on digoxin disposition were found in wild-type mice, but not in P-glycoprotein knockout mice. However, it is not clear from these initial studies what mechanisms were responsible for decreased pump expression and whether changes in tight junctional permeability can also be linked to altered P-glycoprotein expression. Second, using a well-characterized experimental model of chronic, peripheral inflammatory pain in rats, Seelbach et al. (Seelbach et al., 2007) found increased P-glycoprotein expression in Western blots of brain capillary membranes, reduced uptake of injected morphine into the brain and reduced antinociceptive effects of injected morphine in a tail-flick assay. These changes came at a time when tight junction permeability to sucrose had increased (Wolka et al., 2003). Thus, for the weak P-glycoprotein substrate, morphine, the inflammation-induced increase in efflux trumped the increase in junctional leakiness. Certainly, changes in the expression of other transporters could be involved. Note that since a large part of the general population suffers from chronic pain, this study has important implications for CNS pharmacotherapy. At present the signals that underlie this “action at a distance” have not been characterized, but elements of the peripheral and CNS inflammatory responses are likely participants.
Recent studies with isolated rat brain capillaries show both a complex time course of P-glycoprotein response to proinflammatory mediators and a complex chain of signaling (Figs. 3A and 3B). Exposure of capillaries to low levels of LPS, TNF-α or ET-1 causes a rapid, reversible loss of P-glycoprotein transport function with no change in protein expression; inhibitors of protein synthesis are without effect and tight junctional permeability is not affected (Hartz et al., 2004; 2006). Signaling is complex, involving ligand binding to TLR4, TNF-R1 and ETB receptors followed by activation of NOS and PKC (Fig. 3B). A minor component of LPS signaling bypasses all steps in the main pathway except NOS activation; this might be initiated by LPS acting through for example a scavenger receptor. All steps in the main signaling sequence occur on or in the capillary endothelial cells in a single sequence of events. Thus, signaling by TNF-α and ET-1 is downstream of LPS binding to TLR4, which in turn causes release of TNF-α and ET-1 by the endothelial cells (Fig. 3B).
Figure 3.
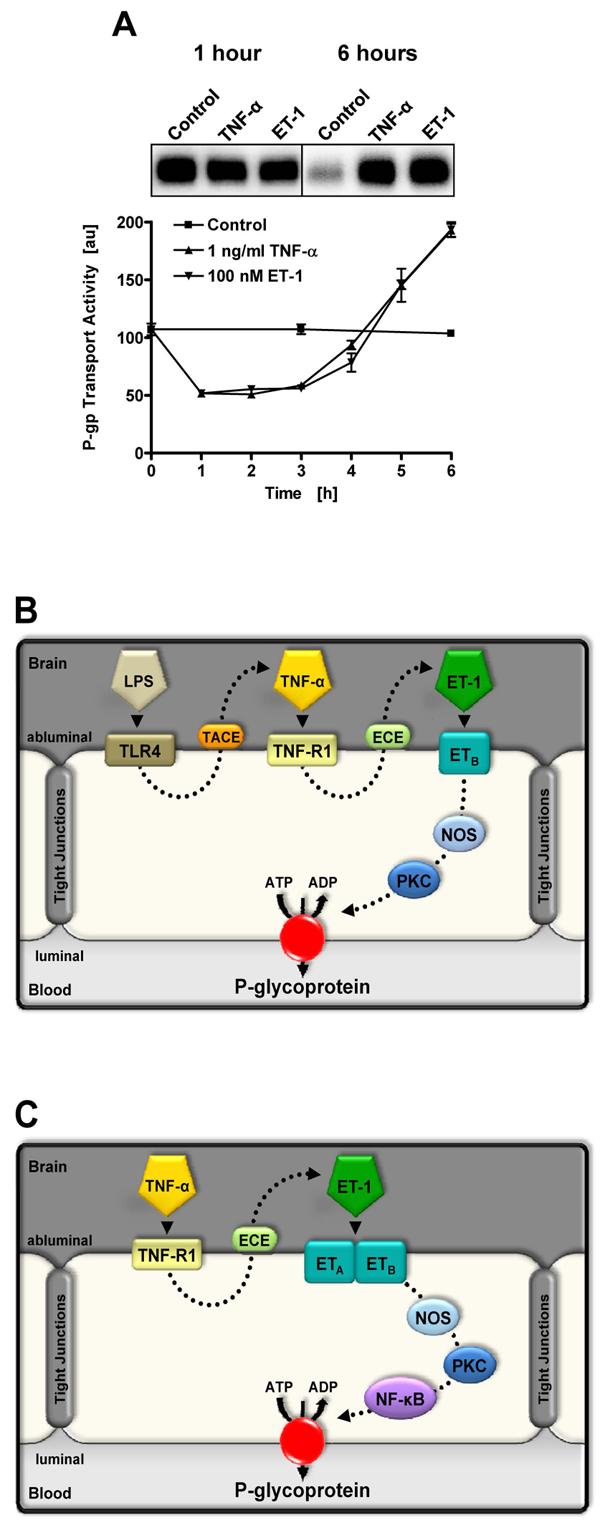
Proinflammatory signaling to P-glycoprotein in rat brain capillaries. (A) Time course showing changes in P-glycoprotein-mediated transport activity in isolated rat brain capillaries exposed to TNF-α and ET-1. Note the rapid reduction in activity and the delayed increase over control levels. The inset shows Western blots of capillary membranes after 1 h and 6 h of exposure. Note the absence of change in P-glycoprotein expression after 1 h and the dramatic change after 6 h (Bauer et al., 2007a). These blots were processed using different exposure times and expression levels cannot be compared across blots. We showed constant P-glycoprotein expression in capillaries over 6 h of incubation in control medium (Bauer et al., 2007a). (B) LPS, TNF-α and ET-1 signaling to P-glycoprotein in the short-term (rapid reduction of transport activity (Hartz et al., 2004; 2006)). (C) TNF-α and ET-1 signaling to P-glycoprotein in the long-term (increased transport activity and transporter expression (Bauer et al., 2007a)).
In this brain capillary model, experiments with microtubule disruptors suggest that changes in P-glycoprotein trafficking underlie both loss of transport function and its restoration (Hartz et al., unpublished data). This is very much like the situation in hepatocytes where one mechanism that drives changes in bile flow is the rapid, regulated trafficking of ABC transporters (P-glycoprotein, MRP2, MDR2 and the bile salt export pump) between intracellular compartments and the canalicular plasma membrane (Kipp et al., 2001). Both microtubules and the products of phosphoinositide-3-kinase have been implicated in membrane vesicle trafficking. Striking movies of hepatocytes expressing a P-glycoprotein-green fluorescent protein show intermittent, tubulovesicular movement of the fusion protein between the pericanalicular region and the bile canalicular membrane (Kipp and Arias, 2000; Kipp et al., 2001).
Note that the ability to manipulate P-glycoprotein trafficking suggests exciting possibilities with regard to drug delivery. Rapid and reversible loss of specific transport activity in the capillary endothelium could provide the window in time needed to deliver P-glycoprotein substrates to the CNS with only minor disruption of protection. At present this mechanism of modulation has only been demonstrated in vitro. Whether this will happen in an in vivo model remains to be seen.
In contrast to short-term exposure, longer-term exposure of the capillaries to low levels of TNF-α or ET-1 eventually causes increased P-glycoprotein expression signaled through TNF-R1, ETB and ETA receptors, NOS, PKC and the transcription factor, NF-κB (Fig. 3C (Bauer et al., 2007a)). After 6 h of exposure, P-glycoprotein transport activity and protein expression are about twice that of controls (which do not change over the time course of the experiment). Inhibiting protein synthesis blocks these increases. As one might expect, expression of other proteins also changes. At the same time P-glycoprotein expression is increased, expression of Mrp2 and Mrp4 is decreased, expression of Glut-1 and Na+/K+-ATPase is increased and expression of BCRP and tight junctional proteins does not change (Bauer et al., 2007a). At present, it is not clear whether the same signaling pathway is responsible for the changes observed in all transporters, but it is clear that the effects of TNF-α and ET-1 exposure are not restricted to P-glycoprotein.
Taken together, these data for brain capillaries show that certain proinflammatory stimuli first decrease P-glycoprotein transport activity with no change in expression and then increase expression and activity (Fig. 3A). A similar overall pattern of change in drug efflux transporter activity and expression (Mrp2 and P-glycoprotein) was previously demonstrated in renal proximal tubule, where ETB receptor, NOS and PKC signaling first decreased transporter activity and then increased expression and activity (Miller, 2002; Terlouw et al., 2003). In these tubules, signaling was initiated by low levels of tubular nephrotoxicants. One consequence of long-term exposure of tubules to low concentrations of ET-1 or the nephrotoxic aminoglycoside, gentamicin, was reduced sensitivity to acute gentamicin toxicity. It was suggested that the initial decrease in transporter activity was an attempt to preserve ATP in the face of acute injury and that the longer-term increase in transporter expression was protective. One could invoke the same explanation for TNF-α action in our experiments with rat brain capillaries.
3. Oxidative Stress/Ischemia
Cellular stress, e.g., exposure to heavy metals, hypoxia, reactive oxygen species and some chemotherapeutics as well as heat stress, upregulates expression of P-glycoprotein and some MRP isoforms in tumor cells and some epithelial tissues (Sukhai and Piquette-Miller, 2000; Thevenod, 2003; Thevenod et al., 2000). Increases in pump expression, along with increases in expression of heat shock proteins can be seen as a response designed to repair cellular damage and to remove from the cells both the stress-causing agents and the products of their actions.
In stroke, ischemia/hypoxia followed by reperfusion leads to generation of a complex cocktail of reactive oxygen species, proinflammatory cytokines and chemokines. Damage to the infarct region is calamitous and damage to the peri-infarct region can be variable, determining largely long-term functional losses and thus the full extent of recovery. In brain capillary endothelial cells, exposure to hydrogen peroxide over a period of 1-2 days, increases both P-glycoprotein expression and P-glycoprotein-mediated transport (Felix and Barrand, 2002). This in vitro exposure is followed by activation of intracellular signaling through protein kinases (ERK1/2 and stress activated protein kinase, PKC, Akt (PKB)) and transcription factors (c-Jun, a component of AP-1) which in turn activate NF-κB and transcription (Nwaozuzu et al., 2003). All of these signals are indeed upregulated in animal models of stroke. Consistent with these results on oxidative stress, GSH depletion causes similar upregulation of P-glycoprotein expression in rat brain capillary endothelial cells in primary culture (Hong et al., 2006). These effects are reversed by the ROS scavenger, N-acetylcysteine, suggesting that loss of GSH leads to elevated ROS, which induces P-glycoprotein expression.
What happens to P-glycoprotein expression following stroke in vivo is largely an unsettled issue. In an early study employing a rat brain focal ischemia model, Samoto et al. (Samoto et al., 1994) observed a loss of P-glycoprotein expression in the ischemic lesion followed by recovery of expression during the subsequent post-ischemic period when angiogenesis was likely occurring. More recently Spudich et al. (Spudich et al., 2006) showed upregulation of endothelial cell P-glycoprotein expression (mRNA and protein) 3-24 h following focal cerebral ischemia (MCAO) in a mouse model. Importantly, this group found that inhibition of P-glycoprotein improved the efficacy of neuroprotective drugs and thus stroke outcome. In contrast, Langmade et al. (Langmade et al., 2006) surveyed several blood-brain barrier transporters in the peri-infarct region of rats after MCAO and found that although expression of endothelial BCRP and Oatp2 increased (mRNA and protein), there was no change in P-glycoprotein expression (mRNA and immunostaining of tissue sections). These disparate results may reflect differences in the models chosen, the stress applied and the regions of the brain assayed. Certainly, because of the critical role P-glycoprotein plays in limiting access of therapeutic drugs to the CNS and the need for continued pharmacotherapy during recovery, it is important to know where, to what extent and under what circumstances expression of this blood-brain barrier transporter is altered following stroke.
4. Seizures
Although a variety of drugs have proven useful in controlling seizure activity, a substantial fraction of epileptic patients do not respond to commonly prescribed AEDs that act through a range of mechanisms. Since limited drug delivery to the brain is a common cause of therapeutic failure, one suggested underlying basis for this pharmacoresistance in epilepsy is the over expression of ATP-driven drug efflux pumps at the blood-brain barrier, including P-glycoprotein, Mrp1, Mrp2 and BCRP (Loscher and Potschka, 2005). Evidence connecting transporter overexpression with pharmacoresistance to AEDs is strongest for P-glycoprotein and can be summarized as follows: First, P-glycoprotein is overexpressed in epileptogenic brain tissue, including capillary endothelial cells, from AED-resistant patients. In rodent models, experimentally induced seizures upregulate P-glycoprotein expression. Second, uptake of several AEDs into rodent brain is increased by specific P-glycoprotein inhibitors and in P-glycoprotein-null mice. Third, inhibition of P-glycoprotein reduces seizure severity in animal models of epilepsy. Fourth, limited clinical data suggest that adjunct therapy with a P-glycoprotein inhibitor improves seizure control in a patient with refractory epilepsy (reviewed in (Loscher, 2007; Loscher and Potschka, 2005). In spite of these findings, the role of P-glycoprotein in AED resistance is controversial. Indeed, it is still not clear which (if any) commonly prescribed AEDs are substrates for human P-glycoprotein (Baltes et al., 2007a; Baltes et al., 2007b). In addition, a case can be made that the pharmacokinetics of many of these drugs are not consistent with P-glycoprotein being a primary barrier to uptake into the CNS (Anderson and Shen, 2007). Finally, there is still substantial controversy over whether polymorphisms in the P-glycoprotein gene affect AED uptake and seizure frequency (Basic et al., 2008; Siddiqui et al., 2003; Sills et al., 2005; Tan et al., 2004b).
We have investigated the chain of events connecting seizure activity and increased P-glycoprotein expression. During seizures, the neurotransmitter, glutamate, accumulates in the brain interstitium (Holmes, 2002). Importantly, glutamate has been shown to increase P-glycoprotein expression in rat brain endothelial cells (Zhu and Liu, 2004). It is known that glutaminergic signaling increases COX-2 expression and that at least in rat mesangial cells COX-2 activation leads to increased P-glycoprotein expression (Sorokin, 2004). We thus hypothesized that glutamate signaled through NMDA receptors on brain capillary endothelial cells to activate COX-2 and in turn increase P-glycoprotein expression (Bauer et al., 2007b). To test this hypothesis, we exposed isolated rat brain capillaries to glutamate for 15-30 min and found that specific P-glycoprotein transport activity and protein expression had roughly doubled 5.5 h later. These increases were blocked by cycloheximide, actinomycin D, NMDA receptor antagonists and specific inhibitors of COX-2, but not COX-1. In capillaries from COX-2 knockout mice glutamate did not increase P-glycoprotein-mediated transport or expression. In rats, intracerebral microinjection of glutamate locally increased brain capillary P-glycoprotein expression and in an animal model of epilepsy (pilocarpine-induced status epilepticus in rats), seizure-induced increases in capillary P-glycoprotein expression were reduced by indomethacin, a non-selective COX inhibitor (Bauer et al., 2007b) and by celecoxib, a specific COX-2 inhibitor (Potschka et al, unpublished data). All of these findings are consistent with glutamate signaling through an NMDA receptor and COX-2 to increase P-glycoprotein expression in brain capillaries. These initial studies suggest one practical approach to increasing drug penetration of AEDs that are P-glycoprotein substrates: inhibition of endothelial cell signaling through COX-2. They show how important it is to understand at the molecular level the signaling basis for changes in transporter expression, since elements of signaling pathways are potential therapeutic targets.
5, Brain Cancer
Despite dramatic advances in the ability to treat cancers in the periphery, the CNS still remains a sanctuary for primary and metastatic disease. This is largely due to the two elements of the blood-brain barrier, with entry of macromolecule-based drugs (antibodies and SiRNA) being limited by the structural barrier and entry of lipophylic, small-molecule drugs being limited by the biochemical barrier. Certainly, chemotherapeutics were among the earliest identified P-glycoprotein substrates (Ford and Hait, 1993) and P-glycoprotein is a major (if not the major) factor in the inability of these drugs to enter the CNS (Breedveld et al., 2006). Defining blood-brain barrier function within and around tumors is an active area of research that has provided evidence for both increases and decreases in tight junctional properties, perhaps suggesting opportunities for chemotherapy with certain metastatic brain tumors (Gerstner and Fine, 2007).
One can ask whether important tumor-induced changes are also seen for the elements of the biochemical barrier and what signals might underlie these changes. Gerstner and Fine (2007) summarized available literature on P-glycoprotein expression patterns in neovasculature supplying metastatic brain tumors and gliomas and found substantial differences. P-Glycoprotein expression could be detected in neovasculature from both sources, but at substantially reduced levels than in normal brain. In neovasculature from metastatic brain tumors it appeared that transporter expression was consistently lower than in gliomas. It is not clear from these studies to what extent reduced expression levels of P-glycoprotein can confer multidrug resistance. Nor is it clear how tumor signals to neovasculature modulate P-glycoprotein expression. Given the apparent increased incidence of metastatic brain tumors (Palmieri et al., 2007), this is an important and evolving area of research where an understanding of signaling to multidrug resistance transporters could be of practical benefit.
It is well-documented that P-glycoprotein contributes to multidrug resistance in certain tumors, and that blood-brain barrier P-glycoprotein plays a key role in limiting chemotherapeutics from accessing tumors within the CNS. This raises the question why inhibitors of transporter activity are not in routine use in the clinic. Certainly, potent and specific P-glycoprotein inhibitors with acceptable toxicity profiles are now available. Moreover, as mentioned above, proof of principle studies in animal models have shown that specific inhibition of P-glycoprotein can both increase brain levels of chemotherapeutics and improve drug efficacy against implanted human tumors (Fellner et al., 2002; Kemper et al., 2003; Kemper et al., 2004).
Early attempts to incorporate first generation P-glycoprotein inhibitors, e.g., cyclosporine A, verapamil and tamoxifen, into chemotherapy protocols showed some promise in patients with non-CNS tumors. However, results of later studies with first, second and third generation P-glycoprotein inhibitors have been either contradictory or disappointing (Fox and Bates, 2007; Leonard et al., 2003). Although more limited in number, studies in which P-glycoprotein inhibitors were used in the hope of improving response to chemotherapeutics in patients with brain tumors have also not shown any positive results (Chen et al., 2006; Fine et al., 2006).
There are several possible explanations for this disconnect between promising animal studies and disappointing clinical trials. Certainly, there are many variables that have to be considered in choosing a dosing protocol. Thus it is important to carefully select both inhibitor and chemotherapeutic and to adjust dose levels of both to balance efficacy vs systemic and CNS toxicity. In addition, one must consider the possible contributions of other drug efflux pumps expressed on the luminal side of brain capillary endothelial cells, e.g., BCRP and MRP2, which may undergo compensatory increases when P-glycoprotein is inhibited (Deeken and Loscher, 2007).
6. HIV-1
Use of highly active antiretroviral therapy (HAART) since the mid-1990s has changed dramatically the clinical picture of HIV-1 infection. However, it has also emphasized the importance of the CNS as a sanctuary for the virus. Virtually all of the drugs used to fight HIV infection poorly penetrate the CNS. Indeed, HIV protease inhibitors are substrates for P-glycoprotein and several Mrps and a number of reverse transcriptase inhibitors are substrates for BCRP (Owen et al., 2005). Both ritonavir and saquinavir are ligands for the nuclear receptor, PXR (Dussault et al., 2001), and induce P-glycoprotein expression in lymphocytes (Owen et al., 2005) and in brain capillary endothelial cells (Perloff et al., 2007), although it is not clear that this occurs solely through PXR activation. HIV transactivator protein, TAT, can also upregulate expression of P-glycoprotein and Mrp1 in brain capillary endothelial cells by activating NF-κB and a mitogen-activated protein kinase pathway, respectively (Hayashi et al., 2006; Hayashi et al., 2005). At this time, it is not clear to what extent the drugs used in HAART or the viral proteins themselves affect blood-brain barrier P-glycoprotein expression in patients.
Microglia and astroglia, the cells that harbor the virus within the CNS, express P-glycoprotein (Bendayan et al., 2002). At present, we know little about regulation of ABC transporter expression in microglia, the primary HIV sanctuary within the CNS. However, recent experiments with a primary culture of rat astrocytes indicate that expression and transport function of P-glycoprotein is downregulated following exposure to the HIV viral envelope protein, gp120 (Ronaldson and Bendayan, 2006). In these cells, signaling appeared to involve proinflammatory cytokines, with a major role for IL-6. Taken together, these findings suggest that a dynamic glial barrier behind the blood-brain barrier can further restrict the ability of HIV drugs to access sites of infection within the CNS and that the glial barrier may be further strengthened by inflammation.
7. Neurodegenerative Diseases
It is clear that the blood-brain barrier is affected in virtually every CNS disease. Indeed, it has been postulated in many instances that this tissue is not merely a spectator, but also an active contributor to disease (Zlokovic, 2008). A role for P-glycoprotein in neurogenerative diseases has been examined for Alzheimer's disease and to a lesser extent for Parkinson's disease. For the latter, there appears to be positive and negative associations between specific MDR1 haplotypes and disease incidence (Drozdzik et al., 2003; Furuno et al., 2002; Tan et al., 2005; Tan et al., 2004a), although mechanisms underlying these associations are still unclear.
In Alzheimer's disease, P-glycoprotein may play a role in the movement of β-amyloid protein from brain to blood. Unlike other ATP-driven drug efflux pumps (e.g., the Mrps), P-glycoprotein has no identified endogenous substrates and no clearly defined diseases associated with the knockout phenotype or functionally important SNPs. Given the transporter's broad substrate specificity, the range of possible substrates should be large, but that supposition is inconsistent with reality. In this regard, recent evidence suggests that β-amyloid protein may be one such endogenous substrate, since P-glycoprotein-mediated transport has been reported in cell lines that over-express the transporter (Kuhnke et al., 2007; Lam et al., 2001).. To move from brain to blood, β-amyloid protein must cross both the basolateral and apical plasma membranes of the capillary endothelium. Current thought is that LRP1 is responsible for the first step in β-amyloid transport (Deane and Zlokovic, 2007). There is suggestive evidence from animal models that P-glycoprotein could mediate β-amyloid efflux to blood. That is efflux from brain is reduced in P-glycoprotein-null mice and in mice treated with a specific p-glycoprotein inhibitor (Cirrito et al., 2005), It should be noted, however, that in one study with rats P-glycoprotein inhibitors did not appreciably reduce β-amyloid efflux from the brain (Ito et al., 2006). Based on these few reports, there is no consensus in the literature concerning this matter.
Interestingly, brain samples taken at autopsy, show a significant negative correlation between β-amyloid levels and P-glycoprotein expression (Vogelgesang et al., 2002). It is not clear whether this reflects a cause and effect relationship, however, our preliminary rodent data indicate that β-amyloid exposure signals increased P-glycoprotein degradation in brain capillaries in vitro and in vivo (Hartz et al, unpublished data). If P-glycoprotein does indeed mediate β-amyloid efflux from the brain, our findings suggest a vicious positive feedback loop, in which increasing brain β-amyloid levels reduce P-glycoprotein expression which leads to further elevation of β-amyloid levels. Even if this supposition is not true, the data suggest altered blood to brain transport of therapeutic drugs in Alzheimer's patients. Further studies are needed to resolve the many uncertainties with regard to the relationship between β-amyloid transport and blood-brain barrier P-glycoprotein.
V. Perspectives
It is now clear that the brain capillary endothelium is not a static barrier. Rather, both the structural/physical and selective/biochemical elements of the barrier have proven to be dynamic, responding to pathophysiological signals from both the periphery and the CNS. Given its multispecificity and potency as a drug efflux pump, the importance of P-glycoprotein as a critical element of the selective blood-brain barrier is unquestioned. Indeed, the research focus on P-glycoprotein at the blood-brain barrier is beginning to shift from questions related to where the transporter is and what it does to those focusing on when and how function changes. In this regard, five important questions must be answered.
1) What extracellular and intracellular signals modulate blood-brain barrier P-glycoprotein?
2) Which signaling pathways are responsible for changes in P-glycoprotein?
3) To what extent do epigenetic mechanisms establish basal levels of transporter expression in individuals and contribute to changes seen in disease?
4) How does disease affect P-glycoprotein?
5) Will disease-induced changes in P-glycoprotein warrant a shift in how we therapeutically target the transporter?
We can now provide a partial map of important signals and their relationship to certain diseases (Figure 4). This map shows that in brain capillaries P-glycoprotein activity changes in response to xenobiotics including therapeutic drugs, inflammation, disease and chemical stress and that such changes occur through altered transporter trafficking, increased synthesis and degradation. Signaling is complex, involving multiple surface receptors, intracellular messengers and transcription factors. At several points in the map discrete signaling pathways intersect, suggesting divergence of signaling through PKC and NOS isoforms and possibly context-dependent signaling switches. Moreover, we now know that these same signaling pathways can modulate expression of other ABC transporters at the blood-brain barrier, e.g, Mrps and BCRP, as well as drug metabolizing enzymes (Bauer et al., 2004; Bauer et al., 2008; Bauer et al., 2007a; Bauer et al., 2007b; Bauer et al., 2006). Most of this work has been carried out using isolated brain capillaries, but some has already been validated in vivo using transgenic and knockout animals (Bauer et al., 2004; Bauer et al., 2007b; Bauer et al., 2006). Certainly, an emphasis on a combined in vitro/in vivo approach is needed here, especially when one wants to relate signals and mechanisms that alter transporter activity to specific diseases and therapy.
Figure 4.
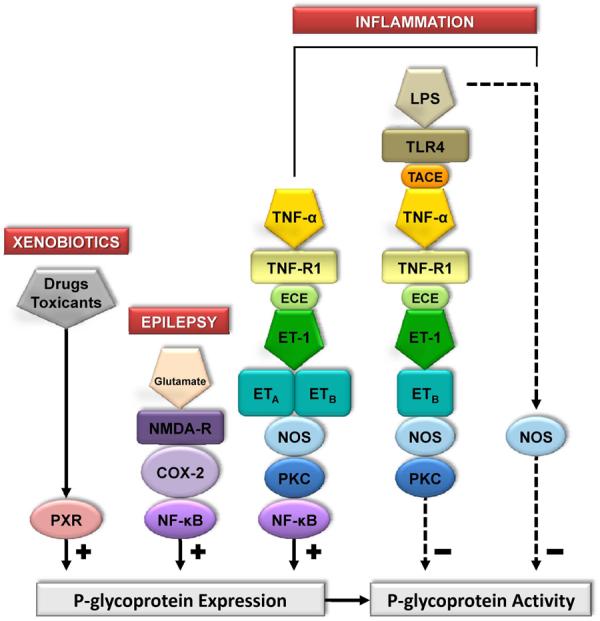
Network of signaling mechanisms that modulate P-glycoprotein activity in rodent brain capillaries.. Shown is a compilation of signaling pathways disclosed over the past several years by our studies on rat and mouse brain capillaries. Each step has been validated using pharmacological tools or knockout mice. At this time, two of these pathways, PXR and glutamate, have been validated in vivo. For details see: (Bauer et al., 2004; Bauer et al., 2007a; Bauer et al., 2007b; Bauer et al., 2006; Hartz et al., 2004; 2006).
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences.
ABBREVIATIONS
- ABC
ATP-binding cassette
- AED
anti-epileptic drug
- BCRP/ABCG2
breast cancer resistance-associated protein
- BSEP/ABCB11
bile salt export pump
- CAR
constitutive androstane receptor
- COX
cyclooxygenase
- ECE
endothelin converting enzyme
- ERK1/2
extracellular signal-regulated kinase 1/2
- ET-1
endothelin-1
- ETA
endothelin A receptor
- ETB
endothelin B receptor
- FXR
farnesoid X receptor
- GLUT-1
glucose transporter 1
- GSH
glutathione
- HNF-1α
hepatocyte nuclear factor 1 α
- HAART
Highly active antiretroviral therapy
- hPXR
human pregnane X receptor
- INF
interferon
- IL
interleukin
- LPS
lipopolysaccharide
- LRP1
low density lipoprotein receptor 1
- LXR
liver X receptor
- Mrp
multidrug resistance-associated protein
- NF-κB
nuclear factor-κB
- NMDA-R
N-methyl-D-aspartic acid receptor
- NOS
NO synthase
- Oatp
organic anion transporting polypeptide
- PCN
pregnenolone-16α-carbonitrile
- PKC
protein kinase C
- PXR
pregnane X receptor
- ROS
reactive oxygen species
- RXRα
retinoid X receptor α
- SNP
single nucleotide polymorphism
- TACE
tumor necrosis factor-α converting enzyme
- TLR4
toll-like receptor 4
- TNF-R1
tumor necrosis factor receptor 1
- TNF-α
tumor necrosis factor-α
- VDR
vitamin D receptor
References
- Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007a;100:158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res. 2007b;100:174–190. doi: 10.1161/01.RES.0000255690.03436.ae. [DOI] [PubMed] [Google Scholar]
- Anderson GD, Shen DD. Where is the evidence that p-glycoprotein limits brain uptake of antiepileptic drug and contributes to drug resistance in epilepsy? Epilepsia. 2007;48:2372–2374. doi: 10.1111/j.1528-1167.2007.01260_3.x. [DOI] [PubMed] [Google Scholar]
- Babakhanian K, Bendayan M, Bendayan R. Localization of P-glycoprotein at the nuclear envelope of rat brain cells. Biochem Biophys Res Commun. 2007;361:301–306. doi: 10.1016/j.bbrc.2007.06.176. [DOI] [PubMed] [Google Scholar]
- Baker EK, El-Osta A. The rise of DNA methylation and the importance of chromatin on multidrug resistance in cancer. Exp Cell Res. 2003;290:177–194. doi: 10.1016/s0014-4827(03)00342-2. [DOI] [PubMed] [Google Scholar]
- Baker EK, El-Osta A. MDR1, chemotherapy and chromatin remodeling. Cancer Biol Ther. 2004;3:819–824. doi: 10.4161/cbt.3.9.1101. [DOI] [PubMed] [Google Scholar]
- Baker EK, Johnstone RW, Zalcberg JR, El-Osta A. Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene. 2005;24:8061–8075. doi: 10.1038/sj.onc.1208955. [DOI] [PubMed] [Google Scholar]
- Baltes S, Fedrowitz M, Tortos CL, Potschka H, Loscher W. Valproic acid is not a substrate for P-glycoprotein or multidrug resistance proteins 1 and 2 in a number of in vitro and in vivo transport assays. J Pharmacol Exp Ther. 2007a;320:331–343. doi: 10.1124/jpet.106.102491. [DOI] [PubMed] [Google Scholar]
- Baltes S, Gastens AM, Fedrowitz M, Potschka H, Kaever V, Loscher W. Differences in the transport of the antiepileptic drugs phenytoin, levetiracetam and carbamazepine by human and mouse P-glycoprotein. Neuropharmacology. 2007b;52:333–346. doi: 10.1016/j.neuropharm.2006.07.038. [DOI] [PubMed] [Google Scholar]
- Barakat S, Demeule M, Pilorget A, Regina A, Gingras D, Baggetto LG, Beliveau R. Modulation of p-glycoprotein function by caveolin-1 phosphorylation. J Neurochem. 2007;101:1–8. doi: 10.1111/j.1471-4159.2006.04410.x. [DOI] [PubMed] [Google Scholar]
- Basic S, Hajnsek S, Bozina N, Filipcic I, Sporis D, Mislov D, Posavec A. The influence of C3435T polymorphism of ABCB1 gene on penetration of phenobarbital across the blood-brain barrier in patients with generalized epilepsy. Seizure. 2008 doi: 10.1016/j.seizure.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Bauer B, Hartz AM, Fricker G, Miller DS. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain barrier. Mol Pharmacol. 2004;66:413–419. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- Bauer B, Hartz AM, Lucking JR, Yang X, Pollack GM, Miller DS. Coordinated nuclear receptor regulation of the efflux transporter, Mrp2, and the phase-II metabolizing enzyme, GSTpi, at the blood-brain barrier. J Cereb Blood Flow Metab. 2008 doi: 10.1038/jcbfm.2008.16. [DOI] [PubMed] [Google Scholar]
- Bauer B, Hartz AM, Miller DS. Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol Pharmacol. 2007a;71:667–675. doi: 10.1124/mol.106.029512. [DOI] [PubMed] [Google Scholar]
- Bauer B, Hartz AM, Pekcec A, Toellner K, Miller DS, Potschka H. Seizure-Induced Upregulation of P-glycoprotein at the Blood-Brain Barrier through Glutamate and COX-2 Signaling. Mol Pharmacol. 2007b doi: 10.1124/mol.107.041210. [DOI] [PubMed] [Google Scholar]
- Bauer B, Yang X, Hartz AM, Olson ER, Zhao R, Kalvass JC, Pollack GM, Miller DS. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol Pharmacol. 2006;70:1212–1219. doi: 10.1124/mol.106.023796. [DOI] [PubMed] [Google Scholar]
- Begley DJ. ABC transporters and the blood-brain barrier. Curr Pharm Des. 2004;10:1295–1312. doi: 10.2174/1381612043384844. [DOI] [PubMed] [Google Scholar]
- Belanger MM, Gaudreau M, Roussel E, Couet J. Role of caveolin-1 in etoposide resistance development in A549 lung cancer cells. Cancer Biol Ther. 2004;3:954–959. doi: 10.4161/cbt.3.10.1112. [DOI] [PubMed] [Google Scholar]
- Bendayan R, Lee G, Bendayan M. Functional expression and localization of P-glycoprotein at the blood brain barrier. Microsc Res Tech. 2002;57:365–380. doi: 10.1002/jemt.10090. [DOI] [PubMed] [Google Scholar]
- Bendayan R, Ronaldson PT, Gingras D, Bendayan M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J Histochem Cytochem. 2006;54:1159–1167. doi: 10.1369/jhc.5A6870.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breedveld P, Beijnen JH, Schellens JH. Use of P-glycoprotein and BCRP inhibitors to improve oral bioavailability and CNS penetration of anticancer drugs. Trends Pharmacol Sci. 2006;27:17–24. doi: 10.1016/j.tips.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Chambers TC, McAvoy EM, Jacobs JW, Eilon G. Protein kinase C phosphorylates P-glycoprotein in multidrug resistant human KB carcinoma cells. J Biol Chem. 1990;265:7679–7686. [PubMed] [Google Scholar]
- Chen J, Balmaceda C, Bruce JN, Sisti MB, Huang M, Cheung YK, McKhann GM, Goodman RR, Fine RL. Tamoxifen paradoxically decreases paclitaxel deposition into cerebrospinal fluid of brain tumor patients. J Neurooncol. 2006;76:85–92. doi: 10.1007/s11060-005-4171-7. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115:3285–3290. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordon-Cardo C, O'Brien JP, Casals D, Rittman-Grauer L, Biedler JL, Melamed MR, Bertino JR. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci U S A. 1989;86:695–698. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas S, Miller DS, Bendayan R. Multidrug resistance-associated proteins: expression and function in the central nervous system. Pharmacol Rev. 2006;58:140–161. doi: 10.1124/pr.58.2.3. [DOI] [PubMed] [Google Scholar]
- Deane R, Zlokovic BV. Role of the blood-brain barrier in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2007;4:191–197. doi: 10.2174/156720507780362245. [DOI] [PubMed] [Google Scholar]
- Deeken JF, Loscher W. The blood-brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res. 2007;13:1663–1674. doi: 10.1158/1078-0432.CCR-06-2854. [DOI] [PubMed] [Google Scholar]
- Drozdzik M, Bialecka M, Mysliwiec K, Honczarenko K, Stankiewicz J, Sych Z. Polymorphism in the P-glycoprotein drug transporter MDR1 gene: a possible link between environmental and genetic factors in Parkinson's disease. Pharmacogenetics. 2003;13:259–263. doi: 10.1097/01.fpc.0000054087.48725.d9. [DOI] [PubMed] [Google Scholar]
- Dussault I, Forman BM. The nuclear receptor PXR: a master regulator of “homeland” defense. Crit Rev Eukaryot Gene Expr. 2002;12:53–64. doi: 10.1615/critreveukaryotgeneexpr.v12.i1.30. [DOI] [PubMed] [Google Scholar]
- Dussault I, Lin M, Hollister K, Wang EH, Synold TW, Forman BM. Peptide mimetic HIV protease inhibitors are ligands for the orphan receptor SXR. J Biol Chem. 2001;276:33309–33312. doi: 10.1074/jbc.C100375200. [DOI] [PubMed] [Google Scholar]
- Farrell RJ, Murphy A, Long A, Donnelly S, Cherikuri A, O'Toole D, Mahmud N, Keeling PW, Weir DG, Kelleher D. High multidrug resistance (P-glycoprotein 170) expression in inflammatory bowel disease patients who fail medical therapy. Gastroenterology. 2000;118:279–288. doi: 10.1016/s0016-5085(00)70210-1. [DOI] [PubMed] [Google Scholar]
- Felix RA, Barrand MA. P-glycoprotein expression in rat brain endothelial cells: evidence for regulation by transient oxidative stress. J Neurochem. 2002;80:64–72. doi: 10.1046/j.0022-3042.2001.00660.x. [DOI] [PubMed] [Google Scholar]
- Fellner S, Bauer B, Miller DS, Schaffrik M, Fankhanel M, Spruss T, Bernhardt G, Graeff C, Farber L, Gschaidmeier H, Buschauer A, Fricker G. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J Clin Invest. 2002;110:1309–1318. doi: 10.1172/JCI15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine RL, Chen J, Balmaceda C, Bruce JN, Huang M, Desai M, Sisti MB, McKhann GM, Goodman RR, Bertino JS, Jr., Nafziger AN, Fetell MR. Randomized study of paclitaxel and tamoxifen deposition into human brain tumors: implications for the treatment of metastatic brain tumors. Clin Cancer Res. 2006;12:5770–5776. doi: 10.1158/1078-0432.CCR-05-2356. [DOI] [PubMed] [Google Scholar]
- Fojo AT, Ueda K, Slamon DJ, Poplack DG, Gottesman MM, Pastan I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc Natl Acad Sci U S A. 1987;84:265–269. doi: 10.1073/pnas.84.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford JM, Hait WN. Pharmacologic circumvention of multidrug resistance. Cytotechnology. 1993;12:171–212. doi: 10.1007/BF00744664. [DOI] [PubMed] [Google Scholar]
- Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. 2007;7:447–459. doi: 10.1586/14737140.7.4.447. [DOI] [PubMed] [Google Scholar]
- Francis GA, Fayard E, Picard F, Auwerx J. Nuclear receptors and the control of metabolism. Annu Rev Physiol. 2003;65:261–311. doi: 10.1146/annurev.physiol.65.092101.142528. [DOI] [PubMed] [Google Scholar]
- Furuno T, Landi MT, Ceroni M, Caporaso N, Bernucci I, Nappi G, Martignoni E, Schaeffeler E, Eichelbaum M, Schwab M, Zanger UM. Expression polymorphism of the blood-brain barrier component P-glycoprotein (MDR1) in relation to Parkinson's disease. Pharmacogenetics. 2002;12:529–534. doi: 10.1097/00008571-200210000-00004. [DOI] [PubMed] [Google Scholar]
- Gao B, Stieger B, Noe B, Fritschy JM, Meier PJ. Localization of the organic anion transporting polypeptide 2 (Oatp2) in capillary endothelium and choroid plexus epithelium of rat brain. J Histochem Cytochem. 1999;47:1255–1264. doi: 10.1177/002215549904701005. [DOI] [PubMed] [Google Scholar]
- Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem. 2001;276:14581–14587. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- Gerstner ER, Fine RL. Increased permeability of the blood-brain barrier to chemotherapy in metastatic brain tumors: establishing a treatment paradigm. J Clin Oncol. 2007;25:2306–2312. doi: 10.1200/JCO.2006.10.0677. [DOI] [PubMed] [Google Scholar]
- Goralski KB, Hartmann G, Piquette-Miller M, Renton KW. Downregulation of mdr1a expression in the brain and liver during CNS inflammation alters the in vivo disposition of digoxin. Br J Pharmacol. 2003;139:35–48. doi: 10.1038/sj.bjp.0705227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman MM, Pastan I. The multidrug transporter, a double-edged sword. J Biol Chem. 1988;263:12163–12166. [PubMed] [Google Scholar]
- Hartley DP, Dai X, He YD, Carlini EJ, Wang B, Huskey SE, Ulrich RG, Rushmore TH, Evers R, Evans DC. Activators of the rat pregnane X receptor differentially modulate hepatic and intestinal gene expression. Mol Pharmacol. 2004;65:1159–1171. doi: 10.1124/mol.65.5.1159. [DOI] [PubMed] [Google Scholar]
- Hartz AM, Bauer B, Fricker G, Miller DS. Rapid regulation of P-glycoprotein at the blood-brain barrier by endothelin-1. Mol Pharmacol. 2004;66:387–394. doi: 10.1124/mol.104.001503. [DOI] [PubMed] [Google Scholar]
- Hartz AM, Bauer B, Fricker G, Miller DS. Rapid modulation of P-glycoprotein-mediated transport at the blood-brain barrier by tumor necrosis factor-alpha and lipopolysaccharide. Mol Pharmacol. 2006;69:462–470. doi: 10.1124/mol.105.017954. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Pu H, Andras IE, Eum SY, Yamauchi A, Hennig B, Toborek M. HIV-TAT protein upregulates expression of multidrug resistance protein 1 in the blood-brain barrier. J Cereb Blood Flow Metab. 2006;26:1052–1065. doi: 10.1038/sj.jcbfm.9600254. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Pu H, Tian J, Andras IE, Lee YW, Hennig B, Toborek M. HIV-Tat protein induces P-glycoprotein expression in brain microvascular endothelial cells. J Neurochem. 2005;93:1231–1241. doi: 10.1111/j.1471-4159.2005.03114.x. [DOI] [PubMed] [Google Scholar]
- Hennessy M, Spiers JP. A primer on the mechanics of P-glycoprotein the multidrug transporter. Pharmacol Res. 2007;55:1–15. doi: 10.1016/j.phrs.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Ho EA, Piquette-Miller M. Regulation of multidrug resistance by pro-inflammatory cytokines. Curr Cancer Drug Targets. 2006;6:295–311. doi: 10.2174/156800906777441753. [DOI] [PubMed] [Google Scholar]
- Holmes GL. Seizure-induced neuronal injury: animal data. Neurology. 2002;59:S3–6. doi: 10.1212/wnl.59.9_suppl_5.s3. [DOI] [PubMed] [Google Scholar]
- Hong H, Lu Y, Ji ZN, Liu GQ. Up-regulation of P-glycoprotein expression by glutathione depletion-induced oxidative stress in rat brain microvessel endothelial cells. J Neurochem. 2006;98:1465–1473. doi: 10.1111/j.1471-4159.2006.03993.x. [DOI] [PubMed] [Google Scholar]
- Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001;24:719–725. doi: 10.1016/s0166-2236(00)02004-x. [DOI] [PubMed] [Google Scholar]
- Ito S, Ohtsuki S, Terasaki T. Functional characterization of the brain-to-blood efflux clearance of human amyloid-beta peptide (1-40) across the rat blood-brain barrier. Neurosci Res. 2006;56:246–252. doi: 10.1016/j.neures.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NC, LeCluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- Kemper EM, van Zandbergen AE, Cleypool C, Mos HA, Boogerd W, Beijnen JH, van Tellingen O. Increased penetration of paclitaxel into the brain by inhibition of P-Glycoprotein. Clin Cancer Res. 2003;9:2849–2855. [PubMed] [Google Scholar]
- Kemper EM, Verheij M, Boogerd W, Beijnen JH, van Tellingen O. Improved penetration of docetaxel into the brain by co-administration of inhibitors of P-glycoprotein. Eur J Cancer. 2004;40:1269–1274. doi: 10.1016/j.ejca.2004.01.024. [DOI] [PubMed] [Google Scholar]
- Kipp H, Arias IM. Newly synthesized canalicular ABC transporters are directly targeted from the Golgi to the hepatocyte apical domain in rat liver. J Biol Chem. 2000;275:15917–15925. doi: 10.1074/jbc.M909875199. [DOI] [PubMed] [Google Scholar]
- Kipp H, Arias IM. Trafficking of canalicular ABC transporters in hepatocytes. Annu Rev Physiol. 2002;64:595–608. doi: 10.1146/annurev.physiol.64.081501.155793. [DOI] [PubMed] [Google Scholar]
- Kipp H, Pichetshote N, Arias IM. Transporters on demand: intrahepatic pools of canalicular ATP binding cassette transporters in rat liver. J Biol Chem. 2001;276:7218–7224. doi: 10.1074/jbc.M007794200. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- Kuhnke D, Jedlitschky G, Grube M, Krohn M, Jucker M, Mosyagin I, Cascorbi I, Walker LC, Kroemer HK, Warzok RW, Vogelgesang S. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer's amyloid-beta peptides--implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 2007;17:347–353. doi: 10.1111/j.1750-3639.2007.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, Lee SK, Shankar P, Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- Labialle S, Dayan G, Gayet L, Rigal D, Gambrelle J, Baggetto LG. New invMED1 element cis-activates human multidrug-related MDR1 and MVP genes, involving the LRP130 protein. Nucleic Acids Res. 2004;32:3864–3876. doi: 10.1093/nar/gkh722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labialle S, Gayet L, Marthinet E, Rigal D, Baggetto LG. Transcriptional regulators of the human multidrug resistance 1 gene: recent views. Biochem Pharmacol. 2002;64:943–948. doi: 10.1016/s0006-2952(02)01156-5. [DOI] [PubMed] [Google Scholar]
- Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, Reiner PB. beta-Amyloid efflux mediated by p-glycoprotein. J Neurochem. 2001;76:1121–1128. doi: 10.1046/j.1471-4159.2001.00113.x. [DOI] [PubMed] [Google Scholar]
- Langmade SJ, Gale SE, Frolov A, Mohri I, Suzuki K, Mellon SH, Walkley SU, Covey DF, Schaffer JE, Ory DS. Pregnane X receptor (PXR) activation: a mechanism for neuroprotection in a mouse model of Niemann-Pick C disease. Proc Natl Acad Sci U S A. 2006;103:13807–13812. doi: 10.1073/pnas.0606218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–424. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- Loscher W. Drug transporters in the epileptic brain. Epilepsia. 2007;48(Suppl 1):8–13. doi: 10.1111/j.1528-1167.2007.00993.x. [DOI] [PubMed] [Google Scholar]
- Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6:591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- Marchetti S, Mazzanti R, Beijnen JH, Schellens JH. Concise review: Clinical relevance of drug drug and herb drug interactions mediated by the ABC transporter ABCB1 (MDR1, P-glycoprotein) Oncologist. 2007;12:927–941. doi: 10.1634/theoncologist.12-8-927. [DOI] [PubMed] [Google Scholar]
- McRae MP, Brouwer KL, Kashuba AD. Cytokine regulation of P-glycoprotein. Drug Metab Rev. 2003;35:19–33. doi: 10.1081/dmr-120018247. [DOI] [PubMed] [Google Scholar]
- Miller DS. Xenobiotic export pumps, endothelin signaling, and tubular nephrotoxicants--a case of molecular hijacking. J Biochem Mol Toxicol. 2002;16:121–127. doi: 10.1002/jbt.10030. [DOI] [PubMed] [Google Scholar]
- Mizuno N, Niwa T, Yotsumoto Y, Sugiyama Y. Impact of drug transporter studies on drug discovery and development. Pharmacol Rev. 2003;55:425–461. doi: 10.1124/pr.55.3.1. [DOI] [PubMed] [Google Scholar]
- Molinari A, Calcabrini A, Meschini S, Stringaro A, Crateri P, Toccacieli L, Marra M, Colone M, Cianfriglia M, Arancia G. Subcellular detection and localization of the drug transporter P-glycoprotein in cultured tumor cells. Curr Protein Pept Sci. 2002;3:653–670. doi: 10.2174/1389203023380413. [DOI] [PubMed] [Google Scholar]
- Nwaozuzu OM, Sellers LA, Barrand MA. Signalling pathways influencing basal and H(2)O(2)-induced P-glycoprotein expression in endothelial cells derived from the blood-brain barrier. J Neurochem. 2003;87:1043–1051. doi: 10.1046/j.1471-4159.2003.02061.x. [DOI] [PubMed] [Google Scholar]
- Ohtsuki S, Takizawa T, Takanaga H, Hori S, Hosoya K, Terasaki T. Localization of organic anion transporting polypeptide 3 (oatp3) in mouse brain parenchymal and capillary endothelial cells. J Neurochem. 2004;90:743–749. doi: 10.1111/j.1471-4159.2004.02549.x. [DOI] [PubMed] [Google Scholar]
- Orlowski S, Martin S, Escargueil A. P-glycoprotein and ‘lipid rafts’: some ambiguous mutual relationships (floating on them, building them or meeting them by chance? Cell Mol Life Sci. 2006;63:1038–1059. doi: 10.1007/s00018-005-5554-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen A, Chandler B, Back DJ. The implications of P-glycoprotein in HIV: friend or foe? Fundam Clin Pharmacol. 2005;19:283–296. doi: 10.1111/j.1472-8206.2005.00324.x. [DOI] [PubMed] [Google Scholar]
- Palmieri D, Chambers AF, Felding-Habermann B, Huang S, Steeg PS. The biology of metastasis to a sanctuary site. Clin Cancer Res. 2007;13:1656–1662. doi: 10.1158/1078-0432.CCR-06-2659. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Blood-brain barrier drug targeting: the future of brain drug development. Mol Interv. 2003;3:90–105, 151. doi: 10.1124/mi.3.2.90. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Blood-brain barrier delivery. Drug Discov Today. 2007a;12:54–61. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Blood-brain barrier delivery of protein and non-viral gene therapeutics with molecular Trojan horses. J Control Release. 2007b;122:345–348. doi: 10.1016/j.jconrel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascussi JM, Gerbal-Chaloin S, Duret C, Daujat-Chavanieu M, Vilarem MJ, Maurel P. The tangle of nuclear receptors that controls xenobiotic metabolism and transport: crosstalk and consequences. Annu Rev Pharmacol Toxicol. 2008;48:1–32. doi: 10.1146/annurev.pharmtox.47.120505.105349. [DOI] [PubMed] [Google Scholar]
- Perloff MD, von Moltke LL, Fahey JM, Greenblatt DJ. Induction of P-glycoprotein expression and activity by ritonavir in bovine brain microvessel endothelial cells. J Pharm Pharmacol. 2007;59:947–953. doi: 10.1211/jpp.59.7.0006. [DOI] [PubMed] [Google Scholar]
- Ronaldson PT, Bendayan R. HIV-1 viral envelope glycoprotein gp120 triggers an inflammatory response in cultured rat astrocytes and regulates the functional expression of P-glycoprotein. Mol Pharmacol. 2006;70:1087–1098. doi: 10.1124/mol.106.025973. [DOI] [PubMed] [Google Scholar]
- Rosenfeld JM, Vargas R, Jr., Xie W, Evans RM. Genetic profiling defines the xenobiotic gene network controlled by the nuclear receptor pregnane X receptor. Mol Endocrinol. 2003;17:1268–1282. doi: 10.1210/me.2002-0421. [DOI] [PubMed] [Google Scholar]
- Sachs CW, Chambers TC, Fine RL. Differential phosphorylation of sites in the linker region of P-glycoprotein by protein kinase C isozymes alpha, betaI, betaII, gamma, delta, epsilon, eta, and zeta. Biochem Pharmacol. 1999;58:1587–1592. doi: 10.1016/s0006-2952(99)00240-3. [DOI] [PubMed] [Google Scholar]
- Samoto K, Ikezaki K, Yokoyama N, Fukui M. P-glycoprotein expression in brain capillary endothelial cells after focal ischemia in rat. Acta Neurochir Suppl (Wien) 1994;60:257–260. doi: 10.1007/978-3-7091-9334-1_68. [DOI] [PubMed] [Google Scholar]
- Schlachetzki F, Pardridge WM. P-glycoprotein and caveolin-1alpha in endothelium and astrocytes of primate brain. Neuroreport. 2003;14:2041–2046. doi: 10.1097/00001756-200311140-00007. [DOI] [PubMed] [Google Scholar]
- Scotto KW. Transcriptional regulation of ABC drug transporters. Oncogene. 2003;22:7496–7511. doi: 10.1038/sj.onc.1206950. [DOI] [PubMed] [Google Scholar]
- Seelbach MJ, Brooks TA, Egleton RD, Davis TP. Peripheral inflammatory hyperalgesia modulates morphine delivery to the brain: a role for P-glycoprotein. J Neurochem. 2007;102:1677–1690. doi: 10.1111/j.1471-4159.2007.04644.x. [DOI] [PubMed] [Google Scholar]
- Siddiqui A, Kerb R, Weale ME, Brinkmann U, Smith A, Goldstein DB, Wood NW, Sisodiya SM. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N Engl J Med. 2003;348:1442–1448. doi: 10.1056/NEJMoa021986. [DOI] [PubMed] [Google Scholar]
- Sills GJ, Mohanraj R, Butler E, McCrindle S, Collier L, Wilson EA, Brodie MJ. Lack of association between the C3435T polymorphism in the human multidrug resistance (MDR1) gene and response to antiepileptic drug treatment. Epilepsia. 2005;46:643–647. doi: 10.1111/j.1528-1167.2005.46304.x. [DOI] [PubMed] [Google Scholar]
- Smith QR. A review of blood-brain barrier transport techniques. Methods Mol Med. 2003;89:193–208. doi: 10.1385/1-59259-419-0:193. [DOI] [PubMed] [Google Scholar]
- Sorokin A. Cyclooxygenase-2: potential role in regulation of drug efflux and multidrug resistance phenotype. Curr Pharm Des. 2004;10:647–657. doi: 10.2174/1381612043453117. [DOI] [PubMed] [Google Scholar]
- Spudich A, Kilic E, Xing H, Kilic U, Rentsch KM, Wunderli-Allenspach H, Bassetti CL, Hermann DM. Inhibition of multidrug resistance transporter-1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat Neurosci. 2006;9:487–488. doi: 10.1038/nn1676. [DOI] [PubMed] [Google Scholar]
- Sukhai M, Piquette-Miller M. Regulation of the multidrug resistance genes by stress signals. J Pharm Pharm Sci. 2000;3:268–280. [PubMed] [Google Scholar]
- Tan EK, Chan DK, Ng PW, Woo J, Teo YY, Tang K, Wong LP, Chong SS, Tan C, Shen H, Zhao Y, Lee CG. Effect of MDR1 haplotype on risk of Parkinson disease. Arch Neurol. 2005;62:460–464. doi: 10.1001/archneur.62.3.460. [DOI] [PubMed] [Google Scholar]
- Tan EK, Drozdzik M, Bialecka M, Honczarenko K, Klodowska-Duda G, Teo YY, Tang K, Wong LP, Chong SS, Tan C, Yew K, Zhao Y, Lee CG. Analysis of MDR1 haplotypes in Parkinson's disease in a white population. Neurosci Lett. 2004a;372:240–244. doi: 10.1016/j.neulet.2004.09.046. [DOI] [PubMed] [Google Scholar]
- Tan KH, Purcell WM, Heales SJ, McLeod JD, Hurst RD. Evaluation of the role of P-glycoprotein in inflammation induced blood-brain barrier damage. Neuroreport. 2002;13:2593–2597. doi: 10.1097/00001756-200212200-00042. [DOI] [PubMed] [Google Scholar]
- Tan NC, Heron SE, Scheffer IE, Pelekanos JT, McMahon JM, Vears DF, Mulley JC, Berkovic SF. Failure to confirm association of a polymorphism in ABCB1 with multidrug-resistant epilepsy. Neurology. 2004b;63:1090–1092. doi: 10.1212/01.wnl.0000137051.33486.c7. [DOI] [PubMed] [Google Scholar]
- Teng S, Jekerle V, Piquette-Miller M. Induction of ABCC3 (MRP3) by pregnane X receptor activators. Drug Metab Dispos. 2003;31:1296–1299. doi: 10.1124/dmd.31.11.1296. [DOI] [PubMed] [Google Scholar]
- Terlouw SA, Masereeuw R, Russel FG. Modulatory effects of hormones, drugs, and toxic events on renal organic anion transport. Biochem Pharmacol. 2003;65:1393–1405. doi: 10.1016/s0006-2952(03)00036-4. [DOI] [PubMed] [Google Scholar]
- Theron D, Barraud de Lagerie S, Tardivel S, Pelerin H, Demeuse P, Mercier C, Mabondzo A, Farinotti R, Lacour B, Roux F, Gimenez F. Influence of tumor necrosis factor-alpha on the expression and function of P-glycoprotein in an immortalised rat brain capillary endothelial cell line, GPNT. Biochem Pharmacol. 2003;66:579–587. doi: 10.1016/s0006-2952(03)00340-x. [DOI] [PubMed] [Google Scholar]
- Thevenod F. Nephrotoxicity and the proximal tubule. Insights from cadmium. Nephron Physiol. 2003;93:p87–93. doi: 10.1159/000070241. [DOI] [PubMed] [Google Scholar]
- Thevenod F, Friedmann JM, Katsen AD, Hauser IA. Up-regulation of multidrug resistance P-glycoprotein via nuclear factor-kappaB activation protects kidney proximal tubule cells from cadmium- and reactive oxygen species-induced apoptosis. J Biol Chem. 2000;275:1887–1896. doi: 10.1074/jbc.275.3.1887. [DOI] [PubMed] [Google Scholar]
- Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci U S A. 1987;84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari SB, Amiji MM. A review of nanocarrier-based CNS delivery systems. Curr Drug Deliv. 2006;3:219–232. doi: 10.2174/156720106776359230. [DOI] [PubMed] [Google Scholar]
- Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W, Kunert-Keil C, Walker LC, Warzok RW. Deposition of Alzheimer's beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics. 2002;12:535–541. doi: 10.1097/00008571-200210000-00005. [DOI] [PubMed] [Google Scholar]
- Wilk JN, Bilsborough J, Viney JL. The mdr1a−/− mouse model of spontaneous colitis: a relevant and appropriate animal model to study inflammatory bowel disease. Immunol Res. 2005;31:151–159. doi: 10.1385/IR:31:2:151. [DOI] [PubMed] [Google Scholar]
- Wolka AM, Huber JD, Davis TP. Pain and the blood-brain barrier: obstacles to drug delivery. Adv Drug Deliv Rev. 2003;55:987–1006. doi: 10.1016/s0169-409x(03)00100-5. [DOI] [PubMed] [Google Scholar]
- Zhang H, LeCulyse E, Liu L, Hu M, Matoney L, Zhu W, Yan B. Rat pregnane X receptor: molecular cloning, tissue distribution, and xenobiotic regulation. Arch Biochem Biophys. 1999;368:14–22. doi: 10.1006/abbi.1999.1307. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wu JY, Hait WN, Yang JM. Regulation of the stability of P-glycoprotein by ubiquitination. Mol Pharmacol. 2004;66:395–403. doi: 10.1124/mol.104.001966. [DOI] [PubMed] [Google Scholar]
- Zhu HJ, Liu GQ. Glutamate up-regulates P-glycoprotein expression in rat brain microvessel endothelial cells by an NMDA receptor-mediated mechanism. Life Sci. 2004;75:1313–1322. doi: 10.1016/j.lfs.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]