

Abstract
The rate of spontaneous change from ψ to the ψ+ condition, determined in yeast by states of the Sup35p protein, is briefly discussed together with the conditions necessary for such change to occur. Conditions that promote and which affect the rate of induction of ψ+ in Sup35p and of other prion-forming proteins to their respective prion forms are also discussed. These include the influence of the amount of non-prion protein, the presence of other prions, the activity of chaperones, and brief descriptions of the role of native sequences in the proteins and how alteration of sequences in prion-forming proteins influences the rate of induction of [prion+] and amyloid forms.
The second part of this article discusses the conditions which affect the reversion of ψ+ to ψ-, including factors which affect the copy-number of prion “seeds” or propagons and their partition. The principal factor discussed is the activity of the chaperone Hsp104, but the existence of other factors, such protein sequence and of other, less well-studied agents is touched upon and comparisons are made, as appropriate, with studies with other yeast prions.
We conclude with a discussion of models of maintenance, in particular that of Tanaka et al. published in Nature (2006),6 which provides much insight into the phenotypic and genetic parameters of the numerous “variants” of prions increasingly being described in the literature.
Key Words: Saccharomyces cerevisiae, [PSI], [URE3], [PIN], prion induction, prion curing, prion inheritance, Hsp104
Introduction
It is a commonplace observation that the prion forms of proteins are stable in inheritance; indeed it is implicit in the definition of a prion and distinguishes them from such forms as conformers, aggregates, polymers and amyloids which are neither infectious nor heritable. In this context, stability implies more than mere chemical stability and more than just synthesis, since the alternative nonprion state is equally stable. This implies self-reproduction, that is a requirement for a pre-existing template. The relevance of fungal prions to the mammalian variety which cause infectious diseases arises through the commonality of infection and heredity, which has been remarked upon by many distinguished biologists. One nice example of this commonality was pointed out by Francois Jacob, talking about the temperate bacteriophage phage λ, which may be observed either as a fatal disease of its host E. coli, or as one of its genes with a unique location, conferring resistance and lysogeny. The difference lies in the particular mode of reproduction and transmission of the alternative states. In the world of prions, both properties, of infection and heritability, are illustrated by het-s, a prion of the fungus Podospora anserina.1 When two mycelia of this fungus fuse, the [Het-s] prion will migrate from one to the other and spread through its new host mycelium. That is infection, but the prion may also be inherited through the spores produced in the asci by meiosis. In each case, the need and, with reservations, sufficiency of a template is characteristic of the genetic nature of the phenomena.
There are three established native prions of the yeast Saccharomyces cerevisiae whose genes are known: ψ (SUP35), [URE3] (URE2) and [PIN] (RNQ1).2–5,14 There is also a gene, NEW1, coding for a protein of unknown function, which has sequences which promote prion-like behavior in gene fusions but the native protein it codes for has not been shown to occur in a prion form.5
There are additionally numerous other synthetic prions, consisting of sequences derived from those of prion-forming proteins modified by deletion, mutagenesis or by fusion with heterologous natural or artificial sequences for functional modification or as reporters. Of the native prions of yeast, ψ has, perhaps because of its relative stability and easily scored phenotype, lent itself to studies of factors affecting reproductive stability. This article will summarize some of these studies and we will review studies on the cellular events and variables that might affect its stability. We will also consider the kinetic constraints underlying stable transmission of alternative [prion+] or [prion-] states, which have recently been elegantly analyzed by Tanaka et al.6
Yeast cells may exist in either of two states of considerable stability-ψ+ and ψ-. Either may revert to the other spontaneously, but in normal circumstances they do so very rarely.
Changes from ψ- to ψ+
Spontaneous reversion.
The ψ state is stable in growing cultures, in stored cultures under a variety of conditions and in the course of sporulation. Few measurements are available for reversion to ψ+ in any of these conditions. In one study, a less than rigorous fluctuation test suggested a rate of ∼1 x 10-7 per cell division.7 An earlier study in which ψ- states had been induced by various treatments of ψ+ strains showed that many, but not all of these ψ- revertant strains could change spontaneously to ψ+.8 The treatments causing the initial ψ+ to ψ- reversion included growth in the presence of methanol, KCl, DMSO or guanidine hydrochloride and treatment with the conventional mutagens EMS, nitrosoguanidine and UV. At least some of the ψ- strains from all of the treatments, with one exception, could change spontaneously from ψ- back to ψ+. The frequencies observed ranged from 2.8 x 10-3 to 8.5 x 10-8, with a median frequency of ∼6 x 10-6.a The exceptions were 18 ψ- revertants induced by 5 mM guanidine HCl, none of which yielded ψ+ among 108 cells challenged from each.8
These experiments were performed under the paradigm of the time, namely that genetic determinants were nucleic acid. It was deduced that guanidine HCl caused deletions in the ψ determinant as it does in mitochondrial DNA.9 It has since become clear that ψ- is a hyperstable state and simply does not convert to ψ+ unless another prion ([PIN+]) is present.b,10 Although [PIN+] and ψ+ are lost more or less independently when Hsp104 activity is inhibited,18 under most curing protocols, including that used by Lund and Cox,8 guanidine HCl almost invariably cures both ψ+ and [PIN+] together. It follows that all experiments on the spontaneous or induced conversion of the ψ- state to ψ+ are conditional upon and must take into account the implications of the presence of another prion.
Dependence of spontaneous conversion on the presence of another kind of prion is not a property of all prions. The prion-negative state of [URE3], for example is much more labile than that of ψ2,11 and is not absolutely dependent on the strain being [PIN+].12 This is discussed at geater length below, but more particularly by Liebman and Derkach.51
Induction.
The rate at which ψ- converts to ψ+ can be affected by various factors. These include:
Amount of normal protein
Presence of other prions (the [PIN] effect)
Activity of chaperones
DNA sequences in the prion-forming domain and outside it
Acquisition of a prion form of Sup35p
The influence of the amounts of protein.
The native yeast prion proteins, ψ, and [URE3], change from the [prion-] to the [prion+] state more frequently if the parent genes are overexpressed.2,11,13 The native Rnq1p and New1p proteins have not been assayed for this effect, but it is observed when their prion-forming domains (PFDs) are fused to an indicator sequence, such as the C-terminal domain of Sup35p.5,14 Both ψ and [URE3] show about a 100-fold increase in the frequency of change from [prion-] to [prion+] states. The effect is even more pronounced when truncated portions of the genes, containing the PFDs are overexpressed.15,16 In these cases conversion to the prion form is marked by the coupled conversion of the native (full-length) protein.
This property was proposed by Wickner as one of the indicators that a heritable non-mendelian state was due to a prion-based determinant and is now regarded as a fundamental property of such systems (example in Roberts and Wickner, ref. 17). This property has been the basis of searches for other prions in yeast, using high-expression gene libraries.4
The rationale behind this argument is that higher concentrations of the native protein enhance the probability that a spontaneous conversion will occur in one or more of the nonprion-form molecules present and that this will trigger the seeded conversion of the remainder. While this turns out to be what is observed, wherever it is tested, matters are not as simple as that, as we discuss in the next section.
The influence of other prions: ψ and [PIN].
We have noted above that when ψ+ is cured by allowing cell division in the presence of guanidine HCl, it does not seem to be revertible to ψ+. With the exception noted above (footnote), this has turned out to be true even when the SUP35 gene is overexpressed and also when only the N or NM domains or other potent ψ+-inducing constructs are overexpressed. This is, of course, an anomaly, incompatible with one of the important criteria defining a prion proposed by Wickner namely that, as long as the gene coding for the prion-forming protein is present and active, the [prion-] state should always be convertible to the [prion+].
Liebman and her coworkers found that the de novo conversion or induction of ψ+ by overexpression required the presence of another factor. This they named it [PIN+] and showed that it too was inherited in a non-mendelian fashion.10 In due course, they identified [PIN+] as the prion of Rnq1p, [RNQ+].4 In the same paper, they showed that [URE3+] and the overexpressed products of ten other genes could also function as [PIN+].
It also seems to be the case that a heritable form of the heterologous prion may not be necessary for this interaction: amyloid-like aggregates may be sufficient. Firstly, Osherovich and Weissman5 constructed fusions of the Asn-Gln-rich sequences from the prion-forming domains of NEW1 and RNQ1, in each case with GFP. When either of these was cooverexpressed with SUP35PFD-GFP in [pin-] strains, both GFP aggregates, visible microscopically, and heritable ψ+ convertants were obtained, but they remained [pin-].5 The rate of induction was comparable to that found when the strains were heritably [PIN+], about 6% when the NEW1 PFD sequences were overexpressed and one tenth of that in overexpression of the RNQ1 PFD sequences.
Secondly, Derkach et al.4 have found that overexpression of poly-Q tracts, up to three times longer than those in huntingtin genes associated with Huntington's Disease, allow the induction of ψ+ in a [pin-] background. The longer tracts form amyloid readily in yeast cells, and in cells with amyloid from these longer tracts, ψ+ may be induced by overexpression of the NM domains of SUP35.19
[PIN+] is not only necessary for prion formation by Sup35p, but is also necessary for amyloid formation when Sup35p is overexpressed. Most of this amyloid appears not to be heritable: 90% of the cells remain [ψ-], although they allow read-through of nonsense codons.20 It would seem that the formation of amyloid is not sufficient to make a prion: either amyloid is a precursor and a further change in protein conformation must occur to convert it to prion or amyloid and prion are all part of a range of conformers most of which present as amyloid, but with some being susceptible to fragmentation by Hsp104 and being therefore prions. It has been noted that the new “spontaneously-induced” ψ+ strains display a wide range ‘variant’ types.4,5,15,18–20 It may be that in a cell overexpressing Sup35p, provided some [PIN+]-like function exists, a wide variety of refolded type of molecule is produced. If these possess various susceptibilities to the action of Hsp104, then each would present a ‘variant’ phenotype (see Tanaka et al.,6 and discussion below). The ψ+ variant dependent on elevated levels of Hsp104 described by Borchsenius et al.58 is an example of a conformer intermediate between those qualifying as amyloid or as prion.
It is not known what the cause is of these effects. Current heterological speculations lean to the idea that amyloid polymers assist each others' condensation even if they have not enough structural similarities to form copolymers. What does seem to be the case is that the conformational change needed to form self-replicating prions, at least of Sup35p, is, if not wholly impossible in vivo, at least extremely rare in the absence of some amyloid cofactor. In living cells, the normal conformer of Sup35p is hyperstable.
In summary, the presence of any of a wide range of heterologous amyloid proteins, either in prion form or otherwise, seems to be a necessary precondition for the formation of amyloid from Sup35 protein. Conversion of this amyloid to prion is relatively infrequent, clearly showing that a further, or perhaps a completely different kind of conformational change is required to make the Sup35p amyloid heritable. A similar situation may apply to Rnq1 protein, at least in the hybrid RNQ-GFP version, but the absolute requirement for heterologous amyloid probably has not been so rigorously tested. Certainly, ψ+ prions greatly facilitate its conversion. Ure2 protein responds in a similar way to heterologous prions, except that ψ+ has an antagonistic effect on its conversion. However, the presence of another prion ([PIN+]) is not an essential condition for reversion from [ure3-0] to [URE3+].
Sequence.
The sequences defining prion behavior of proteins have been the subject of much analysis (see Tuite and Cox52). The question we address here is whether any specifically affect the frequency of spontaneous reversion from the [prion-] to the [prion+] state. One question is whether any exist which determine a permanent [prion+] condition. The criterion for this would be that the inheritance of the prion condition would be non-mendelian, but there would be a Mendelian segregation for curability.c A less extreme situation would be that the spontaneous rate of prion conversion would simply be very much higher in proteins with one sequence than another. There are surprisingly few examples of this. It seems to be true that the nonprion-forming domains, M and C, of Sup35p inhibit the rate of formation of ψ+ de novo, at least in overexpression experiments.10,21 These experiments served to identify the prion-forming domains of this protein. Similarly, specific fragments of Ure2p yield [URE3+] on overexpression.11 The differences can be quite dramatic: when full length Ure2p is overexpressed, Masison et al.11 found 1.1 x 10-4 [URE3+] revertants, but the numbers were 1.7 x 10-3 and 1.758 x 10-2 for overexpression of a residue 1–65 and a residue 1–80 fragment respectively.
Fernandez-Bellot et al., found a mutated allele of Ure2 which elevated spontaneous rates of reversion from [ure3-0] to [URE3+] some 1000-fold.22 The mutant gene turned out to have 14 point mutations, two of them in the PFD. These two alone did not much increase the frequency of reversion above that found in the wild-type, but adding one of the mutations found in the functional domain raised the frequency 500x. Such cis-interactions implicate non-PFD domains in prion formation and stability and echo the influence of polymorphisms in PrP on susceptibility to CJD in humans. It is interesting, given this result, that mistranslation per se may elevate spontaneous prion-forming rates in Ure2p.23 They compared the effects of a general mistranslation drug, G418, with cycloheximide. In similar experiments with ψ- reversion, Koloteva-Levin et al. found no effect with a drug which targets proline specifically.24
Naturally, much attention has been given to the details of the PFD sequences and their role in [prion-] to [prion+] conversion. The first of these was by Liu and Lindquist, who set out to mimic the effect noted in humans that expanded numbers of the oligopeptide repeats found at positions 50–94 in PrP render subjects more prone to developing CJD.25 Liu and Lindquist found that two extra copies of one of the analogous repeats in the Sup35p sequence led to a 5000x increase in the spontaneous frequency of ψ+ formation.
This was complemented by Parham et al. who one at a time removed repeats and found that first replication and then inclusion in aggregates were affected as more and more repeats were removed.26
This was followed by a random PCR-mutagenesis study by De Pace et al. of the N-terminal prion-forming domain of Sup35.27 They screened the mutant libraries by transforming a ψ+ strain and looking for transformants which lost the ability to suppress the ade1-14 nonsense mutation. Failure to suppress is a signal of the presence of significant amounts of soluble, active Sup35p (eRF3p). They classified these into two categories—those which failed to suppress because they prevented replication of the native ψ+ prion (PNM) and those which did so because they failed to form or be recruited into wild-type amyloid in the first place (ASU). All the mutants they picked up fell into the region coding for the N-terminal 33 residues, which covers the Q/N-rich region of the protein. Rather than test each mutant for the ability to form prion spontaneously de novo, they replaced the whole stretch with poly-Q, and found that indeed overexpression of such constructs induced the ψ+ state.d This effectively identifies the Q/N-rich region as critical for prion formation and propagation and Osherovich and Weissman subsequently showed that almost any poly-Q/N-rich sequence in this region supports de novo reversion.5
Sup35 proteins of many other yeasts have PFDs very similar, but not identical to that of S. cerevisiae. When these heterologous sequences are substituted for the bases coding for first 39 residues of S. cerevisiae Sup35p, although they themselves can form stable prions on overexpression, they cannot induce prion formation in native, resident Sup35p unless it too has the same sequence.28 There is thus a firm species barrier to the transmission of the prion form and it resides in the residues responsible for initiating aggregation, as defined by De Pace et al.27 It is beginning to look as if there are two functional domains in the PFD, one affecting the initial formation of amyloid and the other the replication necessary to make amyloid heritable. A domain-swapping analysis by Osherovich et al. seems to confirm this idea,29 as does the study by Borchsenius et al. in which a deletion which removes part of the oligopeptide repeat (OR) region reduces the ability of the protein to propagate stably, without greatly affecting its ability to aggregate or induce ψ+ formation.55 Furthermore, a recent paper by Crist et al., describes experiments in which oligopeptide repeats from different yeast were substituted for the native S. cerevisiae repeats.30 They show that derivative Sup35p formed aggregates and inactivated Sup35p function just as the native protein does, i.e., mimicked ψ+. The pseudo-ψ+ state could be induced by native ψ+, but these prions did not require Hsp104 activity for replication. In a negative way, this is consistent with the idea that the target for Hsp104 replication of “is the oligopeptide repeat region in Sup35. It was also observed that the pseudo-ψ+, which the authors call [PHI+] could arise spontaneously at 1000x the rate of ψ+, and without benefit of [PIN+].
Acquisition of a prion form of Sup35p.
In addition to de novo conversion, ψ+ prion forms may be acquired by mating, inherited through cell division or by transfection.31,32 Tanaka et al.6 and see below, have also provided evidence that as little as a single molecule of the prion form is sufficient to convert or start the rapid conversion of a substantial majority of soluble Sup35p prion form in vitro. For just how rapidly this can happen in vivo, see Satpute-Krishnan and Serio.33 When cells start with only one or very few prion molecules, conversion is, as expected, Malthusian and occurs with a doubling time of eighteen minutes.6,7,42 It is exactly this property that provides the switch between the two stable states of prion-forming proteins and lies at the basis of the stability of [prion+] forms which we discuss next.
Changes from ψ+ to ψ-
Spontaneous loss.
ψ+ is very stable, sometimes almost as stable as ψ-. “was first discovered as a ψ- mutation in a handful of tetrads which had been expected, being homozygous for a weak super-suppressor, SUQ5 suppressing the red colour of an ade2-1 mutant, all to be white. Instead, three or four of the segregants had red sectors. The red reversions failed to segregate when crossed back to white SUQ5 ade2-1 parents. With one notable exception, neither I nor my colleagues have seen anything like this in our strains since, four decades and thousands of tetrads later. ψ+ is equally stable in mitosis: once again it is very uncommon to see a red sector or colony on plates growing colonies, at least of strong ψ+ strains.
Nevertheless they occur. The rate of spontaneous loss of ψ+ has never been adequately quantified although good systems exist for selecting for the change in phenotype.
Observation of great stability in inheritance raises the question of how it is achieved. There are three situations which might affect it. One is the copy number of a putative determinant. Another is a system for partitioning a low copy-number determinant, as exists for chromosomes, low-copy number bacterial plasmids and, indeed for the relatively high copy-number yeast 2 µ plasmid.34 A third possibility is a feedback regulatory system which switches on a pathway in response to changes in quantity of a significant component. The yeast 2 µ has a feedback system regulating its amplification, for example.56 None of these systems is mutually exclusive and underlying all of them is a requirement to increase the numbers of the determinants at the same rate as the cells multiply.
Curing studies and the origins of genetic stability.
The discovery that the inheritance of yeast prions is entirely (but not exclusively) dependent on the activity of the chaperone heat shock protein, Hsp10435 has made possible a number of studies on the factors maintaining prion stability.
The kinetics of curing with guanidine hydrochloride and the quantification of prion molecules.
Sometime before 1981, Tuite discovered that mM quantities of the denaturing agent guanidine hydrochloride “cured” ψ+ cells to ψ- with 100% efficiency.36,37 It has since been suggested that because such concentrations of guanidine HCl also inhibit Hsp104 activity in vitro and in vivo that ψ+ curing results also from this inhibition.38–40 The curing occurs only in actively growing cultures. It is not immediate upon addition of the guanidine HCl, but shows a considerable lag before ψ- cells begin to appear.41 There are three features of the kinetics of this process that are worth notice. Firstly, once curing starts to be observable, the decline in the proportion of ψ+ cells is exponential and halves at each cell generation. In fact, the entire curve, lag and all, can be fitted with models assuming a halving of average propagon numbers with each generation, a Poisson distribution of “seeds” among cells and that a single “seed” or propagon is sufficient to render a cell genetically ψ+, as recently demonstrated by infection experiments,31 and by further experimental and theoretical development of the Eaglestone model7,42,43 (Fig. 1A).
Figure 1.
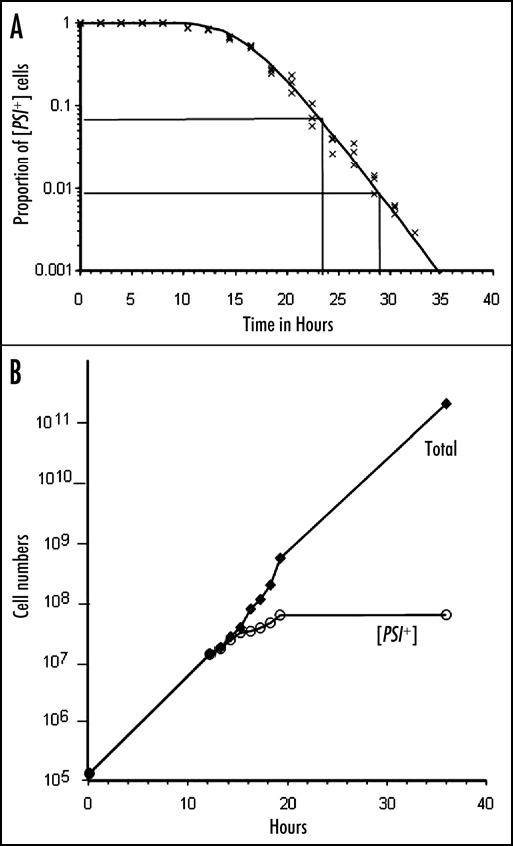
(A) The proportion of γ+ cells left in a growing population following the addition of 5 mM guanidine hydrochloride. The slope of the exponential part of the fitted curve involves a halving of the proportion every 1.8 hours. This was derived from the doubling time of the cell numbers in this culture. The fitted line is from the model described by Cole et al. 43 (B) Data from a curing experiment as described in (A) replotted to show that the numbers of γ+ cells in the culture reaches a maximum but then stays constant as the culture continues to grow at the normal rate (compare ref. 44).
Secondly, propagons do not appear to be destroyed during growth in guanidine HCl. The number of genetically ψ+ cells in the population reaches a maximum asymptotically during the “curing” phase and thereafter remains constant indefinitely44 (Fig. 1B). We suppose this number represents ψ+ cells that are still dividing, but unable to segregate more than one prion molecule at a time, thereby giving rise to one ψ- and one ψ+ daughter at each division.
The “copy-number” of the ψ+ prion has been estimated making an assumption that there is some particulate determinant acting as a “seed,” i.e., a propagon. Numbers of these propagons are supposed to be distributed at random between mother and daughter cells at cell division. Eaglestone et al.41 suggested that the reason for the kinetics of guanidine-promoted curing was that Hsp104 was required for the replication or division of seeds in a cycle involving the accretion of soluble Sup35p molecules to a seed, their consequent conversion to the prion form followed by Hsp104-mediated fragmentation, as suggested by Kushnirov and Ter-Avanesyan in 1998.45 The lag observed when curing is effected by growth in guanidine HCl,41 by competition through overexpression of an Hsp104 double mutant39 or by loss of the Hsp104 gene,46 would be a segregation lag dependent on the dilution of the seeds by a half in every cell generation. A simple model was developed whereby the length of the lag could be used to estimate the average copy-number of propagons.41
The implications of this model have been tested both by growth kinetic experiments and at the molecular level,7,42 and more refined mathematical models have been developed to arrive at estimates of propagon (seed) number.43 These numbers have turned out to be very variable and often strain-dependent but they are always quite high, ranging from 100 to over 1,000 per cell. Similar experiments with [URE3] suggest the copy number of that, less stable prion, is about 20.47
ψ- -inducing agents.
The stability of the ψ+ prion has also been found to be affected by growth in a variety of stress conditions.37,48 However, the stress most commonly applied in studies of this organism, namely heat shock, has no effect on ψ stability.
On the other hand Tuite et al. also found that reversion to ψ- was induced by conventional mutagens such as UV and nitrosoguanidine or EMS and was subject to DNA repair mechanisms, including photoreactivation of UV damage and excision repair.49 Since we must now accept that the determinant of ψ is not DNA, it would seem that there are, nevertheless, uninherited, forms of DNA damage which affect its stability.
Models of Maintenance
Variants and the steady state.
Stability is a property of the particular ψ+ “variant” being considered. So far, most people have observed a positive correlation between stability and the “strength” of a ψ+ variant.15 “Strength” is estimated by the level of suppression, usually by eyeball analysis of the development of the red color that is due to deficiency in adenine biosynthesis. White is strong (Fig. 2). There has also been noted a positive correlation between the copy number of propagons and stability (Fig. 2 and Table 1). Thirdly, it has also been noted that weaker ψ+ variants have larger SDS-resistant “oligomers” as defined by SDS-agarose electrophoresis6,50 and, as expected, more soluble Sup35p.57 It is clear there is an inter-relationship between these four properties: number and size of oligomers, read-through of stop codons and stability in mitosis.
Figure 2.
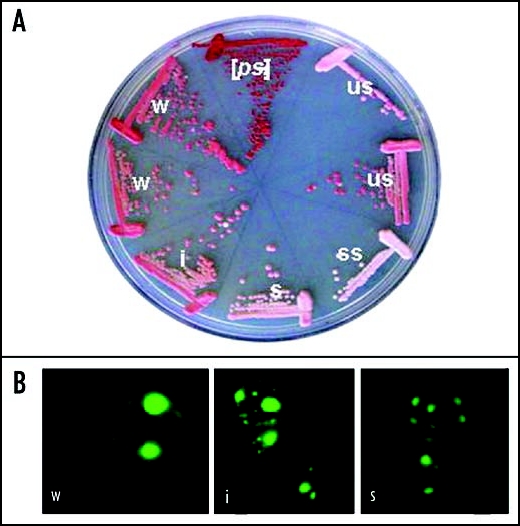
(A) γ+ variants of strain 74-D694 identified by Eric Fernandez-Bellot. W, weak; i, intermediate; s, strong; ss super-strong; us, unstable. (B) Sup35-GFP aggregates in different variants.
Table 1.
Numbers of propagons in ψ+ variants identified by Eric Fernandez-Bellot (see Fig. 2)
| Strain | Propagons Count |
| Weak 2 | 70 ± 10 |
| Weak1 | 105 ± 14 |
| Intermediate | 126 ± 15 |
| Strong | 171 ± 21 |
Tanaka et al. have developed an elegant statistical model which convincingly correlates them.6 There are three quantities, namely the concentrations of monomeric (functional) Sup35p [x], the mass of aggregated Sup35p [z] and the concentration, that is numbers, of fibres or aggregates [y]. In real life, [x] determines the level of suppression: the more functional monomer there is, the less read-through of stop codons; and [y] is the copy-number, which affects stability. These values are governed by four rate constants, namely β, the rate of synthesis of Sup35p; (β) the rate of fibre growth, i.e., the rate at which monomers are incorporated into aggregates (which includes and involves the conversion to the prion form); (γ), the rate at which aggregates are broken down to smaller sizes (by the action of Hsp104) and the rate of cell growth (R) (Fig. 3). β and γ are relevant only to the ψ+ states and are, as determined by these workers' earlier experiments, determined by the structural features peculiar to each ψ+ variant.31
Figure 3.
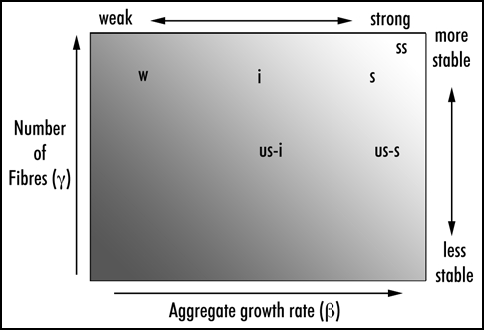
A diagram of the parameters determining ψ+ variant strength and stability: adapted from Figure 1B in Tanaka et al.6 Strength is determined by the concentration of monomeric Sup35p: the less there is, the stronger the suppression. In ψ strains that is determined by the growth rate of aggregate (β) relative to the rate of synthesis of Sup35p (α). Stability is related to the numbers of fibres (vertical axis) which is determined by their fragmentation by Hsp104 etc. (γ). Possible position of the variants found by Fernandez-Bellot Figure 2 are suggested. Tanaka et al. suggest “real” positions of the three variants they describe, calculated from their measured parameters of fibre growth (γ), fragility (γ) and number.
Equations are derived which define steady-state conditions for the ψ state, when all the Sup35p is present as functional monomer, and for ψ+ states which significantly reduce the amount of monomer by maintaining a significant proportion of the Sup35p as aggregate.
They find, interestingly, that the ψ- steady state is not stable: it only persists because of the very high kinetic barrier to spontaneous folding or refolding to the prion form of the protein. (Natural or artificial prion proteins other than Sup35p might, in some cases, have much lower intrinsic stability in the nonprion form). This stability persists only as long as there is no prion conformer present: once present, β (fibre growth) and γ, fibre division ensure a ψ+ steady state. Tanaka determine that ψ+ steady states are stable, but this is probably because they make the assumption in developing their equations (in the absence of contrary evidence) that aggregate degradation to nonprion monomers is negligible in vivo. The actual amounts of monomer, aggregate and numbers of aggregates (fibres) defining variant strength, stability and the size of aggregates, can vary with β and γ independently over quite wide ranges but still tolerate a considerable spectrum of relatively stable ψ+ variants, and β and γ are properties determined for any variant by its structure. Stability is then solely a property of fibre number and partition.
The kinetic instability of the γ- state is a mathematical way of defining a prion as a molecular state requiring a template for its propagation. Tanaka et al. ask whether ψ requires one, two, three or four molecules for a template and find from their infection assay that one will do (ref. 6, supplementary information). Their analysis is supported by a comparison of three phenotypically different ψ+ variants and they relate the measured properties of the variants (propagon number, level of suppression, in vitro fibre fragility and growth rate, and in vivo regeneration rates) to their phenotypes.
The beauty of this analysis is that it allows one to recognize the properties of prions in all their variety as manifestations of a few definable, measurable properties. For example, the strange variant described by Borchsenius et al.58 can be understood as a conformer of Sup35p which, its OR region being perhaps rather inaccessible to Hsp104, has a very low γ rate constant, and so occupies a position on the phanerogram in Figure 3, low down and perhaps towards the right-hand edge. The analysis also allows and accounts for the occurrence of variants which do not exhibit the conventionally accepted correlations of, for example, “strong” with “stable.” In this figure, adapted from Figure 1B in Tanaka et al., we suggest with no experimental evidence apart from suppression and stability, positions for some of the variants described by Fernadez-Bellot (Fig. 2). In their paper, however, Tanaka et al. define positions on the diagram for three variants based on measurements of both the β and γ rate constants.
While this elegant analysis defines the metabolic rate constants and concentration conditions needed to provide the foundations for establishing stable alternative prion states, it stops short of discussing mechanisms of inheritance. This we do in the next section.
Partition, feedback and destruction.
Given genuinely random segregation, a copy-number of 20 such as that found for one of Tanaka et al.'s weak variants, Sc37, should be enough for ψ- segregants to appear at a frequency of only 10-6 x generation-1: comparable to most gene mutation rates. Other variants with typically much higher propagon numbers should be much more stable. However, Cox et al., measuring the distribution of ψ+ propagons at cell division found that mothers acquired twice as many as daughters.7 This ratio does not necessarily imply nonrandomness: it is directly proportional to the relative volumes of mother and daughter at cytokinesis. Nevertheless, if perpetuated, the discrepancy could lead to much greater instability than with 50:50 partition. Five generations of successive malsegregations of this degree without correction would generate about 3% of cells with one or fewer propagons, and it gets worse. Instability of this order is not observed, even in weak variants. Correction may be inherent in the physiology of yeast cell division. Lee Byrne has observed that daughter cells take twice as long to enter the next round of cell division as do their mothers and if propagon synthesis is continuous, that would neatly balance the discrepancy in numbers from unequal partition (Bryne L.S., unpublished observations).
Conditions can arise, or be imposed, which affect partition. Ness et al. report that when curing by guanidine HCl occurs, towards the end of the lag period partition becomes very inefficient and daughters seldom inherit ψ+: the prion or prions stay in the mother in >90% of cell divisions7,42 (Fig. 4). This is understandable in terms of the Tanaka model, since inactivation of Hsp104 affects only γ (fiber division) but not, as Ness et al. show, the other two rate constants. Thus, fibres continue to grow under the curing regime, and after several generations may reach a size which interferes with their passage to daughter cells.
Figure 4.
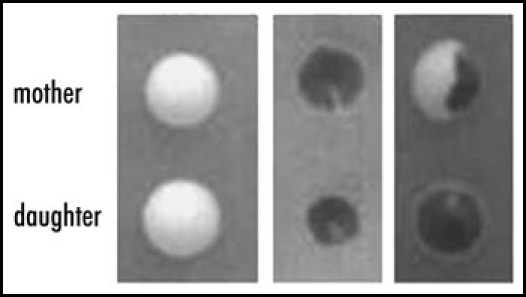
Segregation of ψ+ and ψ- in mother-daughter pairs taken from a culture growing in guanidine hydrochloride in which only 67% of the cells remained ψ+ . Budded cells were selected by micromanipulation on a YEPD agar plate and the daughter bud separated from the mother, each being left to grow into a colony at a marked place on the plate. The third pair illustrates segregation of ψ+ from ψ- both in the chosen pair of cells and at the next division of the mother cell. It is evident that the ψ+ prion remained in the mother at the first division, as it did in 93% of the mother daughter pairs taken from this culture.
Similarly, partition may be affected by the overexpression of Hsp104 itself. The ψ prion is unique in its destabilization by high levels of Hsp104: no other yeast prion is affected by this treatment. It is a common assumption that excess Hsp104 breaks down ψ aggregates and so destroys propagons. However, it is also observed that serious partition defects appear in these conditions and these could well explain the curing effect (Fig. 5). Fernandez-Bellot also showed that sensitivity of ψ+ variants to excess Hsp104 was inversely proportional to their strength (Fig. 6) and this is perhaps more consistent with a partition defect, it being more apparent in low copy-number variants with larger aggregates (Fig. 6; Fernandez-Bellot, unpublished observations).
Figure 5.
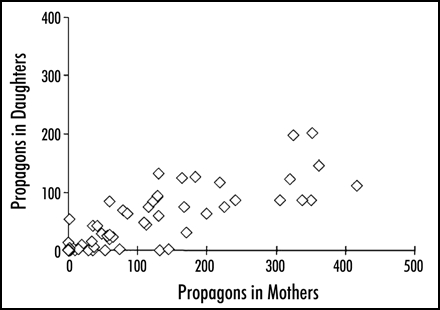
Propagons in pairs of mother and daughter cells taken from a culture of ψ+ cells in which Hsp104 had been overexpressed for 20 hours. The mothers and daughters were separated after cytokinesis and placed on 3 mM guanidine hydrochloride as described in reference 6. The figure illustrates malsegregation in a minority of the dividing cells, never observed in normal cultures. Thirty-six percent of the colonies from this culture were ψ- or sectored.
Figure 6.
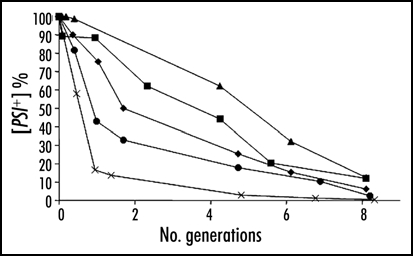
Weak variants are more readily destabilized by overexpressed Hsp104 than are strong variants. The symbols correspond to: ▲, strong; ■, unstable; ◆, intermediate;● and X, weak.
There is no indication, apart from these few departures from normal distribution in unusual circumstances, that there may be any mechanism that promotes equal partition of propagons of ψ+.
Equally, evidence of feedback systems maintaining numbers of propagons sufficient for stability is light. Tanaka et al. state that stability of ψ+ is achieved when αβγ/R3 is equal to or greater than 1.6 However, ratios greater than 1 would imply an accumulation with time of both monomer and aggregate, whereas it is apparent that the totals stay quite constant. If the ultimate regulator is the rate of synthesis of monomer, then the only feedback is from, and inherent in, those systems coupling protein synthesis with cell growth and division.
In experiments that measure the rate of synthesis of new propagons in cells depleted of them by growth in guanidine HCl, Ness et al. have found that there is an exponential increase in numbers with a doubling time of about 20 minutes and that the numbers level out after about two hours.42 This would appear to suggest a feedback control of some sort, but Tanaka et al. show that it is a consequence of the balance between the four rate constants as they define them and the saturation of the terms. The kinetics of the recovery of propagon numbers that they calculate from estimates of the rate constants for a strong ψ+ variant closely match those observed by Ness et al. (ref. 6 and supplementary material).
The effects of chaperones: Prion degradation.
Although it alone is essential for the replication of prions, the chaperone Hsp104 interacts with both Hsp70 and Hsp40 chaperones in prion maintenance. Rikhvanov et al. discusses their roles and link them in a schematic model involving them in generation, reproduction and inactivation of the ψ prion.53 Recently conditions have been described which suggest that propagons may be lost by means other than inactivation of their reproduction and dilution by cell division.54 These authors found that during a prolonged inhibition of cell growth by α-factor in the presence of guanidine HCl, curing of ψ+ with kinetics apparently similar to those observed when cured during growth could be observed. They suggest that this is brought about by destabilisation of prion aggregates as a result of their growth unfettered by the activity of Hsp104.
Although Wu et al. record no observations of such destabilisation, it is entirely possible that in these highly abnormal conditions prion reproductive activity may be lost by their degradation, sequestration or occlusion, and Rikhvanov et al.53 include such a pathway in their model, exploiting the known properties of chaperones in disaggregating, refolding or degrading nonnative proteins. For [URE3+] also, two systems by which it might be cured to the [ure3-0] state have been described.47 One, as with [ψ+], is brought about by growth in guanidine hydrochloride and is accounted for by dilution through cell division. The other is observed when the Ure2p PFD (residues 1–93) is overexpressed. If this is done in [URE3+] cells, large aggregates are formed (containing both full-length protein and PFD) concomitantly with the loss of [URE3+] cells. The aggregates evidently have no propagon activity: they are to all intents and purposes dead and whether they are passed to daughter cells or not, they are not destroyed over a period of 40 hours. Quite possibly this also happens to some extent with Sup35, since blocking Hsp104 activity still allows soluble Sup35p to accrete to aggregates and we have noted above the effect of these treatments on partition (ref. 42 and Fig. 4). This effect may be exaggerated in α-factor arrested cells where dispersion of large aggregates cannot be ameliorated by cell division. It has yet to be established what is going on in the yeast strain treated as is described by Wu et al.54 Whatever it is it clearly plays a minor role in normally dividing cells (see Note).
Summary
The Sup35 protein of yeast has several hyperstable states: ψ-, in which all the Sup35 protein in the cell is in a soluble native form, active as the only and therefore essential, eRF3 in translation; and prion variants of ψ+ in which up to 95% of the protein is in an aggregated form unable, as far as is known, to function in polypeptide chain termination. There are, in addition, conditions in which nonstable states may exist, which are not heritable. For example, when Sup35 or C-truncated versions of it are overexpressed, high molecular weight oligomeric aggregates can be detected in cell cultures and it is speculated that there may be soluble monomeric or oligomeric intermediates formed transiently in the conversion of the native monomer to ψ+ forms. Conversion in vivo of ψ- into any of the ψ+ variants requires the presence of another heterologous prion, commonly that of the Rnq1 protein; or of amyloid. This is also true of the nonheritable aggregates produced by overexpression.
The stable ψ- conformer may owe its stability to the intrinsically low entropy characteristic of folded functionally evolved proteins, but this is overcome, with some difficulty by interactions, whose nature is unknown, with heterologous prions; and with great ease when any variant ψ+ conformer is present. A single prion molecule is sufficient to initiate the switch from the stable ψ- state of a cell, which contains 100% of soluble active monomers, to a stably ψ+ cell with 95% of those molecules being in prion form, in aggregates large or small.
This is the basis of the chemical stability of ψ- and ψ+ conditions. ψ+ stability in dividing cells needs further conditions, since the maintenance of the state in vivo critically depends on the presence of a pre-existing prion molecule.e Firstly, stability requires that such “seeds” or propagons interact with nonprion native conformers to promote their conversion. Secondly, it requires that propagons are inherited by both progeny of every cell division and this means that their numbers have to be maintained at the same rate at which the cells divide and that they be partitioned efficiently. At least two of these conditions depend on apparently distinct sequences in the prion-forming domain of the proteins. Interaction of propagons and native monomer, identified in biochemical assays as the formation of aggregates, depends in Sup35 on the QN-rich region in the first 40 residues at the N-terminal. This is indicated by the fact that this is the domain specifying the “species barrier” and also by the fact that it can be substituted without much affecting ψ+ propagation by known aggregation-promoting sequences from heterologous sources or by synthetic sequences. The maintenance of propagon numbers depends on the activity of Hsp104 interacting with another domain in the Sup35 protein, namely the oligopeptide repeat region. Again there are heterologous sequences from other prion-forming proteins that can substitute for this function.
Acknowledgements
Brian S. Cox wishes to acknowledge the award of an Emeritus Fellowship by the Leverhulme Trust, during the tenure of which this work was done.
These frequencies do not necessarily represent different inherent instabilities, and are more likely to be technical artifacts.
As usual there is an exception.18 ψ can convert to ψ+ in a [pin-] background when a truncated version of SUP35 with a small extension picked up from its vector is overexpressed.
One of the characteristics so far observed of prions in fungi is that all authentic native prions are dependent on Hsp104 activity for reproduction. Inactivating or eliminating Hsp104 leads to their being “cured”. A sequence which automatically forms “prion” would not show curing. The only indication that the phenotype was due to a prion form of a protein would be if it were to convert an alternative, different, sequence of the protein to a prion form, dependent on Hsp104. This emphasises the requirement for seeding as a property of a prion protein to a critical role in defining the phenomenon.
All these experiments were done in the presence of the wild-type SUP35 gene, so whether the effects are dependent on its presence or not is not clear.
It should of course be noticed that this condition, by its absence, is what maintains the stability of the ψ-state and defines prions. If there were no requirement for a template, there would be no “switch,” only a distribution, potentially in every cell, between forms depending on the energetic parameters of each form, the proportions being conditioned by their relative entropies. These in turn may depend on sequence. For example, the propensity of huntingtin to form amyloid is directly related to the number of glutamine residues in the polyQ region. There might also be intermediate situations, in which the likelihood of amyloid formation depends on the previous presence of amyloid: this would be prion-like, at least as far as the conversion part of the process was concerned. Inheritance or infection, however, would depend in addition on reproduction and transmission of the amyloid. It might be quite difficult in some cases to distinguish between the necessity of a template, defining a prion and mere kinetic instability. [PHI+], might for example be one such case.30
Previously published online as a Prion E-publication: http://www.landesbioscience.com/journals/prion/article/4839
Note
Since this article was written, it has been shown that some results described by Wu et al.54 incorporated an artifact due to lethality during emergence from α-factor arrest in the strain used by them. This lethality masked cell division and so gave the illusion of curing in its absence. It has also been shown that there was no destruction of propagons in their conditions and that curing depended on and correlated exactly with cell division (Byrne LJ, Cox BS, Cole DJ, Ridout MS, Morgan BJ, Tuite MF. Cell division is essential for elimination of the yeast [PSI+] prion by guanidine hydrochloride. Proc Natl Acad Sci USA 2007; 104:11688–93.)
This manuscript has been previously published: Cox BS, Byrne L, Tuite MF. Prion Stability. In: Protein-Based Inheritence. Chernoff, Y ed. Austin and New York: Landes Bioscience and Kluwer Academic Press, 2007; 39–46.
References
- 1.Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci USA. 1997;94:9773–9778. doi: 10.1073/pnas.94.18.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wickner RB. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 3.Cox BS. Prion-like factors in yeast. Curr Biol. 1994;4:744–748. doi: 10.1016/s0960-9822(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 4.Derkach IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: The story of [PIN+] Cell. 2001;106:171–172. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 5.Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell. 2001;106:183–184. doi: 10.1016/s0092-8674(01)00440-8. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 7.Cox BS, Ness F, Tuite MF. Analysis of the generation and segregation of propagons: Entities that propagate the [PSI+] prion in yeast. Genetics. 2003;165:23–33. doi: 10.1093/genetics/165.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lund PM, Cox BS. Reversion analysis of [psi-] mutants in Saccharomyces cerevisiae. Genet Res. 1981;37:173–182. doi: 10.1017/s0016672300020140. [DOI] [PubMed] [Google Scholar]
- 9.Juliani MH, Gambarini AG, Da Costa MOP. Induction of rho-minus mutants in Saccharomyces cerevisiae by guanidine hydrochloride. I. Growth analysis. Mutation Res. 1975;29:67–75. [PubMed] [Google Scholar]
- 10.Derkach IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics. 1997;147:507–519. doi: 10.1093/genetics/147.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masison DC, Maddelein ML, Wickner RB. The prion model for [URE3] of yeast: Spontaneous generation and requirements for propagation. Proc Natl Acad Sci USA. 1997;94:12503–12508. doi: 10.1073/pnas.94.23.12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. Interactions among prions and prion “strains” in yeast. Proc Natl Acad Sci USA. 2002;99:16392–16399. doi: 10.1073/pnas.152330699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chernoff YO, Derkach IL, Inge-Vechtomov SG. Multicopy SUP35 gene induces the de novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae. Curr Genet. 1993;24:268–270. doi: 10.1007/BF00351802. [DOI] [PubMed] [Google Scholar]
- 14.Sondheimer N, Lindquist S. Rnq1: An epigenetic modifier of protein function in yeast. Molec Cell. 2000;5:163–172. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 15.Derkach IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics. 1996;144:1375–1386. doi: 10.1093/genetics/144.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masison DC, Wickner RB. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2- in prion-containing cells. Science. 1995;270:93–95. doi: 10.1126/science.270.5233.93. [DOI] [PubMed] [Google Scholar]
- 17.Roberts BT, Wickner RB. A new kind of prion. Cell Cycle. 2004;3:100–103. [PubMed] [Google Scholar]
- 18.Derkach IL, Bradley ME, Masse SVL, Zadorsky SP, Polozkov GV, Inge-Vechtomov SG, Liebman SW. Dependence and independence of [PSI+] and [PIN+]: A two-prion system in yeast? EMBO J. 2000;19:1942–1952. doi: 10.1093/emboj/19.9.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derkach IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci USA. 2004;101:12934–12939. doi: 10.1073/pnas.0404968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salnikova AB, Kryndushkin DS, Smirnov VN, Kushnirov VV, Ter-Avanesyan MD. Nonsense suppression in yeast cells overproducing Sup35 (eRF3) is caused by its nonheritable amyloids. J Biol Chem. 2005;280:8808–8812. doi: 10.1074/jbc.M410150200. [DOI] [PubMed] [Google Scholar]
- 21.Kochneva-Pervukhova NV, Poznyakovski AI, Smirnov VN, Ter-Avanesyan MD. C-terminal truncation of the Sup35 protein increases the frequency of de novo generation of a prion-based [PSI+] determinant in Saccharomyces cerevisiae. Curr Genet. 1998;34:146–151. doi: 10.1007/s002940050379. [DOI] [PubMed] [Google Scholar]
- 22.Fernandez-Bellot E, Guillemet E, Cullin C. The yeast prion [URE3] can be greatly induced by a functional mutated URE2 allele. EMBO J. 2000;19:3215–3222. doi: 10.1093/emboj/19.13.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatin I, Bidou L, Cullin C, Rousset JP. Translational errors as an early event in prion conversion. Cell Mol Biol (Noisy-le-grand) 2001;47:23–28. [PubMed] [Google Scholar]
- 24.Kolotova-Levine N, Merritt G, Tuite MF. Unpublished results.
- 25.Liu JJ, Lindquist S. Oligopeptide-repeat expansions modulate ‘protein-only’ inheritance in yeast. Nature. 1999;400:573–576. doi: 10.1038/23048. [DOI] [PubMed] [Google Scholar]
- 26.Parham SN, Resende CG, Tuite MF. Oligopeptide repeats in the yeast protein Sup35p stabilize intermolecular prion interactions. EMBO J. 2001;20:2111–2119. doi: 10.1093/emboj/20.9.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Pace AH, Santoso A, Hillner P, Weissman JS. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Nature Struct Biol. 2002;9:389–396. [Google Scholar]
- 28.Santoso A, Chien P, Osherovich LZ, Weissman JS. Molecular basis of a yeast prion species barrier. Cell. 2000;100:277–288. doi: 10.1016/s0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- 29.Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biology. 2004;2:0442. doi: 10.1371/journal.pbio.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crist CG, Nakayashiki T, Kurahashi H, Nakamura Y. [PHI+], a novel Sup35-prion variant propagated with non-Glu/Asn oligopeptide repeats in the absence of the chaperone protein, Hsp104. Genes and Cells. 2003;8:603–618. doi: 10.1046/j.1365-2443.2003.00661.x. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:265–267. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 32.King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319–323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 33.Satpute-Krishnan P, Serio TR. Prion protein remodelling confers an immediate phenotypic switch. Nature. 2005;437:262–265. doi: 10.1038/nature03981. [DOI] [PubMed] [Google Scholar]
- 34.Mehta S, Yang XM, Chan CS, Dobson MJ, Jayaram M, Velmurugan S. The 2-micron plasmid purloins the yeast cohesin complex: A mechanism for coupling plasmid partitioning and chromosome segregation. J Cell Biol. 2002;158:625–637. doi: 10.1083/jcb.200204136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;68:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 36.Tuite MF. Oxford: 1979. D. Phil Thesis. [Google Scholar]
- 37.Tuite MF, Mundy CJ, Cox BS. Agents that cause a high frequency of genetic change from [psi+] to [psi-] in Saccharomyces cerevisiae. Genetics. 1981;98:691–711. doi: 10.1093/genetics/98.4.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glover JR, Lindquist SL. Hsp104, Hsp70 and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 39.Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. The elimination of the [psi+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol. 2001;40:1357–1369. doi: 10.1046/j.1365-2958.2001.02478.x. [DOI] [PubMed] [Google Scholar]
- 40.Jung G, Masison DC. Guanidine hydrochloride inhibits Hsp104 activity in vivo: A possible explanation for its effect in curing yeast prions. Curr Microbiol. 2001;43:7–10. doi: 10.1007/s002840010251. [DOI] [PubMed] [Google Scholar]
- 41.Eaglestone S, Ruddock LW, Cox BS, Tuite MF. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI+] of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2000;97:240–244. doi: 10.1073/pnas.97.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ness F, Ferreira P, Cox BS, Tuite MF. Guanidine hydrochloride inhibits the generation of prion seeds but not prion protein aggregation in yeast. Mol Cell Biol. 2002;22:5593–5605. doi: 10.1128/MCB.22.15.5593-5605.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cole DJ, Morgan BJT, Ridout MS, Byrne LJ, Tuite MF. Estimating the number of prions in yeast cells. Mathematical Medicine and Biology. 2004;21:369–395. doi: 10.1093/imammb21.4.369. [DOI] [PubMed] [Google Scholar]
- 44.Cox BS. In: Psi Phenomena in Yeast: Early Days of Yeast Genetics. Hall MD, Linder P, editors. NY: Cold Spring Harbor Laboratory Press; 1993. p. 477. [Google Scholar]
- 45.Kushnirov VV, Ter-Avenasyan MD. Structure and replication of yeast prions. Cell. 1998;94:13–16. doi: 10.1016/s0092-8674(00)81216-7. [DOI] [PubMed] [Google Scholar]
- 46.Wegrzyn RD, Bapat K, Newnam GP, Zink AD, Chernoff YO. Mechanisms of prion loss after Hsp104 inactivation in yeast. Molec Cell Biol. 2001;21:4656–4669. doi: 10.1128/MCB.21.14.4656-4669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ripaud L, Maillet L, Cullin C. The mechanisms of [URE3] prion elimination demonstrate that large aggregates of Ure2p are dead-end products. EMBO J. 2003;22:5251–5259. doi: 10.1093/emboj/cdg488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh A, Helms C, Sherman F. Mutation of the nonmendelian suppressor, Psi, in yeast by hypertonic media. Proc Natl Acad Sci USA. 1979;76:1952–1956. doi: 10.1073/pnas.76.4.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tuite MF, Cox BS. Ultraviolet mutagenesis studies of [psi], a cytoplasmic determinant of Saccharomyces cerevisae. Genetics. 1980;95:611–630. doi: 10.1093/genetics/95.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49636–49643. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 51.Liebman SL, Derkach IL. Prion-Prion interactions. In: Chernoff Y, editor. Protein-Based Inheritence. Austin and New York: Landes Bioscience and Kluwer Academic Press; 2007. pp. 39–46. [Google Scholar]
- 52.Tuite MF, Cox BS. The genetic control and propagation of the [PSI+] prion in yeast. In: Chernoff Y, editor. Protein-Based Inheritence. Austin and New York: Landes Bioscience and Kluwer Academic Press; 2007. pp. 14–26. [Google Scholar]
- 53.Rikhvanov EG, Romanova NV, Chernoff YO. Chaperone effects on prion and nonprion aggregates. In: Chernoff Y, editor. Protein-Based Inheritence. Austin and New York: Landes Bioscience and Kluwer Academic Press; 2007. pp. 83–89. [Google Scholar]
- 54.Wu YX, Greene LE, Masison DC, Eisenberg E. Curing of yeast [PSI+] prion by guanidine inactivation of Hsp104 does not require cell division. Proc Natl Acad Sci USA. 2005;102:12789–12794. doi: 10.1073/pnas.0506384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borchsenius AS, Wegrzyn RD, Newnam GP, Inge-Vechtomov SG, Chernoff YO. Yeast prion protein derivative defective in aggregate shearing and production of new ‘seeds’. EMBO J. 2001;20:6683–6691. doi: 10.1093/emboj/20.23.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Futcher AB. The 2 micron circle plasmid of Saccharomyces cerevisiae. Yeast. 1988;4:27–40. doi: 10.1002/yea.320040104. [DOI] [PubMed] [Google Scholar]
- 57.Zhou P, Derkach IL, Uptain SM, Patino MM, Lindquist S, Liebman SW. The yeast nonmendelian factor [ETA+] is a variant of [PSI+], a prion-like form of release factor eRF3. EMBO J. 1999;18:1182–1191. doi: 10.1093/emboj/18.5.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Borchsenius AS, Muller S, Newnam GP, Inge-Vechtomov SG, Chernoff YO. Prion variant maintained only at high levels of the Hsp104 disaggregase. Curr Genet. 2006;49:21–29. doi: 10.1007/s00294-005-0035-0. [DOI] [PubMed] [Google Scholar]