

Abstract
Yeast prion determinants are related to polymerization of some proteins into amyloid-like fibers. The [PSI+] determinant reflects polymerization of the Sup35 protein. Fragmentation of prion polymers by the Hsp104 chaperone represents a key step of the prion replication cycle. The frequency of fragmentation varies depending on the structure of the prion polymers and defines variation in the prion phenotypes, e.g., the suppressor strength of [PSI+] and stability of its inheritance. Besides [PSI+], overproduction of Sup35 can produce nonheritable phenotypically silent Sup35 amyloid-like polymers. These polymers are fragmented poorly and are present due to efficient seeding with the Rnq1 prion polymers, which occurs by several orders of magnitude more frequently than seeding of [PSI+] appearance. Such Sup35 polymers resemble human nonprion amyloids by their nonheritability, mode of appearance and increased size. Thus, a single protein, Sup35, can model both prion and nonprion amyloids. In yeast, these phenomena are distinguished by the frequency of polymer fragmentation. We argue that in mammals the fragmentation frequency also represents a key factor defining differing properties of prion and nonprion amyloids, including infectivity. By analogy with the Rnq1 seeding of nonheritable Sup35 polymers, the “species barrier” in prion transmission may be due to seeding by heterologous prion of nontransmissible type of amyloid, rather than due to the lack of seeding.
Key Words: amyloid, prion, Rnq1, Sup35, Ure2, translation termination, yeast
Prion and Nonprion Amyloids of Mammals
Prions were originally defined as a unique class of infectious agents, whose infectivity relates solely to protein. In mammals, they cause fatal neurodegenerative diseases, such as Creutzfeldt-Jacob disease of man, sheep scrapie and bovine spongiform encephalopathy (reviewed in refs. 1 and 2). All these diseases are related to the PrP protein, whose conformationally altered form (PrPSc) is able to convert the normal host-encoded protein (PrPC) into this altered prion form. While only one prion protein is known in mammals, the prions appear to represent just a part of a much wider phenomenon, amyloidoses.
Amyloid diseases represent a group of more than 30 human diseases, which are characterized by deposition in different tissues of fibrous aggregates of conformationally altered proteins.3 Some of these diseases, like Alzheimer's and Parkinson's disease, represent a major challenge for the public health care in the developed countries. Although amyloidogenic proteins are structurally and functionally unrelated, they form morphologically similar amyloid fibers which reproduce the key properties of prions: the normal and polymer forms of amyloidogenic proteins are structurally different and the latter can promote polymerization of normal proteins into amyloid fibers. Unlike prion diseases, amyloid diseases are generally not transmissible.
Lately, the prion and amyloid phenomena acquired a more general significance for biology due to the finding of prions in lower eukaryotes and observations that in multicellular organisms they may be used in important biological mechanisms. It was shown that amyloid polymers of Pmel17 template and accelerate the polymerization of melanin precursor into melanin.4 Melanin precursor is toxic, while its polymers protect cells from a broad range of cytotoxic insults including UV and oxidative damage. A very important candidate for prion-like mechanism relates to long-term memory in animals. The translational regulator CPEB of Aplysia californica, which plays a key role in the long-term synaptic changes associated with memory storage, demonstrated prion-like properties in yeast.5
Yeast Prions
Prion-like protein behavior underlies the inheritance of some phenotypic traits in the yeast Saccharomyces cerevisiae6 and filamentous fungus Podospora anserina.7 In yeast, there are several proteins, which can undergo prion-like structural conversion. The most studied of them are translation termination factor eRF3, also called Sup35, and Ure2 involved in regulation of nitrogen metabolism.6 The prion state of these proteins can propagate stably for many cellular generations. This may be observed by characteristic phenotypes, [PSI+] and [URE3], which reflect reduced function of the respective proteins, Sup35 and Ure2, due to aggregation of their prion form. In particular, the prion state of Sup35 results in low levels of soluble functional Sup35 and impaired translation termination, which is manifested as a nonsense-suppressor [PSI+] phenotype. Unlike [PSI+] and [URE3], the phenotypic manifestation of the third yeast prion, [PIN+], is not related to inactivation of the corresponding protein. This prion determinant was originally described as a factor required for the [PSI+] de novo generation by transient Sup35 overproduction.8 Later it was found that it also facilitates the de novo appearance of [URE3].9 Unlike the appearance, propagation of [PSI+] and [URE3] does not depend on the presence of [PIN+].
The prion properties of the described yeast proteins rely on their areas rich in glutamine and/or asparagine (QN), which apparently reflects an increased ability of QN-rich sequences to form prions. Analysis of selected yeast proteins with QN-rich regions uncovered Rnq1 prion, but other candidates showed only some of prion properties. When overproduced, the Ybr016w and Hrp1 proteins form aggregates evident by coalescence of their GFP fusions, but their prion abilities were not reported.10 QN-rich region of the New1 protein formed prion when fused to Sup35, but prion formation by New1 itself was not shown.11 A screen for a protein responsible for the [PIN+] phenotype revealed Rnq1, Ure2, New1 and nine other candidate proteins. Overproduction of these proteins, together with Sup35, allowed [PSI+] appearance in [pin-] [psi-] cells.12 This relied on the observation that overproduction of a prionogenic protein greatly increases the probability of its switch into prion form. In this experiment, a prion-like switch of a candidate protein facilitated an analogous switch of Sup35. All candidates possessed QN-rich regions, but only Rnq1 and Ure2 are known to exist in a prion mode. Possibly, other candidates aggregate stably only when they are overproduced. [PIN+] in most cases is related to the prion state of Rnq1, for which Rnq1 is usually implied as a protein underlying the [PIN+] determinant. However, it should be kept in mind that the Ure2 prion also exhibits the Pin+ phenotype. Interestingly, both [URE3] and [PSI+] facilitate the appearance of the prion form of Rnq1,12 when Rnq1 is overproduced. Thus, QN-rich prions promote one another's de novo appearance.
Modular Structure of Yeast Prion Proteins
The Sup35 protein has a clear three-domain structure. The nonessential N-terminal domain is responsible for [PSI+] appearance and maintenance.13,14 The charged middle (M) domain, is important, though not required for [PSI+] propagation.15,16 The C-terminal domain performs the essential translation termination activity.17 In [PSI+] cells, Sup35 polymerizes via its prion domain, which strongly reduces its function in termination (Fig. 1). The levels of soluble Sup35 decrease, in some cases to less than 1% of the [psi-] level.18,19 This causes increased nonsense codon readthrough, which may be conveniently detected using the ade2-1 UAA or ade1-14 UGA nonsense mutations. In these mutants the [psi-] cells are adenine requiring and accumulate red pigment related to impaired adenine biosynthesis. [PSI+] cells are adenine-independent and form white (strong suppressor [PSI+] variants) or pink (weak variants) colonies.
Figure 1.
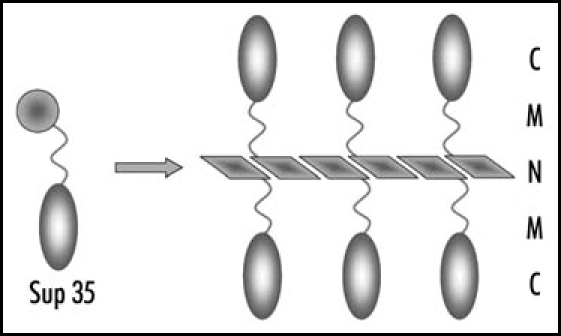
Structure of Sup35 polymers. Amyloid-like fiber is formed by the Sup35N domains. N, M and C, domains of Sup35.
The Ure2 protein has a similar structure, which includes the N-terminal prion-forming domain and the C-terminal functional domain structurally similar to glutathione transferases.20 In contrast to Sup35 and Ure2, Rnq1 lacks conservative or presumed functional domains. The prion-forming region involves C-terminal two thirds of Rnq1.10
The modular structure of Sup35 allows convenient adaptation of the yeast [PSI+] system to test the prionogenic potential of various proteins or protein domains. This is performed by replacing the Sup35 prion domain, via DNA manipulations, for polypeptide sequences of interest, and studying the ability of fusion proteins to cause the [PSI+]-like phenotype. In another type of fusion, the C domain of Sup35 is replaced for green fluorescent protein (GFP), which allows visualization of prion aggregates by the appearance of bright fluorescent foci.21
Two-Level Structure of Prion Aggregates
The available data strongly suggest that yeast prions are composed of amyloid-like fibers. In vitro, purified Sup35, Ure2 and Rnq1 form fibers of uniform structure, which share the key properties of amyloid fibers.22–24 Furthermore, such fibers of Sup35 and Ure2 can transform yeast cells from [psi-] to [PSI+] and from [ure3] to [URE3] states, respectively.25–27 A simple treatment was discovered, which allows distinguishing prion particles from the vast majority of other cellular protein complexes. Unlike these complexes, prion polymers of Sup35 and Rnq1 are insoluble in the presence of SDS at room temperature,28,29 which is likely to represent a general property of amyloids.
The treatment of Sup35 aggregates with SDS reduced their size about 35-fold. This led to the conclusion that the aggregates contain multiple SDS-resistant particles, prion polymers, which are likely to represent amyloid-like fibers. In addition, the aggregates should contain a significant amount of associated nonprion proteins and complexes, since even in a [psi-] state a large portion of Sup35 is found in high molecular mass fraction, being associated with polyribosomes28 (Fig. 2).
Figure 2.
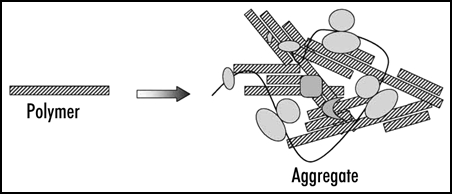
Prion polymers and aggregates. The aggregates represent irregular complexes containing multiple prion polymers and some additional proteins. In case of Sup35 these are presumably Sup35 functional partners, polyribosomes and chaperones.
A procedure for analysis of the size of prion polymers using SDS-agarose gels was developed, called semi-denaturing detergent-agarose gel electrophoresis (SDD-AGE). This method revealed that the Sup35 polymers of [PSI+] cells comprise on average from 10 to 50 Sup35 molecules, depending on the prion variant.28 Thus, yeast prion aggregates represent higher order complexes of relatively short amyloid-like polymers and some nonprion molecules.
Role of Hsp104 Chaperone in Yeast Prion Propagation
[PSI+] propagation may be affected by chaperones of the Hsp70 and Hsp40 families (for review, see ref. 30), but the most significant role is played by Hsp104, which is shown to be strictly required for maintenance of [PSI+]31 and other yeast prions.8,32 Hsp104 breaks large aggregates of denatured protein into smaller pieces, which allows their further refolding by Hsp70 and Hsp40 and solubilization.33,34 A similar action of Hsp104 applied to filamentous prion particles should yield a strikingly different effect. Every break of a filament should accelerate the prion conversion by creating new ends of prion polymers, where the conversion occurs, and multiply prion particles, which is required for their stable inheritance.35 In fact, fragmentation completes the replication cycle of a prion. The other part of this cycle represents prion growth by polymerization, which does not require, at least in vitro, the help of additional factors (Fig. 3).
Figure 3.
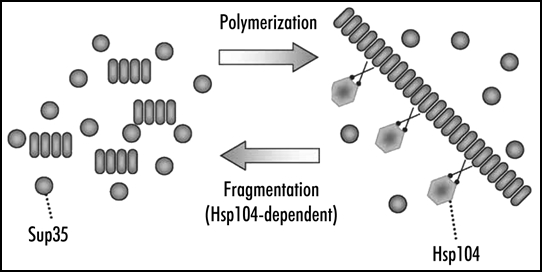
Replication of yeast Sup35 prion polymers. Polymers grow by joining Sup35 monomers and multiply by fragmentation with the Hsp104 chaperone.
An alternative model for the role of Hsp104 was also proposed, presuming that Hsp104 facilitates prion conversion by helping to obtain some unfolded intermediate form of prionogenic protein.21,36 However, studies of Hsp104 inhibition indicate strongly in favor of the former model. The activity of Hsp104 may be inhibited by growing yeast cells in the presence of low concentrations of guanidine-HCl (GuHCl).37 This treatment is known to efficiently cure [PSI+]38 and other known yeast prions. Study of the kinetics of [PSI+] loss in the presence of GuHCl led to the conclusion that it blocks the replication of prion “seeds,” named propagons,39 while not interfering with incorporation of monomers into prions.40
Similar data were obtained using SDD-AGE analysis of the Sup35 polymer size.28 Addition of GuHCl to medium caused a rapid increase in the size of polymers, while not interfering, at least for the first cell generation, with Sup35 polymerization. The size of Sup35 polymers grew twofold per cell generation, a rate that may only be achieved on conditions of the full block of fragmentation and complete incorporation of newly synthesized Sup35 into polymers. The [PSI+] curing by GuHCl could be prevented by mutations in the HSP104 gene.41 Most probably, this indicated that Hsp104 was solely responsible for the block, though did not rule out involvement of other proteins, as such a mutation could affect interaction of Hsp104 with other proteins. Repression of Hsp104 synthesis also increased the size of Sup35 polymers, although repression does not cause immediate inactivation of Hsp104. Thus, Hsp104 is required for polymer fragmentation, but does not affect polymerization.
Different Accessibility of Sup35 Polymers to Fragmentation Defines [PSI+] Prion Variability
Different variants or “strains” were observed for both mammalian and yeast prions. The variation was observed for [PSI+],14 hybrid prion [PSI+ PS] based on Sup35 prion domain from yeast Pichia methanolica,42 [URE3]43 and [PIN+].9 The [PSI+] variants are distinguished by the strength of nonsense suppressor phenotype and mitotic stability. Usually, stronger suppression correlates with higher stability. It was proposed14,44,45 and recently confirmed46,47 that the variation in [PSI+] properties reflects difference in the structure of prion particles. This may result in variation of prion polymerization speed and the frequency of fragmentation of prion polymers, and, therefore, in their different size. A comparison of the size of Sup35 polymers in different [PSI+] isolates indeed revealed a significant variation, with the size generally being inversely related to the strength of [PSI+]. The cells harboring strong [PSI+] have smaller polymers, which means that their number should be higher.28 This explains both the higher mitotic stability of such [PSI+] and their stronger suppressor phenotype, which results from more efficient polymerization and lower levels of soluble functional Sup35.18,19 However, this logic disregards possible variation of polymerization speed. If the speed varies significantly, the correlation of the polymer size and [PSI+] strength would be violated. Of the eight [PSI+] and [PSI+PS] variants tested, one variant violated the correlation: strong [PSI+PS-1] possessed large polymers.28 This shows that the polymerization speed can vary significantly, but in most cases this variation may be neglected compared to variation in fragmentation frequency, which plays a dominant role in defining the variability of [PSI+] properties.
A putative uncertainty in correlation of the polymer number and stability of their inheritance should be noted. Prion stability depends on the number of prion seeds, or propagons, which probably correspond to prion aggregates, rather than to polymers. These considerations are supported by the effects of the SSA1-21 mutation.
This mutation, which alters the Ssa1 chaperone, increases the size of Sup35 aggregates, and, therefore, decreases their number, while not affecting the Sup35 polymers.48 It also greatly reduces [PSI+] stability, which shows that aggregates, rather than polymers, define prion inheritance. However, normally the numbers of polymers and aggregates correlate. Larger polymers seem to have higher propensity to aggregate, so an increase in the polymer size should cause an even greater increase in the size of prion aggregates.28
Nonprion Amyloids of Sup35
It may be proposed that some Sup35 amyloid structures possible in vitro would not propagate in vivo, being harmful to cells or nonheritable. Variants of [PSI+] that are too strong would interfere with the cell viability due to the lack of soluble Sup35. At the other extreme, polymers may be fragmented too rarely, which would interfere with their heritability. Such unconventional Sup35 polymer variants may be uncovered in cells with altered Sup35 levels and/or structure.
Overproduction of Sup35 in cells with conventional [PSI+] causes severe growth inhibition.49 This may be related to the impairment of translation termination due to both the reduced levels of soluble Sup35 and titration of the Sup45 (eRF1) partner termination factor by functionally inactive Sup35 polymers. Any [PSI+] compatible with Sup35 overproduction would represent a new prion variant not existing under standard conditions.
Two such novel types of [PSI+] were obtained in a [PIN+] strain overproducing Sup35: (1) [PSI+] compatible with Sup35 overproduction, but stable at standard Sup35 levels; (2) [PSI+] existing only at increased Sup35 levels. In addition, a category of Sup35 amyloid-like polymers was discovered, which does not manifest itself as [PSI+].50 Surprisingly, these polymers were present in cells before the selection for suppressor phenotype, as revealed by SDD-AGE. Cells containing these polymers did not show suppressor effect due to significant levels of soluble Sup35. About 15% of Sup35 was soluble, which, accounting for 20-fold overproduction, exceeded three-fold the Sup35 levels in [psi-] cells. Increased levels of soluble Sup35 indicate its inefficient polymerization, which may be related to the size of Sup35 polymers increased several-fold compared to conventional [PSI+]. In cells with any given Sup35 levels an increase in polymer size would mean a proportional decrease in the number of Sup35 polymers and polymerization speed.
Another property of these polymers was poor heritability. Their appearance required the presence of [PIN+] determinant. Furthermore, [PIN+] elimination via disruption of RNQ1 eliminated the amyloid-like Sup35 polymers in the cells which already have possessed them. Thus, in contrast to [PSI+] polymers, which propagate very stably in the absence of [PIN+], these polymers are not heritable. This may be related to small number of these polymers due to their inefficient fragmentation, which is evident from their large size. Then, the only reason for existence of these polymers is their efficient appearance de novo. This is likely to occur via seeding by Rnq1 polymers, since about 1/5 of total Rnq1 was bound to Sup35 polymers. This bond was resistant to cold SDS, and thus these Rnq1 and Sup35 belonged to the same polymers. It appears unlikely that Rnq1 was dispersed along the Sup35NM polymers, since homotypic polymerization should be much more efficient than heterotypic. More probably, this Rnq1 represented Rnq1 prion seeds attached to the Sup35NM polymers, which they initiated.
Finally, the nonheritable Sup35 amyloid-like polymers appear with very high frequency compared to its prion variants. A clear inverse correlation between the frequency of appearance and the “strength” was observed: stronger variants appeared less frequently.50 The reason for this correlation is not fully clear. It may be presumed that this is due to the preferential survival of prion seeds corresponding to weaker [PSI+], because they are less recognizable by the cellular chaperones. In this connection, it is noteworthy that among the Sup35 fibers spontaneously formed in vitro, a significant proportion apparently belongs to the prion type, since these fibers could transform yeast cells to the [PSI+] phenotype.26
Thus, Sup35 forms amyloid variants covering the full spectrum of related phenotypes, from highly stable strong suppressor [PSI+] to nonheritable phenotypically undetectable polymers. The key parameter, distinguishing these variants is the frequency of polymer fragmentation, which is highest in strong [PSI+] polymers and lowest in nonheritable amyloid-like polymers. The ability of Sup35 to form both prions and nonheritable amyloids convincingly supports the idea that prion and amyloid phenomena are related. Notably, another amyloid-forming protein does not make full spectrum of variants: the Sup35 fusion protein with its prion-forming domain replaced with a stretch of 66 glutamine residues can form SDS-insoluble amyloid-like polymers, but is unable to form prions.50
Amyloid Cross-Seeding and the “Species Barrier”
Observations that the prion state of Sup35 may be seeded by polymers of Rnq1 and some other proteins lead to the suggestion that cross-seeding may play a role in the appearance of amyloids in mammals.12 Recent work50 showed that the Sup35 prion represents a very small fraction of all seeded Sup35 amyloid-like polymers, and thus the efficiency of cross-seeding is much higher than it was considered previously. This suggests that amyloid cross-seeding in mammals is not just possible, but plays a significant role in amyloid appearance. In agreement with this assumption, injection of synthetic amyloid fibers made of transthyretin or islet amyloid polypeptide caused deposition of amyloid A fibers.51
It is known that the transfer of PrP prion between different species occurs with difficulty or does not occur at all even in the cases when the inter-species difference in the sequence of prion proteins constitutes only few amino acids.2 This effect, known as the “species barrier,” is considered to result from inefficient copolymerization of differing prion proteins. The Rnq1-Sup35 pair provides a good model for this effect and uncovers an additional reason for it. In vitro, Rnq1 fibers seeded Sup35 polymerization, though about 100-fold less efficiently than did Sup35 fibers.24 In vivo, such efficiency of seeding would be sufficient to cross the “species barrier,” because, once appeared, the Sup35 prion will replicate independently. Yeast cell contains more than 1,000 of Rnq1 molecules,52 most of which polymerize per generation. Then, about ten events of Sup35 seeding by Rnq1 may be expected per generation. However, as we mentioned, the majority of these events results in nonprion Sup35 amyloids.50 Thus, the additional barrier for prion transmission is the loss of a specific prion fold, required for efficient fiber fragmentation. It should be noted that the sequence similarity between heterologous PrP proteins is much higher than the similarity between the prion domains of Sup35 and Rnq1. Therefore, in the PrP case the “polymerization” barrier may be lower, while the loss of fragmentable prion fold may become the main reason for prion “species barrier.”
Prions and Nontransmissible Amyloids: Two Modes of the Polymerization Process
Prions of higher eukaryotes are infectious, while other amyloids are not. What are the reasons for this difference? The basic prerequisite of infectivity, the ability of polymers to catalyze further polymerization, is common for all amyloids. Another significant requirement is the ability of polymers to migrate between different organs and from one organism to another. The intracellular location of some amyloids (e.g., formed by α-Synuclein, Huntingtin) should restrict their infectivity, since amyloids, unlike viruses, do not have specific mechanisms for leaving and entering cells. However, the majority of amyloids are extracellular. A reason for noninfectivity may be the inability of consumed amyloid to pass the digestive tract and reach the appropriate organ. Apparently, in the PrP case these tasks are facilitated by its very high protease resistance and association with B-cells, which carry it around an organism.53 Such specific properties are not modeled in yeast. However, usually the lack of infectivity is evident upon direct injection of amyloid material into appropriate tissue. This excludes these properties and allows considering the infectivity in terms of the factors acting in yeast.
For the yeast Sup35, we observed two modes of amyloid-like polymerization. In the “prion” mode, new amyloid particles appear from the existing ones by fragmentation. In the “nonprion amyloid” mode, new particles appear de novo. The fragmentation efficiency appears to represent the key difference between prion and nonprion amyloids in both yeast and mammals. The nonprion amyloid mode is noninfectious, because this process does not replicate old seeds, but generates new ones. The process does not depend on the introduction of external infection but depends on intrinsic propensity of an organism to generate and accumulate new amyloids. This propensity is known to increase with age, presumably due to age-dependent alterations of chaperone and/or protease systems. Consideration of physical properties of these amyloids also reveals reasons for their noninfectivity. Due to infrequent fragmentation, nonprion amyloid fibers are long and tend to precipitate. The commonly observed amyloid plaques represent an evident example of such behavior. If such amyloid polymers can not migrate to new locations, they can appear there only by formation de novo.
The mammalian PrP prion is distinguished by very low frequency of de novo appearance and should multiply via fragmentation. The animal fragmentation factor is not known, since HSP104 homologues are not present in the sequenced animal genomes. Nevertheless, PrP prion particles appear to be fairly small in size,54 which suggests the existence of efficient fragmentation factor. The lack of evident PrP deposits in many cases of Creutzfeldt-Jacob disease2 may also be related to the small size of PrP polymers.
It is not clear why mammalian amyloidogenic proteins do not produce, like Sup35, efficiently fragmented polymer variants. One possible explanation is that the proteins with such ability would form polymers too easy and fast, thus being detrimental for an organism. Such properties should be counterselected by evolution and such proteins “fine tuned” to exclude prion formation by them. An example of such amyloid-only behavior in yeast is given by polyglutamine fusions to Sup35MC, which form only nonheritable polymers.50 Another explanation could be insufficient fragmenting activity in the extracellular space. The efficient fragmentation of PrP may be related then to its specific life cycle, which includes both extracellular and intracellular phases.
The prion and amyloid polymerization modes represent two ideal extremes, while actual amyloids are likely to have intermediate properties, being fragmented, but infrequently. Intermediate properties may allow some amyloids to show infectivity under certain conditions. For example, polymerization of amyloid protein A, known as secondary systemic amyloidosis, may be induced by so-called amyloid enhancing factor, the active ingredient of which was identified as fibers of amyloid protein A.55 However, the induction required additionally an inflammatory stimulus, such as silver nitrate. The mouse senile amyloidosis, related to polymerization of apolipoprotein A-II, showed properties principally similar to prions. This disease was transmitted by oral administration of apolipoprotein A-II fibers,56 whose infectious potential was enhanced by ultrasonic fragmentation.
Acknowledgements
The work in the authors' laboratory was supported by the Wellcome Trust, Howard Hughes Medical Institute, International Science and Technology Center and Russian Foundation for Basic Research.
Note
This manuscript has been previously published: Kushnirov VV, Vishnevskaya AB, Alexandrov IM, Ter-Avanesyan MD. Prion and Nonprion Amyloids: A Comparison Inspired by the Yeast Sup35 Protein. In: Chernoff Y, editor. Protein-Based Inheritence. Austin and New York: Landes Bioscience and Kluwer Academic Press; 2007. pp. 73–79.
Footnotes
Previously published online as a Prion E-Publication: http://www.landesbioscience.com/journals/prion/article/4840
References
- 1.Horwich AL, Weissman JS. Deadly conformations—Protein misfolding in prion disease. Cell. 1997;89:499–510. doi: 10.1016/s0092-8674(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell. 1998;93:337–348. doi: 10.1016/s0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- 3.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 4.Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol. 2005;4:e6. doi: 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Si K, Lindquist S, Kandel ER. A neuronal isoform of the Aplysia CPEB has prion-like properties. Cell. 2003;115:879–891. doi: 10.1016/s0092-8674(03)01020-1. [DOI] [PubMed] [Google Scholar]
- 6.Wickner RB. [URE3] as an altered Ure2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 7.Coustou V, Deleu C, Saupe SJ, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci USA. 1997;94:9773–9778. doi: 10.1073/pnas.94.18.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics. 1997;147:507–519. doi: 10.1093/genetics/147.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. Interactions among prions and prion “strains” in yeast. Proc Natl Acad Sci USA. 2002;99(Suppl 4):16392–16399. doi: 10.1073/pnas.152330699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sondheimer N, Lindquist S. Rnq1: An epigenetic modifier of protein function in yeast. Mol Cell. 2000;5:163–172. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 11.Santoso A, Chien P, Osherovich LZ, Weissman JS. Molecular basis of a yeast prion species barrier. Cell. 2000;100:277–288. doi: 10.1016/s0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- 12.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: The story of [PIN+] Cell. 2001;106:171–182. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 13.Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN. The SUP35 omnipotent suppressor gene is involved in the maintenance of the nonmendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics. 1994;137:671–676. doi: 10.1093/genetics/137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI+] prion factors in Saccharomyces cerevisiae. Genetics. 1996;144:1375–1386. doi: 10.1093/genetics/144.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu JJ, Sondheimer N, Lindquist SL. Changes in the middle region of Sup35 profoundly alter the nature of epigenetic inheritance for the yeast prion [PSI+] Proc Natl Acad Sci USA. 2002;99:16446–16453. doi: 10.1073/pnas.252652099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradley ME, Liebman SW. The Sup35 domains required for maintenance of weak, strong or undifferentiated yeast [PSI+] prions. Mol Microbiol. 2004;51:1649–1659. doi: 10.1111/j.1365-2958.2003.03955.x. [DOI] [PubMed] [Google Scholar]
- 17.Ter-Avanesyan MD, Kushnirov VV, Dagkesamanskaya AR, Didichenko SA, Chernoff YO, Inge-Vechtomov SG, Smirnov VN. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two nonoverlapping functional regions in the encoded protein. Mol Microbiol. 1993;7:683–692. doi: 10.1111/j.1365-2958.1993.tb01159.x. [DOI] [PubMed] [Google Scholar]
- 18.Zhou P, Derkatch IL, Uptain SM, Patino MM, Lindquist S, Liebman SW. The yeast nonmendelian factor [ETA+] is a variant of [PSI+], a prion-like form of release factor eRF3. EMBO J. 1999;18:1182–1191. doi: 10.1093/emboj/18.5.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kochneva-Pervukhova NV, Chechenova MB, Valouev IA, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. [PSI+] prion generation in yeast: Characterization of the “strain” difference. Yeast. 2001;18:489–497. doi: 10.1002/yea.700. [DOI] [PubMed] [Google Scholar]
- 20.Coschigano PW, Magasanik B. The URE2 gene product of Saccharomyces cerevisiae plays an important role in the cellular response to the nitrogen source and has homology to glutathione S-transferases. Mol Cell Biol. 1991;11:822–832. doi: 10.1128/mcb.11.2.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 22.Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 23.King CY, Tittmann P, Gross H, Gebert R, Aebi M, Wüthrich K. Prion-inducing domain 2–114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci USA. 1997;94:6618–6622. doi: 10.1073/pnas.94.13.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ, and nonpolyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci USA. 2004;101:12934–12939. doi: 10.1073/pnas.0404968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319–323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 27.Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: Amyloid of Ure2p is infectious. EMBO J. 2005;24:3082–3092. doi: 10.1038/sj.emboj.7600772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49636–49643. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 29.Bagriantsev S, Liebman SW. Specificity of prion assembly in vivo: [PSI+] and [PIN+] form separate structures in yeast. J Biol Chem. 2004;279:51042–51048. doi: 10.1074/jbc.M410611200. [DOI] [PubMed] [Google Scholar]
- 30.Jones GW, Tuite MF. Chaperoning prions: The cellular machinery for propagating an infectious protein? Bioessays. 2005;27:823–832. doi: 10.1002/bies.20267. [DOI] [PubMed] [Google Scholar]
- 31.Chernoff YO, Lindquist SL, Ono B, et al. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [PSI+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 32.Moriyama H, Edskes HK, Wickner RB. [URE3] prion propagation in Saccharomyces cerevisiae: Requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol Cell Biol. 2000;20:8916–8922. doi: 10.1128/mcb.20.23.8916-8922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 34.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 35.Kushnirov VV, Ter-Avanesyan MD. Structure and replication of yeast prions. Cell. 1998;94:13–16. doi: 10.1016/s0092-8674(00)81216-7. [DOI] [PubMed] [Google Scholar]
- 36.Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 37.Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol. 2001;40:1357–1369. doi: 10.1046/j.1365-2958.2001.02478.x. [DOI] [PubMed] [Google Scholar]
- 38.Tuite MF, Mundy CR, Cox BS. Agents that cause a high frequency of genetic change from [PSI+] to [psi-] in Saccharomyces cerevisiae. Genetics. 1981;98:691–711. doi: 10.1093/genetics/98.4.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eaglestone SS, Ruddock LW, Cox BS, Tuite MF. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI+] of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2000;97:240–244. doi: 10.1073/pnas.97.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ness F, Ferreira P, Cox BS, Tuite MF. Guanidine hydrochloride inhibits the generation of prion “seeds” but not prion protein aggregation in yeast. Mol Cell Biol. 2002;22:5593–5605. doi: 10.1128/MCB.22.15.5593-5605.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jung G, Jones G, Masison DC. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc Natl Acad Sci USA. 2002;99:9936–9941. doi: 10.1073/pnas.152333299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kushnirov VV, Kochneva-Pervukhova NV, Chechenova MB, Frolova NS, Ter-Avanesyan MD. Prion properties of the Sup35 protein of yeast Pichia methanolica. EMBO J. 2000;19:324–3231. doi: 10.1093/emboj/19.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schlumpberger M, Prusiner SB, Herskowitz I. Induction of distinct [URE3] yeast prion strains. Mol Cell Biol. 2001;21:7035–7046. doi: 10.1128/MCB.21.20.7035-7046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kushnirov VV, Kryndushkin DS, Boguta M, et al. Chaperones that cure yeast artificial [PSI+] and their prion-specific effects. Curr Biol. 2000;10:1443–1446. doi: 10.1016/s0960-9822(00)00802-2. [DOI] [PubMed] [Google Scholar]
- 45.King CY. Supporting the structural basis of prion strains: Induction and identification of [PSI+] variants. J Mol Biol. 2001;307:1247–1260. doi: 10.1006/jmbi.2001.4542. [DOI] [PubMed] [Google Scholar]
- 46.Krishnan R, Lindquist SL. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature. 2005;435:765–772. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanaka M, Chien P, Yonekura K, Weissman JS. Mechanism of cross-species prion transmission: An infectious conformation compatible with two highly divergent yeast prion proteins. Cell. 2005;121:49–62. doi: 10.1016/j.cell.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 48.Song Y, Wu YX, Jung G, Tutar Y, Eisenberg E, Greene LE, Masison DC. Role for Hsp70 chaperone in Saccharomyces cerevisiae prion seed replication. Eukaryot Cell. 2005;4:289–297. doi: 10.1128/EC.4.2.289-297.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dagkesamanskaya AR, Ter-Avanesyan MD. Interaction of the yeast omnipotent suppressors SUP1(SUP45) and SUP2(SUP35) with nonMendelian factors. Genetics. 1991;128:513–520. doi: 10.1093/genetics/128.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salnikova AB, Kryndushkin DS, Smirnov VN, Kushnirov VV, Ter-Avanesyan MD. Nonsense suppression in yeast cells overproducing Sup35 (eRF3) is caused by its nonheritable amyloids. J Biol Chem. 2005;280:8808–8812. doi: 10.1074/jbc.M410150200. [DOI] [PubMed] [Google Scholar]
- 51.Johan K, Westermark G, Engström U, Gustavsson A, Hultman P, Westermark P. Acceleration of amyloid protein A amyloidosis by amyloid-like synthetic fibrils. Proc Natl Acad Sci USA. 1998;95:2558–2563. doi: 10.1073/pnas.95.5.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 53.Klein MA, Frigg R, Flechsig E, Raeber AJ, Kalinke U, Bluethmann H, Bootz F, Suter M, Zinkernagel RM, Aguzzi A. A crucial role for B cells in neuroinvasive scrapie. Nature. 1997;390:687–690. doi: 10.1038/37789. [DOI] [PubMed] [Google Scholar]
- 54.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lundmark K, Westermark GT, Nyström S, Murphy CL, Solomon A, Westermark P. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci USA. 2002;99:6979–6984. doi: 10.1073/pnas.092205999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xing Y, Nakamura A, Chiba T, Kogishi K, Matsushita T, Li F, Guo Z, Hosokawa M, Mori M, Higuchi K. Transmission of mouse senile amyloidosis. Lab Invest. 2001;81:493–499. doi: 10.1038/labinvest.3780257. [DOI] [PubMed] [Google Scholar]