

Abstract
Niemann-Pick type C (NPC) disease is an autosomal recessive, lethal neurodegenerative disorder. Although neurodegeneration of Purkinje cells in the mouse model (Npc1−/−) is thought to be autonomous, the basis of neuronal death in other regions of the brain remains elusive. We addressed this issue in vivo by using the glial fibrillary acidic protein (GFAP) promoter to direct astrocyte-specific, replacement expression of Npc1 in Npc1−/− mice. These mice showed enhanced survival, decreased neuronal storage of cholesterol associated with less accumulation of axonal spheroids, lower numbers of degenerated neurons and reactive astrocytes and restoration of myelin tracts. Their death was not associated with the usual terminal decline in weight, but instead with a loss of Purkinje cells and motor coordination. We conclude that neurodegeneration of Npc1−/− mice is greatly affected by the loss of fibrillary astrocyte function.
Keywords: Niemann-Pick C, neurodegeneration, fibrillary astrocytes, Purkinje cell, neuron-glia communication, cholesterol storage
INTRODUCTION
Niemann-Pick (type C) disease is a pan-ethnic autosomal recessive disorder of only partially known pathogenesis (Vincent et al., 2003). The etiology of neuropathology has been studied in Npc1−/− mice (a near perfect model of the human disease) but the role of Npc1 in different neuronal cell types has not been established. It has been established that the neurodegeneration seen in Npc1−/− is an autonomous process in the central nervous system (Loftus et al., 2002). We have furthered these results by studying the effects of expression of Npc1 in fibrillary astrocytes.
Signaling between glia and neurons in the central nervous system may offer therapeutic intervention for neurodegenerative disease (Fields et al., 2002). One glia-derived factor that strongly promotes synapse development in cultured and purified CNS neurons is cholesterol complexed to apolipoprotein E-containing lipoproteins (Mauch et al., 2001). This is relevant to NPC since it is a disorder of intracellular cholesterol trafficking with the accumulation of unesterified cholesterol in endosomes and lysosomes (Patterson et al., 2001). The predicted protein for NPC1 contains a sterol-sensing domain consensus site and other motifs that suggest a direct causative role for a mutant product in this altered cholesterol movement (Carstea et al., 1997). The finding that NPC2 encodes a soluble lysosomal protein with cholesterol-binding properties also implicates cholesterol transport in the disease (Naureckiene et al., 2000).
However, the role of NPC1 in neurons compared to glia has yet to be clarified. NPC1 protein was detected predominantly in peri-synaptic astrocytic processes surrounding axon terminals and dendrites (Patel et al., 1999; Hu et al., 2000) in primates but less so in rodents (Falk et al., 1999; Prasad et al., 2000) although sterol synthesis is most abundant in glia in both classes. Xie et al. (2003) found that most cholesterol turnover in Npc1−/− mice was not due to a 24-hydroxylase activity as it is in normal mice. They concluded that the cholesterol turnover in Npc1−/− mice might primarily reflect glial cell and myelin turnover. Oligodendrocytes have been found to be altered in the Npc1−/− brain: Takikita et al. (2004) found that pre-myelinating oligodendrocytes were abundant in the Npc1−/− brain but pi-glutathione-S-transferase positive mature oligodendrocytes were decreased and polysialylated-NCAM, which is a negative regulator of myelinization, persisted in the Npc1−/− as compared to normals. In addition, Apolipoprotein D, an abundant astrocyte product (Patel et al., 1995) is up-regulated in Npc1−/− mice (Yoshida et al., 1996). Collectively, these data suggest that abnormal NPC1 function in glia may impact lipid trafficking between neurons and glial cells (Ong et al., 2001).
It has been shown that neurodegeneration in Npc1−/− mice is cell autonomous in the cerebellum (Ko et al., 2005) but not in other regions of the brain. That study was performed with chimeras of Npc1−/− and wild type cells. We have studied the function of glia in a possible non-autonomous role and in other parts of the brain by directing Npc1 expression to one class of glial cells, fibrillary astrocytes, using the Glial Fibrillary Acidic Protein (GFAP) promoter that has successfully been used to specifically direct the expression of a number of proteins in astrocytes (Brenner et al., 1994). We found significant improvement in the Npc1−/− mice, suggesting that the lack of NPC1 in astrocytes makes a major contribution to the neuropathology of NPC.
MATERIALS AND METHODS
The GFAP promoter was provided by Dr. Lennart Mucke of the Gladstone Institute of Neurological Disease, UCSF (Johnson et al., 1995). While some cases of aberrant expression have been recorded, a large number of studies have found astrocyte only localization of the promoted gene products (e.g. Nolte et al., 2001; Suzuki et al., 2003; Zhou et al., 1997). The mouse Npc1 cDNA was released by NotI and XhoI digestion from pBSIIKS(+)-Npc1 (gift of Dr. SK Loftus, NIH). Restriction sites of NotI and Xhol were introduced into the C-3123 plasmid containing the GFAP promoter, at the 3′ end of the promoter, by PCR amplification, using a pair of primers: 5′ – ACTAGCGGCCGCGGGTACAATTCCG – 3′ and 5′ –TACCCTCGAGCGGGGATCCAGAC – 3′. Then the Npc1 gene was ligated at the NotI and XhoI sites downstream of GFAP promoter in the C-3123 plasmid. The SV40 splice and polyA sites are maintained. SfiI was used to release the cassette for microinjection into mouse embryos. Injection was performed by the Genetically Modified Mouse Service of the University of Arizona into C57BL/6J X DBA/2J F2 zygotes by standard techniques (F1 parents are used to avoid the 2-cell block to development). Positive transgenics and their progeny were identified by PCR using a GFAP/C-3123 boundary specific forward primer and an Npc1 specific reverse primer. PCR conditions were as follows: 3 min initial denaturation at 95°C and 35 cycles of 95°C for 30 sec, 58°C for 30 sec and 72°C for 1 minute in a Peltier Thermal cycler (MJ Research, INC. USA). The reaction mixture of 25μl final volume contained 25 pmol primers, 2.5 mM MgC12, 1/25 U Taq DNA polymerase (PIERCE). The transgenic positive mice are crossed to Npc1+/− carriers maintained on a BALB/cJ background. Tg positive Npc1+/− are crossed to the Npc1+/− mice maintained on a BALB/cJ background and tg positive, Npc1−/− are studied (about 1/8 of offspring since the transgene and Npc1 are not linked; the background is thus ¾ BALB/cJ).
For quantitative PCR, we designed a primer set in exon 1 of the Npc1 gene, with the reverse primer in the deleted portion of the mutant allele. We used these primers in realtime PCR using iTaq SYBR Green Supermix with ROX (Bio-Rad) and compared the signal to that for ApoB, which we know represents 2 copies. We used the formula 2−ΔCt = fold change to infer the number of copies. A fold change of 1 means that there are 2 copies present. In our initial line and four more, we found the indicated values for our transgene copy number (Table 1).
Table I.
Copy Number of GFAP Npc1 Transgenic Mice
| Line | Fold-Differencea | Copy Numberb |
|---|---|---|
| A | (5) 6.93 ± 1.8 | ~12 |
| B | (3) 36.4 ± 12.9 | ~70 |
| C | (3) 1.26 ± 0.14 | 0c |
| D | (3) 0.94 ± 0.11 | 0c |
| E | (3) 12.9 ± 2.2 | ~24 |
Over diploid wild-type, (n) ± std. dev
Fold difference X 2, −2
There must be at least one
For neuropathological studies, the brains were dissected rapidly on ice, hemisected in the sagittal plane and fixed in fresh 4% paraformaldehyde, 1x PBS for 24 hours before being washed 3x in 1 x PBS. They were paraffin-embedded and sectioned for immunohistochemistry. Paraffin embedded brain sections were stained with NPC1 polyclonal antibody directed to a.a 1254–1273 near the C-terminus. It was peptide-affinity purified to this peptide, and found to react with wild-type Npc1 protein. It cross-reacts with mouse Npc1 and no-reactivity is found in Npc1−/−. It is the generous gift of Dr. William Sherman Garver. Antibody to SM132 (recognizing a dephosphorylated neurofilament epitope) is from Sternberger Monoclonals (Lutherville, MD) and GFAP and Calabrindin-D-28k antibodies were from Sigma (St. Louis, MO). Fluorojade B™ was obtained from Chemicon, Temecula, CA. Brain regions are defined in Zhang et al. (2004). For axonal spheroid quantitation, the numbers of axonal spheroids 8 μm or larger in a 925 × 740 μm field were counted in anatomically matched sections of three brain regions. For each animal, spheroids were counted in two adjacent sections and averaged. The average number of spheroids per animal was then averaged within genotypes.
Motor coordination was tested with the balance beam coordination test. Mice were placed on the center of a horizontal round beam (covered in laboratory tape, 120 cm long, 2 cm in diameter, and sectioned into twelve 10 cm sections) at 50 cm above the bench top level. The number of sections the mouse crossed in 3 minutes was determined. The retention time was also recorded if the mouse failed to stay on the beam for three minutes. This test was repeated for an additional trial and the averages of both trials were recorded.
RESULTS
We initially studied the first transgenic line obtained with the GFAPNpc1 construct, line GFAPNpc1A. This transgene enhanced survival in Npc1−/− mice from 73.2 ± 8.1 days (N = 14) to 87.3 ± 16.6 days (N = 8), p = 0.014 (Fig. 1). These transgenic mice did not have significantly improved weight curves (Fig. 2), but had improved balance beam performance up to 50 days of age (Fig. 3). We subsequently obtained more transgenic lines and determined the number of transgene copies by quantitative real time PCR (Table 1). The initial line, GFAPNpc1A, had approximately 12 copies while the GFAPNpc1E line has approximately 24 copies. Line B, with approximately 70 copies, was not sufficiently fertile to be maintained. The other two lines were not studied since lines GFAPNpc1A and GFAPNpc1E confirmed each other. GFAPNpc1E, Npc1−/− mice have a survival of 172 ± 23.8 days (N = 10), p ≤ 0.0001 – a survival nearly 2.5 times that of Npc1−/− mice (Fig. 1) and improved balance beam performance (Fig. 3). While they did not continue to gain weight after approximately 5 weeks, they did not show a terminal decline in weight (Fig. 2). We also found that the GFAPNpc1E, Npc1−/− mice are fertile, which allowed us to interbreed them. As expected, all offspring were Npc1−/−. Of 10 in 1 litter, 8 were transgene positive. The number of transgene copies was determined in the positive offspring by Quantitative PCR (Table 2). The double transgenics (i.e. homozygotes for the transgene) survived over 230 days (three times the usual survival, see Fig. 1) and also did not show a terminal decline in weight (data not shown).
Figure 1. Survival and weights of transgenic and control Npc1−/− mice.

Kaplan-Meyer survival curves of Npc1−/−, ············; GFAPNpc1A, Npc1−/−, - - - -; GFAPNpc1E, Npc1−/−, ———; and GFAPNpc1E, Npc1−/− double transgenic,  mice.
mice.
Figure 2. Weights of transgenic and control Npc1−/− mice.

Weights of Npc1−/−, ············; GFAPNpc1A, Npc1−/−, - - - -; and GFAPNpc1E, Npc1−/−, ——— transgenic and control (Npc1+/+) mice, .
Figure 3. Balance Beam performance of transgenic and control Npc1−/− mice.

Number of sections crossed in Npc1−/−, ············; GFAPNpc1A, Npc1−/−, - - - -; GFAPNpc1E, Npc1−/−, ——— and control (Npc1+/+) mice, .
Table II.
Copy Number of GFAPNpc1E × GFAPNpc1E Offspring
| Animal Number | Fold-differencea | Copy Numberb |
|---|---|---|
| 8200 | 15.67 | ~28 |
| 8201 | 15.35 | ~28 |
| 8202 | 29.04 | ~56 |
| 8203 | 30.91 | ~56 |
| 8204 | ND | – |
| 8205 | 14.32 | ~28 |
| 206 | 14.93 | ~28 |
| 8208 | 30.91 | ~56 |
Over diploid wild-type
Fold difference X 2, −2
Double labeling of NPC1 and the astrocyte marker, GFAP, confirmed astrocyte-specific NPC1 expression in the GFAPNpc1, Npc1−/− mice (Fig. 4). Immunoreactivity was seen throughout the brains of control wild-type mice (wild-type, only cerebellum shown), in scattered astrocytes and also as halos of astrocytic processes around Purkinje neurons (Fig. 4A). In Npc1−/− mice only a faint background was observed and empty spaces are seen where Purkinje neurons once resided (Fig. 4B). In the GFAPNpc1, Npc1−/− mice, Npc1 immunoreactivity was markedly increased above wild-type and localized to astrocytes (Fig. 4C). No staining was observed when the primary antibody step was omitted (-Npc1 Ab; Fig. 4D). Triple immunofluorescence staining of GFAPNpc1, Npc1−/− mouse brain with Npc1 Ab (red), GFAP Ab as a marker for astrocytes (green), and DAPI for nuclei (blue), showed co-localization of Npc1 expression in some astrocytes, yellow (Fig. 4F), which supports appropriate expression of the wild-type Npc1 transgene in astrocytes.
Figure 4. Wild-type Npc1 expression in GFAPNpc1 mice.

Peptide affinity purified antibody was applied to paraffin embedded sections from wild-type (wt, BALB/cJ, Npc1+/+), Npc1−/− (6 –11.5 weeks), and GFAPNpc1, Npc1−/− (GFAP Tg) mice (6 – 21.5 weeks) to examine levels and distribution of Npc1 protein. Representative sections are shown. A–D sections were counterstained with Meyer’s Hematoxylin (blue) to reveal cellular anatomy. E, F: Npc1 staining is red, GFAP is green, overlay is yellow, DAP1 (nuclei) is blue. White arrows: astrocytes and astrocyte processes. Black arrows: Purkinje neurons. Large boxes, 20X; insets, 40X.
The neuropathology of NPC includes accumulation of lipids in affected neurons and glia, progressive demyelination, neuroaxonal dystrophy with formation of axonal spheroids, and neurodegeneration (Bu et al., 2002; German et al., 2001). GFAPNpc1, Npc1−/− mice demonstrated reduced numbers of axonal spheroids (a well characterized lesion in NPC), decreased numbers of reactive astrocytes, and restoration of myelin tracts but persistent loss of Purkinje cells (Figs. 5 and 7). We also noticed that, in addition to the decrease in axonal spheroids, the normal localization of SM132 immunoreactivity towards neurofilament proteins is restored in the GFAPNpc1E, Npc1−/− mice (Fig. 6A). Fluorojade B™, a marker for degenerating neurons, revealed a significant attenuation of degenerating neurons in the cortex and pons of the GFAPNpc1E, Npc1−/− mice (Fig. 6B and C). There appeared to be an overall decrease in the numbers of Fluorojade B™-positive neurons in GFAPNpc1E, Npc1−/− mice, but the difference was most striking in the cerebral cortex (Fig. 6B). In Npc1−/− mice, nearly all neurons (arrows) were labeled with Fluorojade B™ (green) by 9 weeks of age, but in GFAPNpc1E, Npc1−/− mice, even at 17.5 weeks of age, most neurons were not labeled and appeared as dark empty spaces (arrowheads). Using SMI32, a neurofilament antibody, to decorate the soma of neurons, we found marked reduction of lipid accumulation (stained by filipin, blue) in neurons of the brainstem (arrow heads) of the GFAPNpc1E, Npc1−/− mice relative to (arrows) Npc1−/− mice (Fig. 7).
Figure 5. Neuropathological evaluation in GFAPNpc1 Tg mice.
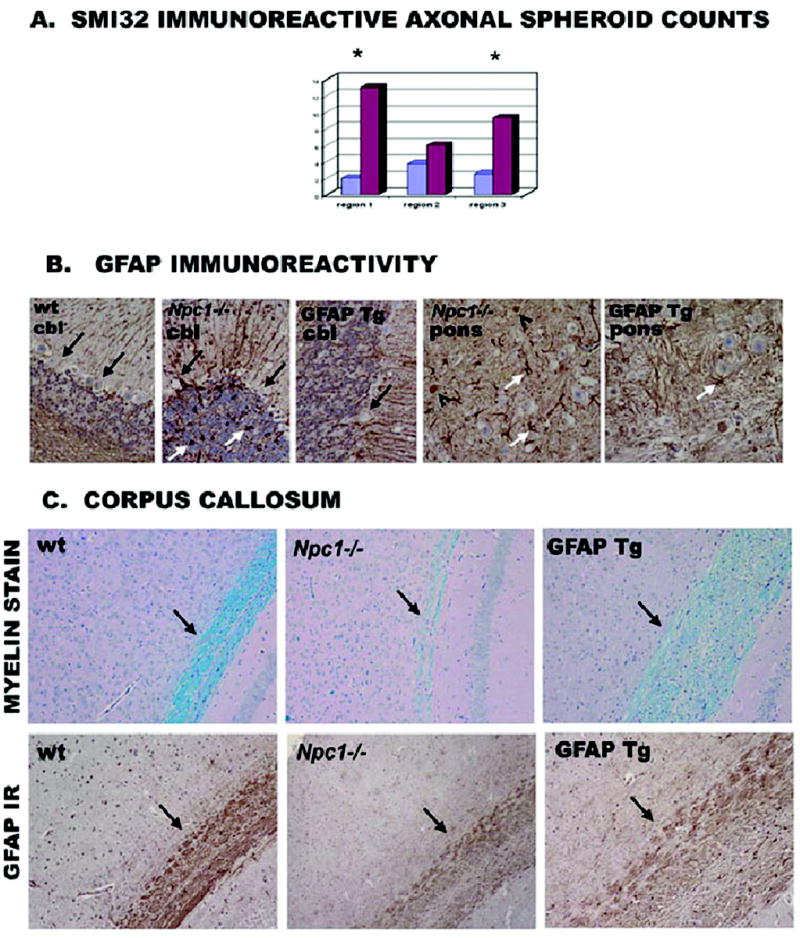
A) SMI32 immunoreactivity for axonal spheroid analysis. Y axis, number of spheroids; blue = GFAPNpc1E, Npc1−/−, purple = Npc1−/−. Sections from GFAPNpc1E, Npc1−/− (N=4) and Npc1−/−(N=5) mice were stained with SM132 non-phospho neurofilament antibody. Region 1: white matter of cerebellum; region 2: dorsal region of pons; and region 3: central regions of pons (* indicates significant [p ≤ 0.05] decrease in spheroid number by students t-test). B) GFAP immuno-reactivity. Sections stained with GFAP antibody showed intensified immunoreactivity in Npc1−/− mice along with numerous reactive astrocytes (white arrows) shown in cerebellum (cbl, left 3 panels) and pons (right 2 panels) wt = Npc1+/+; GFAP Tg = GFAPNpc1A, Npc1−/−. GFAP negative neurons were evident in the Purkinje layer (black arrows) where cell death was apparent in both Npc1−/− and GFAPNpc1A, Npc1−/− mice. GFAP-positive spheroids (black open arrowheads) are evident in Npc1−/− mice (40X power). C) Corpus Callosum, panel designations as in B. Staining of myelin with the histological dye luxol fast blue (black arrows, top row) showed a significant reduction in Npc1−/− mice, but some restoration in GFAPNpc1A, Npc1−/− mice (20X). To visualize long fibers within the corpus callosum, we also stained some sections with GFAP antibody (GFAP IR) which detects astrocytes whose fibers wrap the long axons of passage (black arrow, bottom row). Thus, astrocytic expression of wild-type Npc1 increases GFAP-positive fibers in the corpus callosum (20X power).
Figure 7. Correction of cholesterol storage as detected by filipin (A) and loss of Purkinje cells as detected with Calbindin (B) in Npc1−/− mice.
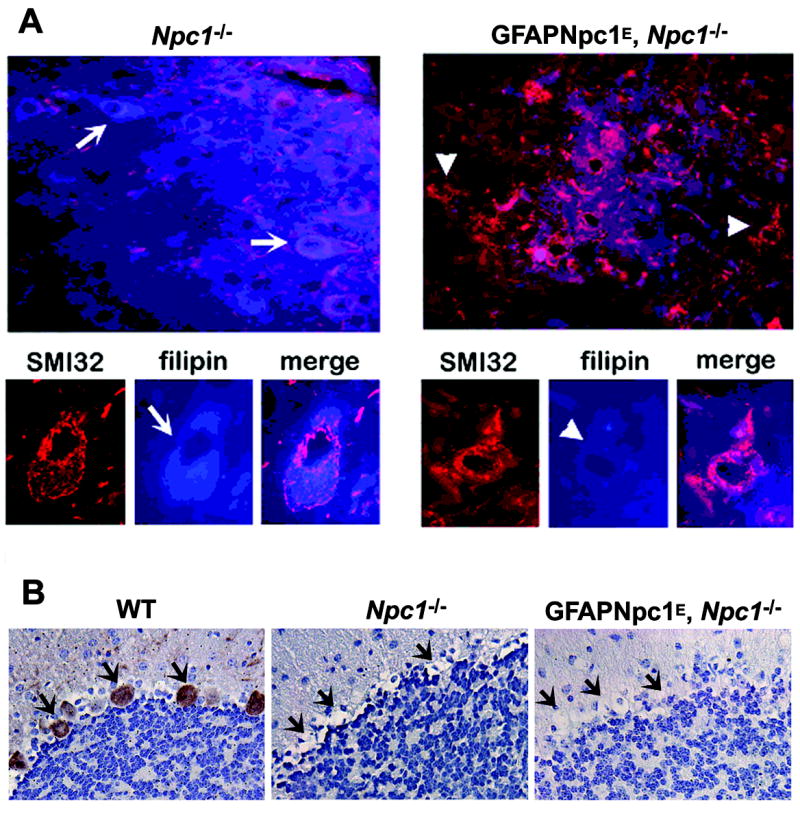
A) Eight week old Npc1−/− and GFAPENpc1, Npc1−/− frozen mouse brain sections (n = 3, each group) were double stained with filipin (blue) and SMI32 antibody (red). Fluorescence images were taken at low (40X, top row) and higher (100X, bottom row) magnification to show specific lipid reduction in SMI32-labeled neurons. Representative sections are shown. The arrowheads point to neurons with less lipid and the arrows point to neurons with massive lipid accumulation. B) Brain tissues from 8 week old Npc1+/+, Npc1−/− and GFAPNpc1, Npc1−/− were analyzed using immunohistochemistry with Calbindin-D-28k antibody to show the presence and absence of Purkinje cells (arrow). Compared to that of wild-type mice, the entire Purkinje cell layer was lost in Npc1−/− and transgenic mice (40x).
Figure 6. Restoration of neurofilament proteins (A) and correction of neurodegeneration as detected by Fluorojade B™ (B, C) in GFAPNpc1E, Npc1−/− mice.
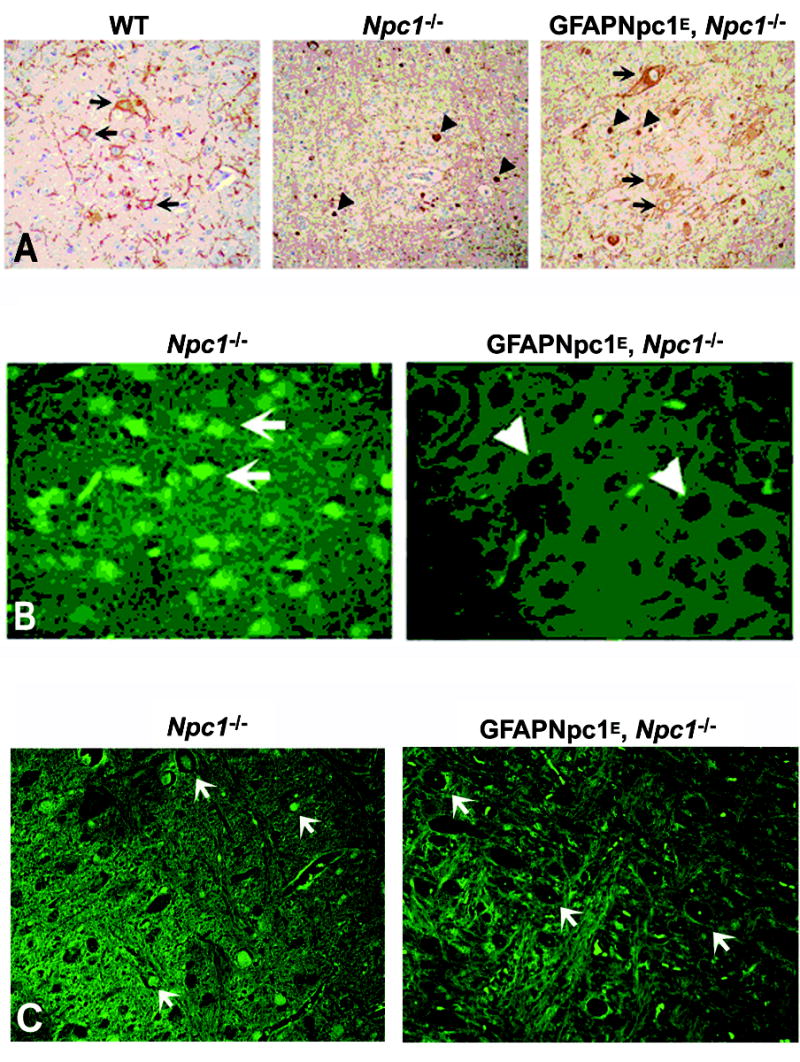
A) Fixed brain sections from 9 week old wildtype (WT = Npc1+/+), Npc1−/− (n = 5), and GFAPNpc1E, Npc1−/− (GFAP Tg; n = 4) mice were immunolabeled with SMI32 antibody (brown). Representative sections are shown. Longer axons and cell bodies (arrows) were seen in WT and GFAPNpc1E, Npc1−/− mice, but not in Npc1−/− mice. Spheroids (arrowheads) were seen in Npc1−/−, and were fewer in number in GFAPNpc1E, Npc1−/− mice. SM132 normally labels neuronal cell bodies (arrows) and axonal processes in the brainstem. In NPC this labeling is absent, and instead SM132 immunoreactivity is observed in spheroids (arrowheads). In the GFAPNpc1E, Npc1 mice, SM132 immunoreactivity reappears in neuronal cell bodies (arrows) and axons. Magnification: 40X. B and C) Frozen brain sections from 9 week old Npc1−/− and 17.5 week old GFAPNpc1E, Npc1−/− mice (n = 3, each group) were stained with Fluorojade B™ for detection of degenerating neurons. Representative sections are shown for cortex (B) and pons (C). Most neurons in Npc1−/− mice displayed Fluorojade B™ fluorescence (arrows), but in GFAPNpc1E, Npc1−/− mice, the neurons were dark in appearance as they are in Npc1+/+ neurons (arrowheads) (40X power).
DISCUSSION
Loftus et al. (2002) studied the impact of visceral pathology on the neurodegeneration in Npc1−/−. The wild-type Npc1 gene was re-introduced into Npc1−/− mice by targeting its expression primarily to the CNS through the use of the prion protein promoter. Interestingly, neurodegeneration was prevented, life span was normalized, and the sterility of Npc1−/− female mice was corrected. The rescue did not completely rectify the accumulation of GM2 or GM3 gangliosides in some neurons and glia (Loftus et al., 2002). We have extended these studies and find that GFAP promoter-driven, replacement expression of wild type Npc1 protein in fibrillary astrocytes ameliorates degeneration, cytoskeletal abnormalities, and pathological lipid accumulation in neurons. Although the genetic background of the transgenics is not 100% pure BALB/cJ (75% BALB/cJ), as is the background of the Npc1−/− mice, genetic modifiers only shift disease onset (or survival) by several weeks (Zhang and Erickson, 2000).
It is likely that the improved behavior and survival of the GFAPNpc1, Npc1−/− mice may be attributed to lower lipid build-up in neurons and restoration of neuronal function. There are many glial functions that could be involved. Secreted factors include neurotrophins such as glial cell line-derived neurotropic factor (GDNF; Gill et al., 2003) and thrombospondins (Christopherson et al., 2005). For instance, direct infusion of GDNF into the brain is found to be of therapeutic value in Parkinson disease (Gill et al., 2003). Glia may also produce toxins affecting neuronal function (Custer et al., 2006; Nagai et al., 2007) and more toxins might be produced by glia with large accumulations of cholesterol. Alternative hypotheses would include restoring glial “normalcy” so that the early (2 week) activation of astrocytes and microglia did not occur (Baudry et al., 2003) or restoration of neurosteroid synthesis (Griffin et al., 2004; Ahmad et al., 2005) which predominantly occurs in glia.
The finding that astrocyte-only over-expression of Npc1 profoundly alters the course of neurodegeneration in Npc1−/− mice strongly implicates astrocytes in the pathogenesis of the neurodegeneration or dysmyelination (Weintraub et al., 1987). Astrocytes are the major site of synthesis of cholesterol in the brain and secrete it via the ABCA1 transporter to make HDL-like particles (Pfrieger, 2003). In vitro studies indicate that glia-secreted cholesterol-laden ApoE is a neurotrophic factor (Mauch et al., 2001). Our data are compatible with the suggestion that abnormal NPC1 function in glia impacts lipid trafficking between neurons and glial cells (Ong et al., 2001). It is likely that receptor recycling, synaptic vesicle dynamics, neuronal plasticity and maintenance of the integrity of the myelin sheath may be critically dependent on intrinsic sterol cycling between glia and neurons. This cycling may be particularly critical for axonal surface cholesterol (Tashiro et al., 2004). ApoE is primarily secreted by astrocytes, both central and peripheral (Boyles et al., 1985), while knockout of the ApoE receptor and VLDL receptor causes severe neurodegeneration (Trommsdorff et al., 1999). NPC1 and NPC2 might be key regulators of this cycling process. However, cultured Npc1−/− astroglia (which may not replicate in vivo astroglia) did not show altered lipoprotein production (Mutka et al., 2004; Karten et al., 2005). Thus, as mentioned, other glial-derived factors could be important.
While our data support the notion that astrocytic expression of wild-type Npc1 can improve the disease state of Npc1−/− mice, it appears to be insufficient for preventing Purkinje cell loss. Purkinje cells are the first neurons known to degenerate in NPC disease (Morris et al., 1982). Using chimeras of Npc1−/− and normal cells, it has been found that Npc1−/− Purkinje cell death was autonomous (Ko et al., 2005). Purkinje cell loss occurred even when wild-type Bergmann glia were in the immediate vicinity and wild-type Purkinje cells survived even if surrounded by mutant glia (Ko et al., 2005). Perhaps astrocytes in other brain regions may influence NPC neuropathology in a different way than in the cerebellum or the very large Purkinje cells may be less responsive to partially corrected astrocyte function. Of note, although cerebellar correction was not found in the GFAPNpc1E mice, there was improvement in motor performance (Fig. 3).
The finding that GFAPNpc1E, Npc1−/− mice did not show a terminal decline in weight was unexpected and may suggest a role of Npc1 in intestinal functions. Patients with NPC frequently have diarrhea (Patterson et al., 2001). Enteric astroglia send processes which envelop axon bundles, potentially affecting function of the neuronal fibers and projections (Gershon et al., 1991). The GFAP promoter has been used to ablate enteric glia in mice carrying a GFAP promoter, herpes simplex TK tg and treated with ganciclovir (Bush et al., 1998). In the past, we have presumed that weight loss in Npc1−/− was secondary to lack of eating and drinking due to motor incoordination. GFAPNpc1E, Npc1−/− transgenics die while still at reasonable weights, suggesting a possible prevention of neurodegeneration in the enteric nervous system.
The recovery in fertility of the GFAPNpc1E, Npc1−/− was also unpredicted. Previous studies have shown that male fertility in Npc1−/− mice can be restored by changing the genetic background on which the mutant is maintained (Erickson et al., 2002). Female infertility was corrected by estrogen treatment which restored the hypothalamic-pituitary-ovarian loop (Gévry et al., 2004) or adding the mdr1a−/− knockout to the Npc1−/− females (Erickson et al., 2002). The first of these 2 studies concluded that “reduction in estrogen synthesis by the ovary was responsible for aberrant expression of prolactin in the pituitary gland” (Gévry et al., 2004). However, our results with astrocyte-only expression suggests that neurodegeneration of cells involved in the hypothalamic-pituitary axis could be primary.
In summary, we describe dramatic in vivo changes after rescue of Npc1 function in astrocytes. The critical role for astrocytes in neurodegeneration may have important implications for designing therapeutic treatment for NPC and other neurodegenerative disorders (Maragakis et al., 2006).
Acknowledgments
We thank Ms. Jessica McVey for administrative support and Ms. Elizabeth Chaitkin for technical support.
Grant information: Research supported by NIH 5RO1 EB000343-05, NCI 2P30 CA023074-26 and the Holsclaw Family Professorship of Human Genetics and Inherited Disease.
References
- Ahmad I, Lope-Piedrafita S, Bi X, Hicks C, Yao Y, Yu C, Chaitkin E, Howison CM, Weberg L, Trouard TP, Erickson RP. Allopregnanolone treatment, both as a single injection or repetitively, delays demyelination and enhances survival of Niemann-Pick C mice. J Neurosci Res. 2005;82:811–821. doi: 10.1002/jnr.20685. [DOI] [PubMed] [Google Scholar]
- Baudry M, Yao Y, Simmons D, Liu J, Bi X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp Neurol. 2003;184:887–903. doi: 10.1016/S0014-4886(03)00345-5. [DOI] [PubMed] [Google Scholar]
- Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest. 1985;76:1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu B, Klunemann H, Suzuki K, Li J, Bird T, Jin L-W, Vincent I. Niemann-Pick disease type C yields possible clue for why cerebellar neurons do not form neurofibrillary tangles. Neurobiol Dis. 2002;11:285–297. doi: 10.1006/nbdi.2002.0551. [DOI] [PubMed] [Google Scholar]
- Bush TG, Savidge TC, Freeman TC, Cox HJ, Campbell EA, Mucke L, Johnson MH, Sofroniew MV. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. 1998;93:189–201. doi: 10.1016/s0092-8674(00)81571-8. [DOI] [PubMed] [Google Scholar]
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ, Dwyer NK, Neufeld EB, Chang TY, Liscum L, Strauss JF, 3rd, Ohno K, Zeigler M, Carmi R, Sokol J, Markie D, O’Neill RR, van Diggelen OP, Elleder M, Patterson MC, Brady RO, Vanier MT, Pentchev PG, Tagle DA. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T, Westrum LE, Sopher BL, La Spada AR. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- Erickson RP, Kiela M, Devine PJ, Hoyer PB, Heidenreich RA. mdr1a deficiency corrects sterility in Niemann-Pick C1 protein deficient female mice. Mol Reprod Dev. 2002;62:167–173. doi: 10.1002/mrd.10093. [DOI] [PubMed] [Google Scholar]
- Falk T, Garver WS, Erickson RP, Wilson JM, Yool AJ. Expression of Niemann-Pick type C transcript in rodent cerebellum in vivo and in vitro. Brain Res. 1999;839:49–57. doi: 10.1016/s0006-8993(99)01678-9. [DOI] [PubMed] [Google Scholar]
- Fields D, Steven-Graham B. New insights into neuron-glia communication. Science. 2002;298:556–562. doi: 10.1126/science.298.5593.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German DC, Quintero EM, Liang CL, Ng B, Punia S, Xie C, Dietschy JM. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J Comp Neurol. 2001;433:415–425. doi: 10.1002/cne.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershon MD, Rothman TP. Enteric glia. Glia. 1991;4:195–204. doi: 10.1002/glia.440040211. [DOI] [PubMed] [Google Scholar]
- Gévry NY, Lopes FL, Ledoux S, Murphy BD. Aberrant intracellular cholesterol transport disrupts pituitary and ovarian function. Mol Endocrin. 2004;18:1778–1786. doi: 10.1210/me.2003-0323. [DOI] [PubMed] [Google Scholar]
- Gill SS, Patel NK, Hotton GR, O’Sullivan K, McCarter R, Bunnage M, Brooks DJ, Svendsen CN, Heywood P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nature Med. 2003;9:589–595. doi: 10.1038/nm850. [DOI] [PubMed] [Google Scholar]
- Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat Med. 2004;10:704–711. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- Hu CY, Ong WY, Patel SC. Regional distribution of NPC1 protein in monkey brain. J Neurocytol. 2000;29:765–773. doi: 10.1023/a:1010942521671. [DOI] [PubMed] [Google Scholar]
- Johnson WB, Ruppe MD, Rockenstein EM, Price J, Sarthy VP, Verderber LC, Mucke L. Indicator expression directed by regulatory sequences of the glial fibrillary acidic protein (GFAP) gene: in vivo comparison of distinct GFAP-lacZ transgenes. Glia. 1995;13:174–184. doi: 10.1002/glia.440130304. [DOI] [PubMed] [Google Scholar]
- Karten B, Hayashi H, Francis GA, Campenot RB, Vance DE, Vance JE. Generation and function of astroglial lipoproteins from Niemann-Pick type C1-deficient mice. Biochem J. 2005;387:779–788. doi: 10.1042/BJ20041694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko DC, Milenkovic L, Beier SM, Manuel H, Buchanan J, Scott MP. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 2005;1:81–95. doi: 10.1371/journal.pgen.0010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus SK, Erickson RP, Walkley SU, Bryant MA, Incao A, Heidenreich RA, Pavan WJ. Rescue of neurodegeneration in Niemann-Pick C mice by a prion-promoter-driven Npc1 cDNA transgene. Hum Mol Genet. 2002;11:3107–3114. doi: 10.1093/hmg/11.24.3107. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Morris MD, Bhuvaneswaran C, Shio H, Fowler S. Lysosome lipid storage disorder in NCTR-BALB/c mice. I. Description of the disease and genetics. Am J Pathol. 1982;108:140–149. [PMC free article] [PubMed] [Google Scholar]
- Mutka A-L, Lusa S, Linder MD, Jokitalo E, Kopra O, Jauhiainen M, Ikonen E. Secretion of sterols and the NPC2 protein from primary astrocytes. J Bio Chem. 279:48654–48662. doi: 10.1074/jbc.M405345200. [DOI] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–662. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- Nolte C, Matyash M, Pivneva T, Schipke CG, Ohlemeyer C, Hanisch UK, Kirchhoff F, Kettenmann H. GFAP promoter-controlled EGFP-expressing transgenic mice: a tool to visualize astrocytes and astrogliosis in living brain tissue. Glia. 2001;33:72–86. [PubMed] [Google Scholar]
- Ong WY, Kumar U, Switzer RC, Sidhu A, Suresh G, Hu CY, Patel SC. Neurodegeneration in Niemann-Pick type C disease mice. Exp Brain Res. 2001;141:218–231. doi: 10.1007/s002210100870. [DOI] [PubMed] [Google Scholar]
- Patel SC, Asotra K, Patel YC, McConathy WJ, Patel RC, Suresh S. Astrocytes synthesize and secrete the lipophilic ligand carrier apolipoprotein D. Neuroreport. 1995;6:653–657. doi: 10.1097/00001756-199503000-00017. [DOI] [PubMed] [Google Scholar]
- Patel SC, Suresh S, Kumar U, Hu CY, Cooney A, Blanchette-Mackie EJ, Neufeld EB, Patel RC, Brady RO, Patel YC, Pentchev PG, Ong WY. Localization of Niemann-Pick C1 protein in astrocytes: implications for neuronal degeneration in Niemann- Pick type C disease. Proc Natl Acad Sci USA. 1999;96:1657–1662. doi: 10.1073/pnas.96.4.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea ED, Neufeld EB, Blanchette-Mackie JE, Pentchev PG. Niemann-Pick disease type C: a lipid trafficking disorder. In: Scriver SR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8. New York: McGraw-Hill; 2001. pp. 3611–3634. [Google Scholar]
- Pfrieger FW. Outsourcing in the brain: do neurons depend on cholesterol delivery by astrocytes? Bioessays. 2003;25:72–78. doi: 10.1002/bies.10195. [DOI] [PubMed] [Google Scholar]
- Prasad A, Fischer WA, Maue RA, Henderson LP. Regional and developemtnal expression of the Npc1 mRnA in the mouse brain. J Neurochem. 2000;75:1250–1257. doi: 10.1046/j.1471-4159.2000.0751250.x. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Watanabe J, Satoru A, Funahashi H, Kikuyama S, Shioda S. A transgenic mouse model for the detailed morphological study of astrocytes. Neurosci Res. 2003;47:451–454. doi: 10.1016/j.neures.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Takikita S, Fukuda T, Mohri I, Yagi T, Suzuki K. Perturbed myelination process of premyelinating oligodendrocyte in Niemann-Pick type C mouse. J Neuropathol Exp Neurol. 2004;63:660–673. doi: 10.1093/jnen/63.6.660. [DOI] [PubMed] [Google Scholar]
- Tashiro Y, Yamazaki T, Shimada Y, Ohno-Iwashita Y, Okamoto K. Axon-dominant localization of cell-surface cholesterol in cultured hippocampal neurons and its disappearance in Nieamann-Pick type C model cells. Euro J Neurosci. 2004;20:2015–2021. doi: 10.1111/j.1460-9568.2004.03677.x. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Vincent I, Bu B, Erickson RP. Understanding Niemann-pick type C disease: a fat problem. Curr Opin Neurol. 2003;16:155–161. doi: 10.1097/01.wco.0000063764.15877.1c. [DOI] [PubMed] [Google Scholar]
- Weintraub H, Abramaovici A, Sandbank U, Booth AD, Pentchev PG, Sela B. Dysmyelination in NCTR-Balb/C mouse mutant with a lysosomal storage disorder. Acta Neuropathol. 1987;74:374–381. doi: 10.1007/BF00687215. [DOI] [PubMed] [Google Scholar]
- Xie C, Lund EG, Turley SD, Russell DW, Dietschy JM. Quantitation of two pathways for cholesterol excretion from the brain in normal mice and mice with neurodegeneration. J Lipid Res. 2003;44:1780–1789. doi: 10.1194/jlr.M300164-JLR200. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Cleaveland ES, Nagle JW, French S, Yaswen L, Oshima T, Brady RO, Pentchev PG, Kulkarni AB. Molecular cloning of the mouse Apolipoprotein D gene and Its upregulating expression in Niemann-Pick disease type C mouse model. DNA Cell Biol. 1996;15:873–882. doi: 10.1089/dna.1996.15.873. [DOI] [PubMed] [Google Scholar]
- Zhang J, Erickson RP. A modifier of Niemann Pick C 1 maps to mouse chromosome 19. Mamm Genome. 2000;11:69–71. doi: 10.1007/s003350010013. [DOI] [PubMed] [Google Scholar]
- Zhang M, Li J, Chakrabarty P, Bu B, Vincent I. Cyclin-dependent kinase inhibitors attenuate protein hyperphosphorylation, cytoskeletal lesion formation, and motor defects in Niemann-Pick Type C mice. Am J Path. 2004;165:843–853. doi: 10.1016/S0002-9440(10)63347-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Sun B, Zhang CL, Fine A, Chiu SY, Messing A. Live astrocytes visualized by green fluorescent protein in transgenic mice. Dev Biol. 1997;187:36–42. doi: 10.1006/dbio.1997.8601. [DOI] [PubMed] [Google Scholar]