

Abstract
We propose models for in vitro grown mammalian prion protein fibrils based upon left handed beta helices formed both from the N-terminal and C-terminal regions of the proteinase resistant infectious prion core. The C-terminal threading onto a β-helical structure is almost uniquely determined by fixing the cysteine disulfide bond on a helix corner. In comparison to known left handed helical peptides, the resulting model structures have similar stability attributes including relatively low root mean square deviations in all atom molecular dynamics, substantial side-chain-to-side-chain hydrogen bonding, good volume packing fraction, and low hydrophilic/hydrophobic frustration. For the N-terminus, we propose a new threading of slightly more than two turns, which improves upon the above characteristics relative to existing three turn β-helical models. The N-terminal and C-terminal beta helices can be assembled into eight candidate models for the fibril repeat units, held together by large hinge (order 30 residues) domain swapping, with three amenable to fibril promoting domain swapping via a small (five residue) hinge on the N-terminal side. Small concentrations of the metastable C-terminal β helix in vivo might play a significant role in templating the infectious conformation and in enhancing conversion kinetics for inherited forms of the disease and explain resistance (for canines) involving hypothesized coupling to the methionine 129 sulfur known to play a role in human disease.
Key words: prion, amyloid fibril, domain swap, beta helix, computational biology
Introduction
Prion disorders such as mad cow and chronic wasting diseases represent significant threats to public health and agriculture.1 They are, in addition, examples of protein aggregation driven disorders (which include Alzheimer's, Parkinson's and Huntington's diseases) in which the aggregates contain substantial β sheet content.2–4 While mechanisms of toxicity remain unclear in these disorders, there is general consensus that β sheet aggregates of prion proteins in either oligomeric or fibrillar form play a critical role.3 Accordingly, it is of considerable interest to study the spectrum of possible aggregate structures with all means possible, including in vitro synthesis and simulation.
With the exception of recent solid state nuclear magnetic resonance studies of synthetic Ab42 fibrils5,6 and HET-s fungal prions,7 there are no studies of sufficiently high resolution (1–2 Å) or with qualitative discriminatory power to rule in/out specific structures. In particular, for the prion aggregates, the best structural information has emerged from electron microscopy with 1–3 nm resolution,8,9 large compared to the typical 4.8 Å spacing between β-strands in a β-sheet conformation.
A candidate conformation, which has received considerable attention, is the β helix, especially the left handed form (LHBH). This structure arises in a number of bacterial enzymes, usually in a trimer configuration (type II, with 18 residues per helical turn in an ideal structure), and in some insect anti-freeze proteins (type I, with 15 residues per helical turn in an ideal structure).10,11 For prions, a trimer of β-helix containing monomers has been proposed as a candidate for the repeat units of a two dimensional crystal,8,12 although a competing model with spiraling β sheet structure has also been developed which agrees with much of the available data.13,14 Fibrils can be formed from LHBH models by stacking the oligomers. An important recent development in this direction is the high resolution fibril structural data from the HET-s fungal prion protein, which show that is a left handed “β-solenoid” with two layer repeats, for which a portion is approximately triangular.8 The β helix has also been proposed as a candidate structure for Ab fibrils in Alzheimer's15 (although this appears ruled out by high resolution solid state NMR studies5,6) and of yeast prions16 (although alternative models with registered β sheet structures have also been proposed17).
The structure has also received attention for Huntington's and other polyglutamine disorders,18,19 in part because the “critical length” of ∼40 glutamine repeats at which disease onset enters the observation window of typical human life spans.20 This roughly corresponds to the minimal LHBH repeat with satisfied internal hydrogen bonds, namely two turns (36 residues). Speculation of the relevance of the LHBH for polyglutamine disorders was furthered by in vitro kinetics measurements at varying concentrations showing a critical nucleus of one monomer.20 Subsequently, all atom MD was used to argue that at least three turn left handed β helices can be stable as monomers.21 Perutz and collaborators22 proposed that the aggregate structure was a circular β helix of approximately 20 residues per turn, which was permeable to water. An important feature of the Perutz model is that there was significant side-chain-to-side-chain hydrogen bonding; others have focused on the stabilization of glutamine β-helices by side-chain-to-backbone bonding. However, some caution should be in order for any tentative assignment of LHBH structure in any form to polyQ aggregates: the Perutz data has been reinterpreted to support a different structure,23 and direct all atom molecular dynamics simulations reveal that the proposed circular structure comes undone to a not fully ordered structure showing “β-turns”.24 Separately, extensive simulations from the disordered side suggest a bottleneck to any cross-β structure at the monomer level.25 Moreover, x-ray diffraction on aggregates of polyQ homopeptides from length 8–45 suggests the formation of slab like geometries.26
Recently, mammalian prion protein fibrils have been grown in vitro at low pH and measured with 30 Å resolution electron microscopy.9 We show in Figure 1 an excerpt from their fibril density plots taken from the electron microscopy data base.27 The monomers of prion protein are truncated at the length corresponding to the proteinase resistant core of the infectious PrP conformation. These fibrils have a helical character, with two strands joined by protein loops. Circular dichroism reveals no substantial α helix content, in contrast to the cellular (normal) prion protein PrP and purified prions. As shown in Figure 1, the evidence suggests that the repeat units are of length about 60 Å, or twelve β strands. The fibril helices have either 10 or 12 repeats per twist. Although the lack of α helices raises questions about the correspondence of these fibrils to in vivo derived infectious material,28,29 the potential relevance of fibrils to disease is bolstered by the observation that inoculation of synthetic fibrils (whole or sonicated) into transgenic mice, which overexpress PrP, induces passable prion disease.30
Figure 1.
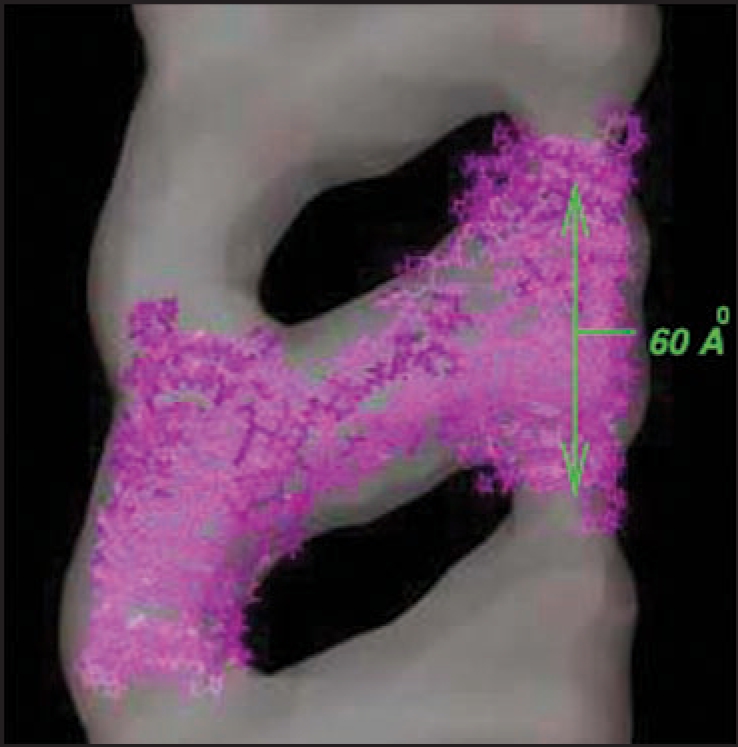
30 Å resolution electron density map of in vitro grown mammalian prion protein fibril from ref. 9 (grey) with model repeat unit of four PrP proteins adopting beta helical conformations in both the N-terminal and C-terminal regions embedded (lavender). Embedding of the tetramer model and image production through Chimera.65
In this paper we propose models for these fibrils based upon prion protein tetramers with LHBHs drawn both form the N-terminal and C-terminal ends of the truncated protein. Such a model is shown as the lavender wire frame structure inserted in the density map of Figure 1, and will be discussed in detail later in the article. In Figure 2, we contrast the cellular prion protein structure PrP (truncated at residue 90, after ref. 31) with a monomer containing C- and N-terminal LHBHs. We draw the N-terminal LHBH from the largely random region, and a 21 residue loop (largely overlapping with the long loop of ref. 8) links this LHBH to the C-terminal LHBH. In this model, the long loop is responsible for the large scale intra-tetramer domain swapping, while the small PKSKP loop between residues 101–105 can be responsible for inter-tetramer domain swapping that holds the fibril together. For the C-terminus LHBH (CLHBH) we show that the requirement of a disulfide bond between cysteines almost uniquely determines the threading; we consider 3 turn and nearly 4 turn versions. This CLHBH appears to be reasonably stable by comparison with three turn sequences from known LHBHs, and has, in particular, excellent side-chain-to-side-chain bonding characteristics. For the N-terminus LHBH (NLHBH) we consider both a three turn model proposed in the aforementioned trimer work, and a new two turn model, different from that previously introduced by Langedijk et al.,32 which has markedly improved stability compared to the earlier model, but not compared to the CLHBH. We identify eight unique arrangements for the tetramer repeat units which could in principle generate fibrils through a combination of LHBH stacking and large hinge domain swapping. We argue that for the fibrils observed in reference 9 that a combination of short hinge domain swapping and large hinge domain swapping33 presents the most likely scenario, which eliminates all but three of the tetramer models. We note that if a small concentration of the CLHBH were present in vivo as a templating element, it might help rationalize the role of some of the mutations in the N-terminal region and in species dependent susceptibility or resistance to infection. Fibrillization34 and oligomerization35 experiments performed in vitro suggest that methionine at codon 129 is critical for aggregation. Assuming to this to be so for the fibrils of reference 9, we select uniquely one of the eight fibril models formed from CLHBHs and NLHBHs. While ours is not the first theoretical36 or experimental work37 to propose b structure formation in the c-terminal region of prion protein fibrils, we believe our model contains a significantly higher level of detail to engender falsifiability than in these important earlier contributions.
Figure 2.

Comparison of PrP with LHBH structure. On the left we show human PrP (adopted from PDB structure 1QM0,31) with three color coded regions: residues 90–145 (orange), residues 146–167 (green), and residues 168–230. In the LHBH structure, which is the building block for tetramers (like that shown in Fig. 1), residues 90–145 go into a LHBH (N3, after ref. 7), 146–167 into a loop, and 168–230 into another LHBH (C4, present work).
Results
Key details about fibrils.
Ref. 9 shows electron microscopy evidence for prion protein fibrils with twinned cross-linked filaments, possessing repeat units with lengths consistent with twelve β strands perpendicular to the fibrillar axis, no α helical structure, and no strong evidence for inter-monomer disulfide bonds between cysteines. The cross sectional diameter of the filaments is of order 25–35 Å, and the linkers between the filaments are of length 50–60 Å. There is a gap of order 8–15 Å between these repeat units, which is thus likely devoid of β sheet bonding. The cross linked regions are electron rich and approximately twice as wide as the empty regions between filaments. These facts alone allow us to begin to build a powerful empirical case for LHBH structure to the filaments. We make the following observations:
With the inclusion of side chains pointing to the outside, it is easy to see that the cross sectional size of LHBH motifs are of the order of 25–35 Å.
The two versions of LHBH trimers proposed already for the prion oligomers of refences 7 and 12 contain LHBH structures in each monomer on the N-terminal side of the truncated prion protein with three β helical turns in residues 90–144.
The remnant α helical portions of the original LHBH trimer model has 63 residues between the structure disrupting proline at residue 165 and the terminal glutamine at residue 228, easily enough to accommodate three full LHBH turns.
The two cysteines with a disulfide bond are at residues 179 and 214 respectively, differing by 35 residues or almost two full turns of a LHBH. Indeed, assuming the cysteines are at LHBH corners, there is no difficulty in accommodating the intra-monomer disulfide bond. This requirement fixes the three turn CLHBH threading to within 1–2 residues. It is important to note that the reducing environment used in reference 9 to generate fibrils could have potentially broken disulfide bonds to allow inter-monomer Cysteine-Cysteine bonding. We cannot assess this possibility in our work, but note that considerable attention in reference 9 is devoted to the establishment that intra-monomer disulfide bonds predominate despite the initial reducing environment. Hence, in our model we assume that the disulfide bonds do not bridge between proteins.
The 21 residue linking region between residues 145 and 165 is, fully extended, approximately 60 angstroms and thus of sufficient length to explain the observed cross links.
We start the CLHBHs past residue 164 to allow full exposure of the YYR motif for scrapies specific antibody binding.38
While the cross-β structure of these fibrils is potentially consistent with LHBH based models, it is at present inconsistent with β-spiral models13,14 for which the β strands are at a non-normal angle to the spiral axis.
It is possible that the fibrils may be consistent with some steric zipper based model;39 we have chosen not to explore such a model here because (1) experimental results for steric zipper based fibrils are so far limited to small peptides, of length 8–10, and it is not clear how this will extrapolate to large peptides such as the truncated prion protein; (2) our model is of interest and falsifiable in its own right independent of what other models may describe prion protein fibrils.
In a separate paper by Surewicz et al., the c-terminal is also found to be the stable core of the fibril formation. Their data derived from site-directed spin labeling shows residues 160–220 as the major participants in conversion from PrPC to PrPSc.40 However, their fibrils possess monomeric layers of single strand thickness, in contrast to the multi-strand LHBH structure. The difference may lie in the different (higher pH) preparation conditions compared to that of reference 9.
C-terminal β helix.
We have threaded (see Figs. 3 and 4) and constructed three and four turn CLHBHs (C3-residues 178–226 of human PrP,C4-residues 166–226 of human PrP) and display the four turn version in Figure 5, along with the modeled two and three turn NLHBHs (both taken from residues 90–145 of the human PrP). The C3 is simply 12 residues shorter on the N-terminal cap of the LHBH. As noted above, the key constraint is that the disulfide bond between the cysteines appear on a corner so that the threading is fixed to within 1–2 residues given the 2 residues per corner of the LHBH. Given this constraint, we simply mutate the residues from three or four turn stretches of a known LHBH to the sequence for the prion. It is also possible to slightly modulate the threading to produce β helices which can bind to 0, 1 or 2 sugars depending upon the position of the N-linking asparagines, as shown in Figure 6.
Figure 3.
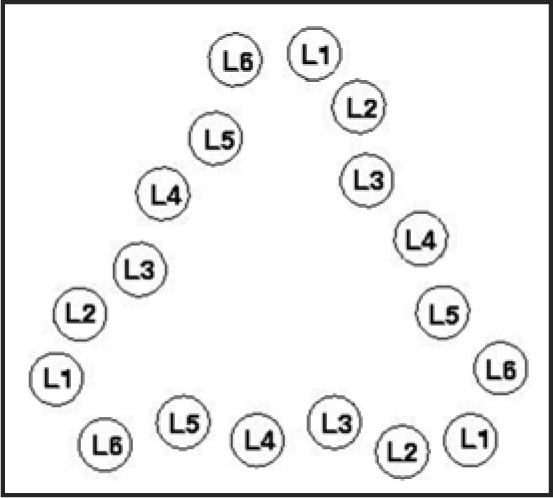
Index scheme for one turn of a Type II LHBH.11 The nomenclature here follows reference 7. Note that L3,L5 point inwards.
Figure 4.

Threads for model β helices discussed in this paper. A single turn with 18 positions is used in the columns. Cysteines are colored gold, asparagines which link to sugars are colored blue, inward pointing acidic or basic residues are painted red. Asterisks denote loops as detailed at the bottom of the Figure, and capital X's denote jumps.
Figure 5.

Left handed β helical structures for model prion tetramers. C4: 4 turn β helix for the C-terminal region (residues 166–226). Note, the cysteines highlighted with yellow spheres, glutamates highlighted in red, and glycans linked asparagines highlighted in green. N3: 3-turn β helix for the N-terminal region drawn from references 7 and 11. N2: New 2-turn N-terminal beta helix. Images produced with VMD.67 Note: PSKPK denotes the small hinge region hypothesized to link the domain swap between tetramers; G-A represents the hydrophobic loop removed from N3 to stabilize N2.
Figure 6.

Different C-terminal β-helical threads accommodating 0 or 1. Depending upon the positions of the N-linking asparagines (pointing in or out, highlighted in green) the C-terminal β helix can accommodate 0 (C4U),1(C4M1) or 2 sugars(C4D, c.f. Fig. 5). Cysteines are highlighted in yellow, glutamates in red.
In order to assess the stability of the CLHBHs, we performed molecular dynamics simulations comparing to three turn sections of LHBHs taken from known proteins (Fig. 7). The key observation in Figure 7 is that within the 10 ns time scale of the simulations, the root mean square deviation (RMSD) from the starting configuration for the CLHBHs is bracketed by that of known segments of the surveyed LHBH proteins. While this does not prove stability, it certainly enforces the viability of the CLHBH candidate structure in building fibril models. Note that the larger RMSD of the C4 model is almost entirely associated with the partial extra layer.
Figure 7.

Root-mean square deviations (RMSD) vs. all atom AMBER8 molecular dynamics64 simulation time to 10 ns from starting structures for LHBH regions of two known proteins (1MR7—Streptogramin A Acetyltransferase68 and 1KGQ—Tetrahydrodipicolinate N-Succinyltransferase69—and two model LHBHs (C4D—diglycoslyated and C4D-D(0)—diglycosylated and protonated glutamate), as shown in Figure 6. In runs up to 1 ns which include five other known LHBH structures we obtain similar RMSDs all bracketed by 1MR7 and 1KGQ. Up to 1 ns, we find similar results for C4M1,C4M2, and N2 (not shown-order 2.5 Å), while the N3 RMSD is much larger (≈6 Å).
We have also assessed three stability characteristics for the CLHBHs to be compared with several of the type II LHBH PDB proteins.11 Figure 8 shows the number of side-chain-to-side-chain hydrogen bonds, volume packing fraction and a frustration index (counting the number of satisfied polar/charged and hydrophobic interactions with water). We find these side-chain/side-chain hydrogen bonds to be predominantly at the corners of known structures, with an average of about 2 per turn. Both the C3,C4 LHBHs score above this. Another characteristic of importance is the packing fraction within the helix. For small side chains, there will be no steric resistance to inward collapse of the helix, and easy penetration of water. The proteins surveyed from the PDB have a mean packing fraction of 0.78, while the C3,C4 LHBHs have packing fractions of 0.71, within the standard deviation of 0.09 for the PDB survey. Finally, we crudely measure frustration of hydrophilic/hydrophobic residues as indicated in the methods section; a positive index indicates low frustration, a negative index high frustration. The mean for the PDB survey is 0.07, and the C3,C4 LHBHs have frustration indices of 0.15 and 0.18 respectively. Hence, on these computational and empirical measures, the model CLHBHs appear to be strong candidates as stable components in the explanation of the fibrils of reference 9.
Figure 8.

Empirical stability measures for known LHBHs and four model LHBHs. We compare side-chain-to-side-chain hydrogen bonding, volume packing fraction and frustration index (see text for definitions), with a positive frustration index indicating good exposure of hydrophilic residues and burial of hydrophobic residues. The C3,C4 models compare favorably to known left handed LHBH values; N3 does badly on packing and side-chain-to-side-chain hydrogen bonding, while N2 fares better. In addition to 1KGQ and 1MR7 (c.f. Fig. 7) we have compared to 1SSM (Serine Acetyltransferase71), 1T3D (Serine Acetyltransferase72), 1G97 (N-acetylglucosamine-1-phosphate uridyltransferase73) and 1LXA (UDP-N-acetylglucosamine acyltransferase74).
(We note that there is overlap of our work with that of reference 11, which observed in particular the importance of good packing and low frustration of the side-chain-to-water interactions).
While there is no direct structural characterization of the PrPb form obtained from either unfolded PrP in high salt or from exposure for long periods of PrPa form to high salt solution, our structure may provide some rationalization for the role of salt.41 Given the cysteine constraint, it is hard to maintain strict LHBH structure in a given thread while avoiding burial of at least one charged residue; our thread shown in Figure 5 and presented in the supplemental section has one buried glutamic acid which is a non-issue for low pH formation as in the experiments of reference 9. However, if the disease inducing structure for a given species or strain requires a different thread, achieving that thread may require burial of basic residues which then necessitates accompanying counter-ion screening.
It is interesting to note that pH-dependent calorimetric data provides evidence for a stable intermediate phase between condensed PrP and random coil prion protein for ovine prions.42 In particular, at pH ≤ 4.0, an intermediate phase containing irreversible aggregates displaying evidence for cross-β structure forms (a similar phase, probably linked to histidine protonation arises at higher pH, above 6.0). Whether there is any correspondence between our C-terminal LHBH and the low pH intermediate is worth further exploration.
N-terminal β helix.
We have threaded (Figs. 3 and 4) two different NLHBHs drawn from residues 90–145 of human PrP, as shown in Figure 5. The three turn model (N3) is the LHBH taken from the mini-prion trimer model of references 7 and 12. For the two turn model (N2), we extract a loop composed mostly of small side-chain alanine and glycine (residues 116–128 of human PrP). Although there is no precedent for such a loop in known LHBH structures,10 this construction does solve the chief problem with the N3 model, which is that the small G,A side chains allow a collapse of the middle layer; the resulting RMSD at one nanosecond is of order 5–6 angstroms, significantly worse than the LHBHs shown in Figure 6. Moreover, as shown in Figure 7, the N3 model has virtually no side-chain-to-side-chain hydrogen bonding, and has as well a poor packing fraction (reflecting the small alanine, glycine volumes in the middle turn of the LHBH). We cannot rule out the possibility that the N3 LHBH is stabilized in contact with, e.g., C3 LHBHs. In contrast, the N2 model is a superior candidate model on its own merits than N3; N2 has (1) a comparable RMSD to the C4 model, (2) improved side-chain-to-side-chain hydrogen bonding compared to the N3 model (though worse than the C3,C4 models), and (3) excellent packing within the LHBH.
We note that Langedijk it et al. previously introduced a two-turn model32 for residues 105–143, which overlaps with our model, running from residues 90–145. Our threads to the LHBH model differ in the following respects: (1) We include residues 90–105 in our first turn; (2) We place the small volume A–G residues in a loop to avoid helix incursions by water with attendant destabilization, while Langedijk et al., include these residues in the helical thread; (3) Langedijk et al., place considerable emphasis upon methionine alignment in the two rungs, which we do not.
Model tetramers.
To compare with the the 30 Å resolution data presented in reference 9 and shown in Figure 1, we can compose tetramers as the repeat unit, where the twelve β strands per repeat unit derive from four β helices (two C-terminal and two N-terminal). Thus each monomer would either contain two three turn LHBHs (N3,C3) or the two-strand/four turn mix (N2,C4). The EM images suggest an approximate mirror symmetry to each fibril strand for these tetramers. There is in addition a possible orientation degree of freedom associated with the triangular cross section of the β helix, which we shall consider briefly near the end of this subsection. With the mirror symmetry, there are eight possible tetramer configurations, illustrated in Figure 9, four in which the strands themselves mirror each other across the fibril axis (models I–IV), and four in which they run in an opposite sense (models I'–IV'). We have color coded each monomer within a tetramer, and provided an arrow on each β helix to indicate the n-to-c terminal sense of the peptide backbone. The tetramers are held together overall intra-strand by β sheet hydrogen bonding and interstrand by domain swapping with the large hinge constructed from residues 145–165 for the C4 case and 145–176 for the C3 case. Note that the hinge regions leave exposed binding epitopes for scrapies sensitive antibodies.38,43,44
Figure 9.

Schematics of proposed tetramer repeat units. Boxes: C- or N-terminal LHBHs viewed from the side (perpendicular to the helix axis). The arrow denotes the n-terminal to c-terminal progression of the sequence. Lines: Large loop regions (residues 145–166 for N2-C4 pairing, or 145–176 for N3-C3 pairing). Lines and boxes are color coded by monomer. The first row contains filaments with the same N-C sense, while the second row contains filaments with opposite N-C sense.
To further restrict possible models, we consider the impact of three constraints: (1) the requirement that the cross strand region have high electron density at the middle of the repeat unit and a hole at the end. (2) That there be a “notch” or gap at the ends. (3) That the strands have a cross sectional aspect ratio of ∼1.3:1 for the strand-to-strand orientation vs. the perpendicular orientation in the vicinity of maximal inter-strand electron density.
Regarding the “hole” we can immediately rule out models II' and III, which would clearly have density holes both at tetramer ends and middles. Models I, I' may possible satisfy this constraint if built from C4 and N2 β helices, but otherwise there are likely to be holes in the tetramer middles. However, given this constraint, the most likely candidates are models II, III', IV and IV' in which the hinge stretches either cross or are concentrated in the center of the tetramer.
We further note that the lateral aspect ratio of the dense repeat unit midsection to the hole is approximately 2:1. This suggests that the C4/N2 combination for models II, III', IV and IV' are more likely. In each case, this would expand the middle region relative to the hole region.
We now turn our attention to the observed “notch” in density between repeat units of the fibril, which would yield an 8–10 Å gap between β strands. We speculate that intertetramer bonding is mediated by a different mechanism. A natural candidate, when the ends of the tetramers are formed from N2 or N3 LHBHs is that the small 5 residue proline containing loop (residues 101–105 in humans) noted in reference 12 as a possible hinge region for domain swapping mediates domain swapping between tetramers here. In this case, the bottom layer of each N2 or N3 LHBH from one tetramer strand hydrogen bonds in the appropriate β sheet conformation with the upper layers from the adjoining tetramer. Given the relatively large number of basic residues in the bottom layer, this also represents a way to avoid some of the Coulomb cost that would arise if the monomers hydrogen bonded without domain swapping.
With this domain swapping assumption, the relevant models are restricted to III', IV, IV'. We note that if this assumption is correct as necessary for fibril formation, then there would be nothing preventing the other models from forming, but they would be off pathway to fibril formation. We summarize the features of our eight tetramer models in the Table 1.
Table 1.
Comparison of eight fibril repeat unit models shown in Figure 7
| Model | Correct fibril periodicity? | Fibril gap? | Possible M129/D178 contact? |
| I | No | No | No |
| II | Yes | No | No |
| III | No | Yes | Yes |
| IV | Yes | Yes | No |
| I' | No | No | No |
| II' | No | No | No |
| III' | Yes | Yes | Yes |
| IV' | Yes | Yes | No |
The second column refers to whether the model can reproduce the observed fibril periodicity from reference 8 via large scale domain swapping in the region of residues 145–166. Half of the models (II, IV, III' and IV') can do this. The third column refers to whether the model can reproduce the ‘notch’ between repeat units via domain swapping in the region of residues 102–106 (the PKSKP loop shown in Figure 3) which appears to correspond to a larger separation (about 7–8 angstroms) than for β-sheets. This is limited to repeat units with N-terminal LHBH on the ends (models III, IV, III', IV'). Finally, the fourth column refers to whether the model can allow direct contact between the M129 residue and the D178 residue implicated in Fatal Familial Insomnia. This only arises when the C-terminal side of the N-terminal LHBH can contact the N-terminal side of the C-terminal LHBH, restricting it to models III, III', of which the latter is the sole model displaying both the key fibril features and a possible mechanistic explanation for FFI.
Finally, the cross section aspect ratio constraint favors that a triangular vertex be oriented pointing towards the opposite strand, with the hinge emerging from that vertex.
Figure 1 illustrates an embedding of the IV model with C4/N2 LHBH units into the 30 Å resolution data from reference 9. Clearly there is a reasonable qualitative match of the structures.
Discussion
Possible relevance to heritable/species related susceptibility/resistance.
The known mutations engendering inherited prion disease are clustered between residues 102 and 145 and between 171 and 238.45 While the first set of mutations is within the region which has β sheet structure by consensus,46 the second is generally assumed to contain the remnant α helix structures of the prion28,29 and it is thus difficult to reconcile the prevalence of these C-terminal region mutations with known attributes of prion disease.
A corollary exists for animals in terms of disease resistance and possible susceptibility. Notably, among animals exposed to bovine spongiform encephalopathy (BSE) prions in England through contaminated protein supplements, canines, cervids and pigs appear to have escaped infection.47 If one looks for sequence differences unique to these animals relative to BSE susceptible species, all but one cluster in the C-terminal region (canines have what corresponds to an S103N mutation in humans). Focusing on canines, dogs possess what corresponds to an H177R mutation in humans.
The apparent stability of our proposed C-terminal LHBH models suggests a possible role for these structures in prion disease and in animal susceptibility/resistance. In particular, the apparently less robust N-terminal LHBHs could perhaps be templated by a small concentration of metastable or out of equilibrium CLHBHs. At the same time, numerous data implicate the N-terminal region of the proteinase resistant core in β sheet formation and toxicity. This is supported by previous unfolding/refolding experiments, which strongly suggest that a β sheet form of the prion protein is stable with respect to the a-rich form similar to wild type41 (although the stable form is probably oligomeric rather than monomeric).
Fatal familial insomnia mutation.
The mutation for fatal familial insomnia (FFI), D178N, finds expression in concert with homozygous methionine at codon 129.45 At first sight, it is remarkable that these residues, separated by 49 backbone units, should interact in some way to induce the disease. A possible rationalization of this result within the current context is as follows: (1) We observe that the M129 residue resides on the C-terminal side of the N-terminal LHBH, precisely on a corner with our N2 LHBH threading to allow easy alignment with the C3 or C4 LHBHs. (2) The D178N mutation replaces a hydroxyl group with an amide group which may promote formation of a hydrogen bond to the sulfur of the methionine group. (3) As shown in Figure 10, it is possible to align the upper layer of the C3 or C4 β helix with a β helix like partial turn containing the M129 such that the M129 sulfur lies in proximity to both the H177 and mutant N178. One can easily find rotamer conformations for the M,H,N residues with ≤2 Å separation between the methionine sulfur and the side chain amides of the histidine and asparagines, hence allowing for a possible double hydrogen bond formation. In contrast, as shown in Figure 10, only one histidine amide-methionine sulfur bond can form for the WT PrP sequence.
Figure 10.

Schematic alignment of M129 with C-terminal LHBH for WT human, FFI human and canine prions. For WT, one S-amide hydrogen bond (with H177) is possible. For FFI, two S-amide hydrogen bonds (with H177 and N178) are possible. For canines (referenced to the human sequence) within the Swiss PDB viewer66 rotamer library, no orientation of the R177 could produce amide hydrogen bonding with M129 sulfur. In each case, alignments are produced within the Swiss PDB viewer66 by sweeping through low score rotamers and images via VMD.67 We identify the small PSKPK loop for domain swapping between tetramers, and the hydrophobic G-A loop pulled from the N2 Model to stabilize the LHBH.
The possibility of a sulfur acceptor for amide donors in hydrogen bonding of course requires careful consideration. While earlier surveys of known protein structures showed potential for the methionine sulfur to play such a role,48 a later examination of a subset of high resolution structural data revealed only three instances of such a role.49 A potential limitation of the latter study is the restriction to a small number (order 70) high resolution protein structures. A more extensive study of complexes of the form Y-S-Z from a data base of chemical structures revealed that C-S-C conformations with a sulfur acceptor are relatively rare (only about 6% of C in the relevant bonding geometry for the neighboring C's permit S as an acceptor) but remain possible49 (reviewed in ref. 51).
More recent theoretical works including electron correlation effects beyond Hartree-Fock or density functional theory approximations suggest that the enthalpy of hydrogen bonding with sulfur acceptors is only modestly weaker than those for oxygen acceptors.52,53 Accordingly, the corresponding force fields in molecular dynamics simulations may require updating.54
Role of M129 vs. V129 in fibril/oligomer growth/disease susceptibility.
The conjectured role of the M129 to residue 177 and/or 178 hydrogen bonding discussed above for FFI is also of relevance to in vitro models of fibril and oligomer growth. Of the potential fibril promoting tetramer models in Figure 9 consistent with the experimental constraints, only one has the C-terminus of the N LHBHs in contact with the N-terminus of the C LHBHs, namely, model III'.
We note that to date the human cases of variant CJD associated with the human form of BSE in Europe have arisen exclusively for methionine homozygotes while for Kuru M129 homozygosity dominated the known shorter incubation time cases.55 In transgenic mice with inserted human prion gene but with the native mouse gene knocked out, valine homozygosity at residue 129 led to fifty percent longer incubation times at expression levels 2–4 times higher than for mice with methionine at residue 129.56 Finally, cases of iatrogenic CJD caused by tainted human growth hormone in Europe appear to be most pronounced for methionine homozygotes.57
With valine at 129, there is no easy side-chain hydrogen bond formation. A potential candidate is the Y128 residue, but there is no simply identifiable rotamer or rethreading which allows the H177 amide to link to the oxygen group of the tyrosine. We note in this context that pathogenesis for the vCJD like strain for chimeric mice is blocked with V129 homozygosity.58 We observe, from the perspective of this model, that the V129 coding may favor a rethreading of the N-terminal β helix to bury the more hydrophobic residue rather than leave it exposed on a turn.
Resistance to infection in canines.
In dogs, what corresponds to the H177 residue is replaced with arginine, as shown in Figure 9. The amide group of the arginine is at a longer distance from the backbone than that of the histidine, and the flexible arginine side chain has a much large number of rotamers than for histidine. Within the Swiss PDB utility, we find no acceptable rotamer conformation with R177 (R140 for dogs) that can allow the mid-side chain amide to hydrogen bond to the M129 sulfur (M92 for dogs). We thus conjecture that the R177 point sequence difference confers protection against disease by inhibiting formation of the hydrogen bonded intermediate conformation.
Estimated impact on conversion kinetics.
Typical hydrogen bond formation energies are estimated to be 3–5 kcal/mole (reviewed in ref. 53). We assume (1) that the locking of a methionine to the C-terminal LHBH constitutes a reaction transition state for templating N-terminal LHBH formation, (2) that there are entropy losses associated with rotamer locking, (3) take the estimate of −5.5 kcal/mole for sulfur hydrogen bond formation enthalpy,51 and (4) account for less stable PrP in the D178N FFI mutation compared to WT by ≈1.9 kcal/mole (see Fig. 6 of ref. 45). Using the observation frequency data from the penultimate rotamer library59 to compute the entropy, we estimate that the FFI transition state is about −2.3 kcal/mole below the WT human transition state, and the canine transition state is about 2.7 kcal/mole above the WT human barrier. If we further assume that this is the rate limiting step in formation of toxic PrP oligomers, then from standard transition state theory we obtain a speedup by a factor of 70 for FFI over WT CJD in humans, and a slowdown of about a factor of 90 for canine conversion over WT CJD in humans. We note that our simple estimate for the WT human-canine transition state barrier difference is within a factor of two of the barrier difference estimated from kinetic studies of membrane catalyzed in vitro fibril growth,60 where dog fibrils assembled 300 times more slowly than WT human sequence fibrils. The FFI-WT barrier difference is within a factor of two of the general estimates of 3–4 kcal/mole for point mutation driven kinetically controlled conversion processes in ref. 46. Finally, the FFI-to-WT spontaneous incubation ratio would give a peak incubation time of about 3400 years for spontaneous CJD, in reasonable agreement with estimates of >1000 years from a two dimensional aggregation model.61,62
Experimental tests.
Some obvious experimental tests of our model include:
The M129V substitution should be carried out for the preparation conditions of reference 9 to see whether fibrillization is blocked or slowed as found in reference 34. If this is maintained, clearly Model III' of Figure 8 is favored from our work.
It is important to repeat the fibril growth experiments of reference 9 with the D178N mutation to test for enhanced fibril growth rates, and with the H177R “canine” mutation to test for inhibition of growth. The latter has been done for membrane catalyzed fibril growth already,48 but a detailed study of the fibrils with EM under the growth conditions of reference 9 is desirable.
To test which model most likely applies to the fibrils, we advocate site directed spin labeling of non-critical residues in the middle of our N and C LHBHs. Pulsed electron-electron double resonance experiments (PELDOR)63 can then be used as a ruler to detect distances between the selected spin labels in the condensed fibril structures.
Any fibrils synthesized under the conditions of reference 9 can be tested with HD exchange to confirm the “protected regions.” If we are correct, in addition to the region overlapping with the work of reference 40, there should be protection not only in the C-terminal region, but also in the N-terminal region, which will definitively rule out the fibril structure proposed in reference 41 for these synthesis conditions.
Proline scanning substitution should of course break fibril formation in the C4 structure away from the LHBH corners and this is an important test of our model.
Methods
Threading.
Our current threading procedure is “manual”: we arrange the amino acids in a spreadsheet matrix with columns indexed by the Type II LHBH positions and rows labeled by turn number of the helix. We require:
charged residues point out, except possibly on top or bottom turns of the helix.
high volume residues point out
prolines are restricted to turn (L6 or L1) positions.
prefer polar residues out and hydrophobic residues in.
allow no more than one loop per turn.
demand that the two cysteines in CLHBH models be stacked above each other two turns away to within ± 1 residue and point out.
Demand for the N2 and N3 models that the proposed proline hinge region be at a corner.
We adjust the LHBH thread to best satisfy the above constraints.
Molecular dynamics.
The MD simulations were carried out using the AMBER8 molecular dynamics package64 with the parm99 force field using TIP3PBOX water. We first minimized the structure by allowing 2,000 steps to relax or find the lowest energy configuration. We then raise the temperature isochorically from 100 K to 300 K over 20 ps time interval unrestrained. Finally, we perform 10 ns production runs isobarically, also unrestrained. We measure the root mean square deviation from the input structure (RMSD) as a figure of merit measuring the stability of the structures. For comparison of the model LHBHs with known structures, we extracted three turn LHBH units from existing pdb files, and subjected these to the above protocol.
We constructed the model tetramer structure of Figure 1 with prion monomers consisting of the C4M1 and N2 threads together with a loop joining them. The initial tetramer structure was run through energy minimization in the AMBER suite for 2,000 moves with waters included to eliminate steric clashes.
LHBH stability characteristics.
Side-chain to side-chain hydrogen bond counting. Hydrogen bonds are calculated using the following criteria: (1) A maximal distance of 3.5 Å between donor and acceptor. (2) An angular cutoff of 60 degrees. (3) No backbone atoms were allowed (i.e., N,O,C,CA), meaning only side chain to side chain bonding. (4) No water mediated bonds are included.
Volume packing ratio. We produce a simple estimate of the packing fraction by summing standard side-chain volumes for those residues with side-chains inside the β-helix and dividing this cumulative residue volume into the ideal triangular prism volume for an eighteen residue/turn helix, with the triangle inscribing the backbone and a spacing of 4.8 Å between layers assumed.
Frustration index. This is defined simply as (per β helix turn) the net number of charged/polar residues pointing out (total out − total in) plus the net number of hydrophobic residues pointing in (total in − total out) ignoring residues on the helix caps and glycine.
Molecular visualization and comparison.
We used Chimera65 to align molecular level renderings of our model structures with the experimental density profiles. To examine the possible role of M129 we employed the Swiss PDB viewer66 to (1) construct a possible structure of residues 128–130 from a LHBH corner, (2) align this structure with residues 174–179 of our proposed C-terminal LHBH structure and (3) explore the rotamer space of residues 129, 177 and 178 for potential hydrogen bonding of the M129 sulfur as an acceptor with amide groups from residues 177 and 178 (for FFI). Only relatively low score rotamer positions (2 or less) were accepted.
Acknowledgements
Supported by U.S. Army Congressionally Directed Biomedical Research grant NP020132, NSF REU Grant PHY-0243904 (S.C.C.), and the Center for Theoretical Biological Physics at UC San Diego (NSF Grant PHY0216576 and 0225630). Figure 1 was produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081). We acknowledge useful discussions with C. Govaerts, H. Wille, G. Millhauser, C. Trevisan, S. Yang, J.N. Onuchic, J.D. Britt and K. Sigurdson. D.L.C. is grateful for substantial assistance from J. Pan and G. Vigil.
Abbreviations
- LHBH
left handed β helix
- MD
molecular dynamics
- CLHBH
C-terminal left handed β helix for the prion protein
- NLHBH
N-terminal left handed β helix for the truncated prion protein
- C3
three turn C-terminal left handed β helix for the prion protein
- C4
four turn C-terminal left handed β helix for the prion protein
- C4U
model unglycosylated C-terminal left handed β helix for the prion protein
- C4M1
model 1 for monoglycosylated C-terminal left handed β helix for the prion protein
- C4D
model diglycosylated C-terminal left handed β helix for the prion protein
- N2
two turn N-terminal left handed β helix for the truncated prion protein
- N3
three turn N-terminal left handed β helix for the truncated prion protein
- CJD
Creutzfeldt-Jakob disease
- BSE
bovine spongiform encephalopathy
- PELDOR
pulsed electron-electron double resonance experiments
- VMD
visual molecular dynamics
- RMSD
root mean square deviation
- FFI
fatal familial insomnia
Footnotes
Previously published online as a Prion E-publication: http://www.landesbioscience.com/journals/prion/article/7059
References
- 1.Prusiner SB. Introduction to Prion biology and diseases. In: Prusiner SB, editor. Prion biology and disease. Cold Spring Harbor, NY: Cold Spring Harbor Press; 2004. pp. 1–88. [Google Scholar]
- 2.Cox DL, Lashuel HA, Lee KY, Singh RRP. The materials science of protein aggregation. MRS Bull. 2005;30:452–457. [Google Scholar]
- 3.Caughey B, Baron GS. Prions and their partners in crime. Nature. 2006;443:803–810. doi: 10.1038/nature05294. [DOI] [PubMed] [Google Scholar]
- 4.Lansbury PT, Lashuel HA. Reversibility of prion-induced neurodegeneration. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- 5.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Doeli H, Schubert D, Riek R. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petkova AT, Yau WM, Tycko R. Experimental contraints on quatemary structure in Alzheimer's amyloid fibrils. Biochemistry. 2005;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Amyloid Fibrils of the HET-s(218–289) Prion form a β Solenoid with a Triangular Hydrophobic Core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 8.Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc Nat Acad Sci USA. 2004;101:8342–8347. doi: 10.1073/pnas.0402254101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tattum MH, Cohen-Krausz S, Kalili-Shirazi A, Jackson GS, Orolova EV, Collinge J, Clarke AR, Saibil HR. Elongated oligomers assemble into mammalian PrP amyloid fibrils. J Mol Biol. 2006;357:75–985. doi: 10.1016/j.jmb.2006.01.052. [DOI] [PubMed] [Google Scholar]
- 10.Jenkins J, Pickersgill R. The architecture of parallel β-helices and related folds. Prog Biophys Mol Biol. 2001;77:111–175. doi: 10.1016/s0079-6107(01)00013-x. [DOI] [PubMed] [Google Scholar]
- 11.Choi JH, Govaerts C, May BCH, Cohen FE. Analysis of the sequence and structural features of the left-handed β-helical fold. Proteins, Structure, Function and Bioinformatics. doi: 10.1002/prot.22051. http://www3.interscience.wiley.com/tools/citex?clienttype=1&subtype=1&mode=1&versio n=1&id=117955077&redirect=/cgi-bin/fulltext/117955077/HTMLSTART. [DOI] [PubMed] [Google Scholar]
- 12.Yang S, Levine H, Onuchic JN, Cox DL. Structure of infectious prions: Stabilization by domain swapping. FASEB J. 2005;19:1778–1782. doi: 10.1096/fj.05-4067hyp. [DOI] [PubMed] [Google Scholar]
- 13.DeMarco ML, Daggett V. From conversion to aggregation: Protofibril formation of the Prion protein. Proc Nat Acad Sci USA. 2004;101:2293–2298. doi: 10.1073/pnas.0307178101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeMarco ML, Silveira J, Caughey B, Daggett V. Structural properties of Prion protein protofibrils and fibrils: An experimental assessment of atomic models. Biochem. 2006;45:15573–15582. doi: 10.1021/bi0612723. [DOI] [PubMed] [Google Scholar]
- 15.Guo JT, Wetzel R, Ying X. Molecular modeling of the core of Abeta amyloid fibrils. Proteins. 2004;57:357–364. doi: 10.1002/prot.20222. [DOI] [PubMed] [Google Scholar]
- 16.Kishimoto A, Hasegawa K, Suzuki H, Taguchi H, Namba K, Yoshida M. Beta-Helix is a likely core structure of yeast prion Sup35 amyloid fibers. Biochem Biophys Res Comm. 2004;315:739–745. doi: 10.1016/j.bbrc.2004.01.117. [DOI] [PubMed] [Google Scholar]
- 17.Shewmaker F, Ross ED, Tycko R, Wickner RB. Amyloids of shuffled prion domains that form Prions have a parallel in-register beta-sheet structure. Biochem. 2008;47:4000–4007. doi: 10.1021/bi7024589. [DOI] [PubMed] [Google Scholar]
- 18.Wetzel R. Ideas of order for amyloid fibril structure. Structure. 2002;10:1031–1036. doi: 10.1016/s0969-2126(02)00809-2. [DOI] [PubMed] [Google Scholar]
- 19.Stork M, Giese A, Kretzschmar HA, Tavan P. Molecular dynamics simulations indicate a possible role of parallel beta-helices in seeded aggregation of poly-gln. Biophys J. 2005;88:2442–2451. doi: 10.1529/biophysj.104.052415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Ann Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 21.Chen SM, Ferrone FA, Wetzel R. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Nat Acad Sci USA. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perutz M, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc Nat Acad Sci USA. 2002;99:5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sikorski P, Atkins E. New Model for Crystalline Polyglutamine Assemblies and their connection with Amyloid Fibrils. Biomacromol. 2005;6:425–432. doi: 10.1021/bm0494388. [DOI] [PubMed] [Google Scholar]
- 24.Zanuy D, Gunasekaran K, Lesk AM, Nussinov R. Computational Study of the Fibril Organization of Polyglutamine Repeats Reveals a Common Motif Identified in β-helices. J Mol Biol. 2006;358:330–345. doi: 10.1016/j.jmb.2006.01.070. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Vitalis A, Wyczlkowski MA, Pappu RV. Characterizing the Conformational Ensemble of Monomeric Polyglutamine. Proteins: Structure, Function and Bioinformatics. 2006;63:297–311. doi: 10.1002/prot.20761. [DOI] [PubMed] [Google Scholar]
- 26.Sharma D, Shinchuk LM, Inouye H, Wetzel R, Kirscher DA. Polyglutamine Homopolymers having 8–45 residues Form Slablike β-Crystallite Assemblies. Proteins. 2005;61:398–411. doi: 10.1002/prot.20602. [DOI] [PubMed] [Google Scholar]
- 27. See the EMD Database at http://www.ebi.ac.uk/msd-srv/emsearch/index.html.
- 28.Safar J, Roller PP, Gajdusek DC, Gibbs CJ. Conformational transitions, dissociation and unfolding of scrapie amyloid (Prion) protein. J Biol Chem. 1993;268:20276–20284. [PubMed] [Google Scholar]
- 29.Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang ZW, Fletterick RJ, Cohen FE, Prusiner SB. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion protein. Proc Nat Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Legname G, Baskakov IV, Nguyen H-oB, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian Prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 31.Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez-Garcia F, Billeter M, Calzolai L, Wider G, Wuthrich NMR solution structure of the human prion protein. Proc Natl Acad Sci USA. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langedijk JPM, Fuentes G, Boshulzen R, Bonvin AMJJ. Two-rung model of a left-handed β-helix for Prions explains species barrier and strain variation in Transmissible Spongiform Encephalopathies. J Mol Biol. 2006;360:907–920. doi: 10.1016/j.jmb.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 33.Bennett MJ, Sawaya MR, Eisenberg D. Deposition diseases and 3D domain swapping. Structure. 2006;14:811–824. doi: 10.1016/j.str.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 34.Lewis PA, Tattum MH, Jones S, Bhelt D, Batchelor M, Clarke AR, Collinge J, Jackson GS. Codon 129 polymorphism of the human prion protein influences the kinetics of amyloid formation. J Gen Virol. 2006;87:2443–2449. doi: 10.1099/vir.0.81630-0. [DOI] [PubMed] [Google Scholar]
- 35.Tahiri-Alaoui A, Gill AC, Disterer P, James W. Methionine 129 variant of human Prion protein oligomerizes more rapidly than the valine 129 variant. J Biol Chem. 2004;279:31390–31397. doi: 10.1074/jbc.M401754200. [DOI] [PubMed] [Google Scholar]
- 36.Dima RI, Thirumalai D. Perturbation of the secondary structure of the scrapie Prion protein under conditions that alter infectivity. Proc Nat Acad Sci USA. 2004;101:15335–15340. doi: 10.1073/pnas.90.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu X, Wintrode PL, Surewicz WK. Beta-sheet core of hman prion protein amyloid fibrils as determined by hydrogen-deuterium exchange. Proc Nat Acad Sci USA. 2007;104:1510–1515. doi: 10.1073/pnas.0608447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paramithiotis E, et al. A prion protein epitope selective for the pathologically misfolded conformation. Nature Med. 2003;9:893–899. doi: 10.1038/nm883. [DOI] [PubMed] [Google Scholar]
- 39.Sawaya MR, et al. Atomic structures of amyloid cross-b spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 40.Cobb NJ, Sonnichsen FD, Mchaourab H, Surewicz WK. Molecular architecture of human prion protein amyloid: A parallel, in register β-structure. PNAS. 2007;104:18946–18951. doi: 10.1073/pnas.0706522104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baskakov IV, Legname G, Prusiner SB, Cohen FE. Folding of prion protein to its native alpha-helical conformation is under kinetic control. J Biol Chem. 2001;23:19687–19690. doi: 10.1074/jbc.C100180200. [DOI] [PubMed] [Google Scholar]
- 42.Rezaei H, Choiset Y, Eghiaian F, Terguer E, Mentre P, Debehy P, Grosclaude J, Haertle T. Amyloidogenic Unfolding Intermediates Differentiate Sheep Prion Variants. J Mol Biol. 2002;322:799–814. doi: 10.1016/s0022-2836(02)00856-2. [DOI] [PubMed] [Google Scholar]
- 43.Moroncini G, Kanu N, Lolforosi L, Abalos G, Telling GC, Head M, Ironside J, Brockes JP, Burton DR, Williamson RA. Motif-grafted antibodies containing the replicative interface of cellular PrP are specific for PrP. Proc Nat Acad Sci USA. 2004;101:10404–10409. doi: 10.1073/pnas.0403522101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Novitskaya V, Makarava N, Bellon A, Bocharova OV, Bronstein IB, Williamson RA, Baskakov IV. Probing the conformation of the Prion protein within a single amyloid fibril using a novel immunoconformational assay. J Biol Chem. 2006;281:15536–15545. doi: 10.1074/jbc.M601349200. [DOI] [PubMed] [Google Scholar]
- 45.Will RG, Alpers MP, Dormont D, Schonberger LB. Introduction to Prion biology and diseases. In: Prusiner SB, editor. Prion biology and disease. Cold Spring Harbor, NY: Cold Spring Harbor Press; 2004. pp. 629–672. [Google Scholar]
- 46.Govaerts C, Wille J, Prusiner SB, Cohen FE. Introduction to Prion biology and diseases. In: Prusiner SB, editor. Prion biology and disease. Cold Spring Harbor, NY: Cold Spring Harbor Press; 2004. pp. 243–282. [Google Scholar]
- 47.Wells GAH, Wilesmith JW. Introduction to Prion biology and diseases. In: Prusiner SB, editor. Prion biology and disease. Cold Spring Harbor, NY: Cold Spring Harbor Press; 2004. pp. 595–628. [Google Scholar]
- 48.Gregoret LM, Rader SD, Fletterick RJ, Cohen FE. Hydrogen bonds involving sulfur atoms in proteins. Proteins. 1991;9:99–107. doi: 10.1002/prot.340090204. [DOI] [PubMed] [Google Scholar]
- 49.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 50.Allen FH, Bird CM, Rowland RS, Raithby PR. Hydrogen-Bond Acceptor and Donor Properties of Divalent Sulfur (Y-S-Z and R-S-H) Acta Cryst. 1997;53:696–701. [Google Scholar]
- 51.Valdes-Martinez J, Hernandez-Ortega S, Rubio M, Li DT, Swearingen JK, Kaminsky W, Kelman DR, West DX. Study of the sulfur atom as hydrogen bond acceptor in N(2)-pyridylmethyl-N'-arylthioureas. J Chem Cryst. 2004;34:533–540. [Google Scholar]
- 52.Rablen PR, Lockman JW, Jorgensen WL. Ab initio study of hydrogen-bonded complexes of small organic molecules with water. J Phys Chem A. 1998;102:3782–3797. [Google Scholar]
- 53.Wennmohs F, Staemmler V, Schindler M. Theoretical investigation of weak hydrogen bonds to sulfur. J Chem Phys. 2003;119:3208–3218. [Google Scholar]
- 54.Wennmohs F, Schindler M. Development of a multipoint model for sulfur in proteins: A new parametrization scheme to reproduce high-level ab initio interaction energies. J Comp Chem. 2005;26:283–293. doi: 10.1002/jcc.20163. [DOI] [PubMed] [Google Scholar]
- 55.Lee HS, Brown P, Cervenakova L, Garruto RM, Alpers MP, Gajdusek DC, Goldfarb LG. Increased susceptibility to Kuru of carriers of the PRNP 129 methionine/methionine genotype. J Infect Dis. 2001;183:192–196. doi: 10.1086/317935. [DOI] [PubMed] [Google Scholar]
- 56.Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 57.Brandel JP. Distribution of codon 129 genotype in human growth hormone-treated CJD patients in France and the UK. Lancet. 2003;362:128–130. doi: 10.1016/S0140-6736(03)13867-6. [DOI] [PubMed] [Google Scholar]
- 58.Wadsworth JDF. Human Prion protein with valine 129 prevents expression of variant CJD phenotype. Science. 2004;306:1793–1796. doi: 10.1126/science.1103932. [DOI] [PubMed] [Google Scholar]
- 59.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- 60.Luehrs T, Zahn R, Wuetrich K. Amyloid formation by recombinant full-length Prion proteins in phospholipid bicelle solutions. J Mol Biol. 2006;57:833–841. doi: 10.1016/j.jmb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 61.Slepoy A, Singh RRP, Pazmandi F, Kulkarni RV, Cox DL. Statistical mechanics of prion diseases. Phys Rev Lett. 2001;87:058101. doi: 10.1103/PhysRevLett.87.058101. [DOI] [PubMed] [Google Scholar]
- 62.Kulkarni RV, Slepoy A, Singh RRP, Cox DL, Pazmandi F. Simulations of oligomeric ontermediates in prion diseases. Biophys J. 2003;85:2213–2223. doi: 10.1016/s0006-3495(03)74647-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prisner T, Rohrer M, MacMillan F. PULSED EPR SPECTROSCOPY: Biological application. Ann Rev Phys Chem. 2001;52:279–313. doi: 10.1146/annurev.physchem.52.1.279. [DOI] [PubMed] [Google Scholar]
- 64.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 66.Guex N, Peitsch MC. Swiss-Model and the Swiss-PDBViewer: an environment for It. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 67.Humphrey W, Dalke A, Schulten K. Visual Molecular Dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 68.Kehoe LE, Snidwongse J, Courvalin P, Rafferty JB, Murray IA. Structural basis of synercid (Quinupristin-Dalfopristin) resistance in gram-positive bacterial pathogens. J Biol Chem. 2003;278:29963–29970. doi: 10.1074/jbc.M303766200. [DOI] [PubMed] [Google Scholar]
- 69.Beaman TW, Vogel KW, Drueckhammer DG, Blanchard JS, Roderick SL. Acyl group specificity at the active site of tetrahydridipicolinate N-succinyltransferase. Protein Sci. 2002;11:974–979. doi: 10.1110/ps.4310102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Olsen LR, Huang B, Vetting MW, Roderick SL. Structure of serine acetyltransferase in complexes with CoA and its cysteine feedback inhibitor. Biochemistry. 2004;43:6013–6019. doi: 10.1021/bi0358521. [DOI] [PubMed] [Google Scholar]
- 71.Pye VE, Tingey AP, Robson RL, Moody PC. The structure and mechanism of serine acetyltransferase from Escherichia coli. J Biol Chem. 2004;279:40729–40736. doi: 10.1074/jbc.M403751200. [DOI] [PubMed] [Google Scholar]
- 72.Kostrewa D, D'Arcy A, Takacs B, Kamber M. Crystal structures of Streptococcus pneumoniae N-acetylglucosamine-1-phosphate uridyltransferase, GlmU, in apo form at 2.33 A resolution and in complex with UDP-N-acetylglucosamine and Mg(2+) at 1.96 A resolution. J Mol Biol. 2001;305:279–289. doi: 10.1006/jmbi.2000.4296. [DOI] [PubMed] [Google Scholar]
- 73.Raetz CR, Roderick SL. A left-handed parallel beta helix in the structure of UDP-N-acetylglucosamine acyltransferase. Science. 1995;270:997–1000. doi: 10.1126/science.270.5238.997. [DOI] [PubMed] [Google Scholar]