

Abstract
The role of water channel aquaporin 1 (AQP-1) in uninjured or injured spinal cords is unknown. AQP-1 is weakly expressed in neurons and gray matter astrocytes, and more so in white matter astrocytes in uninjured spinal cords, a novel finding. As reported before, AQP-1 is also present in ependymal cells, but most abundantly in small diameter sensory fibers of the dorsal horn. Rat contusion spinal cord injury (SCI) induced persistent and significant four- to eightfold increases in AQP-1 levels at the site of injury (T10) persisting up to 11 months post-contusion, a novel finding. Delayed AQP-1 increases were also found in cervical and lumbar segments, suggesting the spreading of AQP-1 changes over time after SCI. Given that the antioxidant melatonin significantly decreased SCI-induced AQP-1 increases and that hypoxia inducible factor-1α was increased in acutely and chronically injured spinal cords, we propose that chronic hypoxia contributes to persistent AQP-1 increases after SCI. Interestingly; AQP-1 levels were not affected by long-lasting hypertonicity that significantly increased astrocytic AQP-4, suggesting that the primary role of AQP-1 is not regulating isotonicity in spinal cords. Based on our results we propose possible novel roles for AQP-1 in the injured spinal cords: (i) in neuronal and astrocytic swelling, as AQP-1 was increased in all surviving neurons and reactive astrocytes after SCI and (ii) in the development of the neuropathic pain after SCI. We have shown that decreased AQP-1 in melatonin-treated SCI rats correlated with decreased AQP-1 immunolabeling in the dorsal horns sensory afferents, and with significantly decreased mechanical allodynia, suggesting a possible link between AQP-1 and chronic neuropathic pain after SCI.
Keywords: aquaporin 1, growth-associated protein 43, hypertonicity, hypoxia inducible factor, melatonin, spinal cord injury
Aquaporin-1 (AQP-1) mediates water transport in several organs where it plays an important role in the formation of urine, aqueous humor in the eye (Zhang et al. 2002; Umenishi and Schrier 2003) or cerebrospinal fluid (CSF) in the brain. Brain AQP-1 is mainly expressed in the CSF-facing membranes of the ventricular choroid plexus, where it regulates the formation of CSF, partly because of its ion channel function (Speake et al. 2003; Oshio et al. 2005; Boassa et al. 2006).
In the spinal cord, AQP-1 is also expressed in the ependymal cells lining the central canal, but more robustly in the sensory fibers of the superficial laminae of the dorsal horn (Oshio et al. 2005; Shields et al. 2007). Considering the expression of AQP-1 in the dorsal horn, it has been suggested that AQP-1 has a role in normal pain processing. Although, Oshio et al. (2003) show that AQP-1-null mice demonstrate a higher pain thresholds for capsaicin and thermal stimuli, Shields et al. (2007) report that AQP-1-null mice do not have altered sensory processing. Therefore, AQP-1 may not have a role in normal pain processing, and its physiological role in the spinal cord remains unknown.
Although it has been shown that various neuropathological conditions stimulate AQP-1 expression, AQP-1 in spinal cord injury (SCI) has not been studied. AQP-1 increases are found in the brains of Alzheimer patients (Pérez et al. 2007); in Creutzfeldt–Jakob disease (Rodríguez et al. 2006); in traumatic brain injury patients (Suzuki et al. 2006), and in human hemangioblastomas (Chen et al. 2006). Interestingly, in all cases the neuropathological conditions stimulated AQP-1 expression in astrocytes residing in diseased brain tissue, even though astrocytes and other glial cells were not known to express AQP-1. In contrast, GFAP-positive Schwann cells in the PNS are known to express AQP-1 (Gao et al. 2006), but not AQP-4, a protein abundant in spinal cord astrocytes (Nesic et al. 2006). The possible roles of increased expression of AQP-1 in the etiology of different neuropathological conditions are still unknown, although it is likely that AQP-1 increases may contribute to edema and cyst formation, typical outcomes in most of these pathological conditions.
In spite of obvious clinical significance, the mechanisms that control AQP-1 gene expression in the CNS, under normal or pathological conditions are still poorly understood. Recently Kim et al. (2007) showed that the thyroid transcription factor-1 up-regulates AQP-1 synthesis and thus facilitates CSF formation in the brain. In tissues other than CNS, the effect of hypertonicity on AQP-1 synthesis via extracellular signal-regulated kinases, p38 kinase, and c-Jun-terminal kinase is better characterized (Umenishi and Schrier 2002, 2003).
Therefore, the aim of this study was to: (i) characterize SCI-induced temporal and spatial changes in protein levels of AQP-1; (ii) identify cellular localization of AQP-1 before and after SCI; (iii) determine signals that contribute to AQP-1 changes after SCI; and (iv) shed light on the roles of AQP-1 in uninjured and injured spinal cords.
Materials and methods
Rat model of spinal cord injury
Male Sprague–Dawley rats weighing 225–250 g were anesthetized by intraperitoneal (i.p.) injection of pentobarbital (50 mg/kg). Anesthesia was considered complete when animals did not display hind limb withdrawal in response to a noxious foot pinch. A laminectomy of the 10th thoracic vertebra was performed before the animals were contused with the Infinite Horizons Impactor (LLC, Lexington, KY, USA) using a force of 150 kdyn with 1 s dwell time. The surgical site was closed by suturing the muscle and fascia and stapling the skin; which was followed by the superficial application of 0.3 mL of 1% lidocaine. Animals were then injected subcutaneously with 2 mL of 0.9% sterile saline and placed on a heating pad to maintain body temperature until they revived. The analgesic buprenorphin (0.1 mg/kg) was administered subcutaneously (s.c.) twice a day for 3 days. Sham and SCI rats also received baytril (2.7 mg/kg) s.c twice a day until bladder function returned, which typically occurred 2 weeks after SCI. Bladders of injured animals were manually emptied twice daily until normal function returned. All procedures complied with the recommendations in the NIH Guide for the Care and Use of Laboratory Animals and were approved by the UTMB Animal Care and Use Committee. We used the minimal number of animals necessary for each experiment and the suffering of animals was minimized.
Intrathecal catheters
Rats were implanted with intrathecal (i.th.) catheters at the time of sham treatment. Catheters were made by attaching 30 mm of PE10 tubing to 40 mm of PE60 tubing with Superglue on the inner surface of the tubes' intersection and epoxy resin on the outer surface of the tubes' intersection. A partial laminectomy of the thirteenth thoracic vertebra was conducted and a small incision was carefully made in the dura surrounding the spinal cord. The catheter was then inserted into the subdural space and slid along the surface of the cord until the tip of the catheter was directly over T11, so that the fluid expelled from the catheter was released over spinal cord segment T10. The catheter was secured by suture and Superglue to both the L1–L2 vertebral junction and a nearby muscle tissue site. The PE60 end of the catheter was connected to an osmotic minipump (2.5 μL/h; Alzet 2ML4, Cupertino, CA, USA). All solutions used for minipumps (isotonic and hypertonic) were sterile and filtered before use.
Protein extraction/subcellular fractionation/western blotting
Animals were perfused with 120–150 mL of 0.9% NaCl containing heparin (1000 U/L), to eliminate blood from the spinal cord tissue. Spinal cord segments were removed and immediately placed on dry ice to freeze fully.
For protein extraction, 200 μL of homogenization buffer (10 mmol/L Tris Base, 300 mmol/L sucrose, 1 mmol/L EDTA, 1 mmol/L dithiothreitol (DTT), 0.5 mmol/L phenylmethylsulfonyl fluoride, 2 μg/mL antipain, 2 μg/mL chymostatin, 2 μg/mL pepstatin, and 2 μg/mL leupeptin, pH 7.5) was added to each individual thoracic spinal cord segment for homogenization on ice with a clear grinder (05-559-27; Fisher, Pittsburgh, PA, USA) that tightly fit inside the 1.5 mL tubes. For extraction of cervical or lumbar segments, 500 μL of homogenization buffer was used. After homogenization samples were vortexed for 5 s, and then centrifuged at 5100 g for 5 min at 4°C. The supernatant was separated from the nuclear pellet and centrifuged at 15 700 g for 1 h at 4°C to pellet the crude plasma membrane. The resulting supernatant contained the cytosolic extract, and the crude plasma membrane was resuspended in 50 μL of homogenization buffer. Nuclear protein was extracted from the nuclear pellet by resuspension in 70 μL of extraction buffer (20 mmol/L HEPES, 400 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L DTT, 1 mmol/L phenylmethylsulfonyl fluoride, 2 μg/mL antipain, 2 μg/mL chymostatin, 2 μg/mL pepstatin, and 2 μg/mL leupeptin) followed by 20 min of vortexing at high speed at 4°C, and subsequent centrifugation at 16 000 g for 10 min at 4°C. The supernatant contained the nuclear protein. Protein concentrations were determined using the BioRad Protein Assay (500–0006; BioRad, Hercules, CA, USA).
Samples containing 40 μg of protein were boiled for 10 min at 100°C with an appropriate volume of 6x sample buffer (350 mmol/L Tris–HCl, pH 6.8, 1 mol/L urea, 1% 2-mercaptoethanol, 9.3% DTT, 13% sodium dodecyl sulfate, 0.06% bromophenol blue, and 30% glycerol). Samples were then placed on ice to cool before being loaded onto a 12% sodium dodecyl sulfate–polyacrylamide gel and separated at 150 V for 4 h. Gels were then transferred overnight onto a polyvinylidene difluoride membrane at 4°C and 25 V. Membranes were reversibly stained with Ponceau S to confirm the transfer of proteins, and destained in water. Membranes were then incubated for 1 h at room temperature (23°C) in blocking solution (5.0% non-fat dry milk in Tris-buffered saline with 0.2% Tween 20). Primary and secondary antibodies were also diluted in blocking solution, and washes were performed with Tris-buffered saline containing 0.2% Tween 20. Peroxidase activity was detected using the Amersham enhanced chemiluminescence lighting system (ECL) (RPN2209; Amersham Biosciences, Piscataway, NJ, USA).
We characterized the purity of our membrane-enriched and cytosolic fractions, using subcellular compartment-specific marker antibodies in western blot assays to evaluate the components present in these fractions. To assess the presence of mitochondria in our fractions, we used an antibody that recognizes protein specifically expressed in the mitochondria, cytochrome oxidase IV (1 : 4000; Abcam, Cambridge, MA, USA) (Liang et al.2003). Membrane-enriched fraction showed the presence of cytochrome oxidase IV, while cytosolic and nuclear fractions were completely devoid of this marker (not shown). Although we believe it unlikely, the possibility cannot be ruled out that some of the AQP-1 that we detect in the membrane fraction comes from mitochondria. AQP-1 has not been reported in mitochondria, but other AQPs have, e.g. AQP-8 and -9 (Amiry-Moghaddam et al. 2005; Calamita et al. 2005). Cytosolic fraction was assessed by using inhibitor kappaβ, a primarily cytoplasmic protein involved in nuclear factor kappaβ signaling (1 : 2000, not shown; Santa Cruz, CA, USA), while nuclear fractions were confirmed using β-lamin (1 : 2000, not shown; Zymed Laboratories, Carlsbad, CA, USA).
Antibodies
AQP-1 (Rabbit anti-AQP-1, 0.5 μg/mL; Chemicon International, Temecula, CA, USA); AQP-4 (Rabbit anti-AQP-4, 1 : 2000; Chemicon International); nitrotyrosine (Mouse anti-nitrotyrosine, 1 : 100; Upstate Biotechnology Inc., Lake Placid, NY, USA); hypoxia inducible factor-1α (Hif-1α) (Mouse anti-Hif-1α, 1 : 1000; Chemicon International); growth-associated protein 43(GAP-43) (Mouse anti-GAP-43, 0.1 μg/mL; Chemicon International). β-Actin is used as a control for loading (anti-β-actin, 1 : 10 000; Sigma-Aldrich Co., St Louis, MO, USA) in all western blots, in addition to checking Ponceau S staining.
Double immunofluorescence staining using confocal laser scanning microscopy
Stained sections were studied with a confocal laser scanning system (Bio-Rad Radiance 2100, K-2 system, Bio-Rad Laboratories, Tokyo, Japan). For double immunofluorescent staining, to avoid ‘bleed through’ of fluorescence signals between adjacent color channels, the data were collected one channel at a time by sequential scanning. Images were collected using the 488 and 568 nm excitation lines of an Argon–Krypton laser. The co-localization of the two antigens was shown by yellow color in the images obtained after overlaying the green and red channels. All presented figures in the manuscript are maximum intensity projections of z-stack series. However, when we tested co-localization of AQP-1 and cell markers, we did not use these maximum intensity projections for the analysis, instead we either analyzed co-localization in single individual sections or we used the Software Imaris, Bitplane Inc., (Saint Paul, MN, USA) to analyze the entire z-stack for co-localization in 3D space.
Digital images were saved and processed with Adobe Photoshop (Adobe Systems Inc., San Jose, CA, USA) for final editing. For quantitative analysis of intensity of immunolabeling, sections were imaged using the same exposure times, and the average density was measured with MetaMorph (Molecular Devices, Downingtown, PA, USA) software. Five randomly selected sections from the spinal cord segment in each animal were analyzed and the relative optical densities were averaged to provide a mean number for each rat.
Antibodies
Class III β-tubulin (TUJ1, mouse monoclonal, 1 : 3000; Covance, Richmond, CA, USA), NeuN (mouse monoclonal, 1 : 5000; Chemicon International), and rat endothelial antigen [rat endothelial cell antigen (RECA)-1; mouse monoclonal, 1 : 200; Serotec Oxford, UK], GFAP (mouse monoclonal, 1 : 800; Chemicon International), oligodendrocytes (CC1, mouse monoclonal, 1 : 200; Oncogene Research Products, Boston, MA, USA), microglia (OX-42, mouse monoclonal, 1 : 200; Serotec), and GAP-43 (mouse monoclonal, 1 : 500; Chemicon International, Raleigh, North Carolina, USA).
Locomotor assessments
Locomotor assessments were performed using open field tests with the Basso, Beattie, and Bresnahan Scale (BBB rank scores; Basso et al. 1995) assigned as measures of hind limb activity to document plantar placement with weight bearing stance.
Mechanical allodynia
Pain-like behavior (allodynia) in contused rats was reflected as a decrease in tactile thresholds, compared with pre-surgery mechanical threshold values measured over 3 days before SCI. We measured the responses (vocalization and body movement) to gradually increasing mechanical stimuli delivered using Von Frey filaments to the back of SCI rats, around the site of injury (T10; Nesic et al. 2005; Crown et al. 2005, 2006). All measurements were performed in the blinded fashion.
Statistical analysis
All statistical tests were evaluated at the alpha level of significance of 0.05, two-tailed. All of the experiments have similar structures in that the effects of a manipulation on the level of the factors were measured. We used parametric methods (t-test) for our analyses. However, if the assumptions for these tests were not met, we proceeded with non-parametric analyses (Mann–Whitney). Likewise, we used non-parametric methods to check all parametric results as a safeguard of assumptions. If the results were not consistent, we reported the results from non-parametric tests. For multiple-group comparisons, data were analyzed using one-way anova. The least significant difference multiple comparisons post hoc test was used to determine p-values. Values of p < 0.05 were considered significant.
Results
Cellular localization of AQP-1 in uninjured and injured spinal cords
In uninjured spinal cords, AQP-1 was expressed in neuronal fibers/cell bodies, ependymal cells, and astrocytic cell bodies, 5 months after sham-treatment (Fig. 1; n = 3 rats per cell marker). The highest levels of AQP-1 in uninjured spinal cords were found in the dorsal horn small diameter fibers in Laminae I and II (Fig. 1a, marked with arrow; n = 21). Ependymal cells lining the central canal also expressed AQP-1 (marked with star in Fig. 1a; n = 21). In Fig 1b, we showed double-labeled AQP-1 and GFAP in the ventral horn where the large motoneurons allowed easier identification of neurons, surrounded by GFAP-labeled astrocytes; the small arrow indicates astrocytic cell bodies in the gray matter weakly expressing AQP-1. Astrocytic cell bodies in uninjured white matter had higher levels of AQP-1 and were easier to identify, as shown in Fig. 1(b1). Similar results were obtained for uninjured spinal cords, analyzed 7 days or 3 months after sham-treatment (n = 3; data not shown). Expression of AQP-1 was not previously reported in neurons and astrocytes likely because the AQP-1 abundance is markedly lower in those cells than in sensory fibers of the uninjured spinal cords.
Fig. 1.
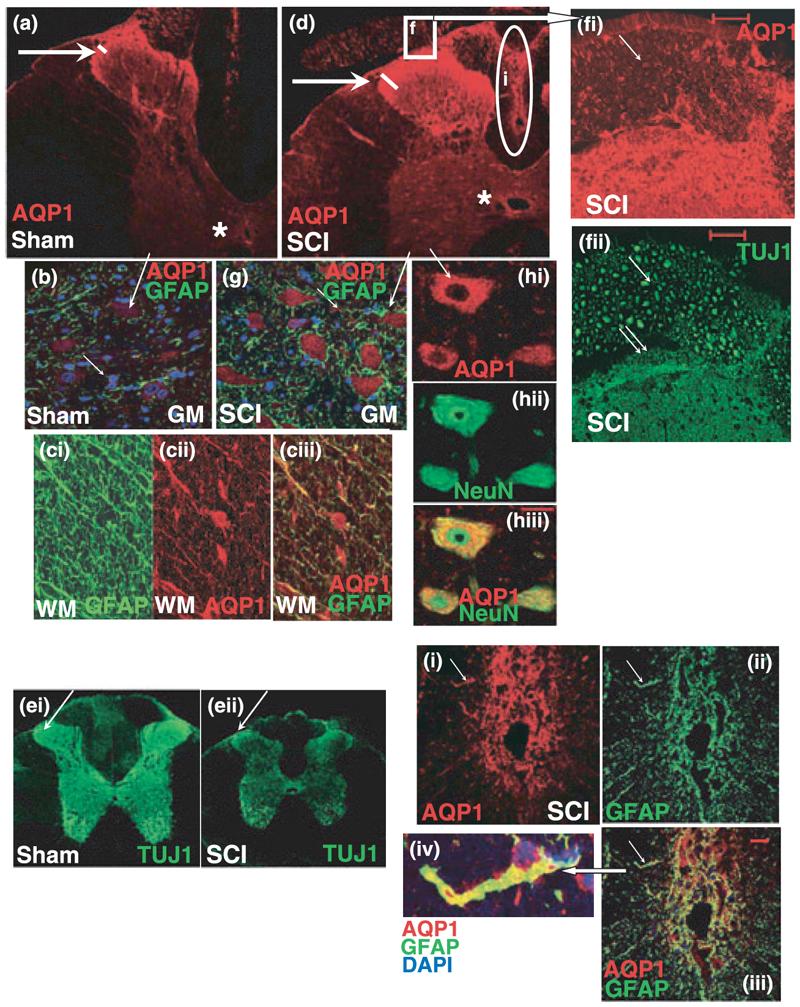
Cellular localization of AQP-1 in uninjured and injured spinal cords. (a) AQP-1 (red) expression in sham spinal cords in T9; 5 months after sham treatment. AQP-1 was primarily expressed in Laminae I and II of uninjured dorsal horns (arrow), but also in ependymal cells lining the central canal (star). (b) AQP-1 (red) was weakly expressed in neuronal cell bodies in uninjured spinal cords, here depicted in motoneuronal somata in the ventral horn of the sham-treated rats presented in (a) (long arrow). Neuronal cell bodies surrounded with GFAP-positive astrocytes (green) were clearly visible. GFAP-labeled astrocytes in the gray mater (GM) also expressed low levels of AQP-1 (marked with the small arrow). (c) GFAP-positive white matter (WM) astrocytes (ci) expressed AQP-1(cii) in cell bodies and processes. Those AQP-1-expressing cell bodies were not identified with CC-1 marker that labels oligodendrocytic somata, so it is likely that AQP-1 is expressed in astrocytic cell bodies in the white matter. Somatic expression of AQP-1 was not observed in gray matter astrocytes. Co-labeing of AQP1 and GFAP was visible in astrocytic processes (ciii). (d) SCI induced increased labeling of AQP-1 in sensory fibers (arrow) and ependymal cells (star; T9 segment 5 months after SCI). AQP-1 positive sensory fibers (arrow) enter dorsal horns via dorsal roots (marked with the square f). Calibration bar: 100 μm. (e) TUJ1 positive axons (green) in uninjured (sham; ei) and injured spinal cords (T9; 5 months after SCI; eii). TUJ-1 labeling markedly decreased in chronically injured spinal cords (SCI), because of the loss of axons throughout spinal cords, including dorsal horn sensory fibers (arrow). Note the shrinkage of chronically injured spinal cords and significant decrease in the overall size of the T9 segment, partly because of the reduced number of axons. (f) AQP-1 expression (red; fi) in TUJ-positive fibers (green; fii) in dorsal root and dorsal horn (spinal cord segment labeled with the square in injured spinal cord, Fig. 1d). Large-diameter fibers expressed low levels of AQP-1 (arrow), in contrast to the small-diameter fibers (<5 μm; double arrow), that expressed significantly higher levels of AQP-1 (calibration bar: 50 μm). (g) Motoneuronal cell bodies expressed visibly more AQP-1 (red) in chronically injured spinal cords (5 months after SCI in T9) than in corresponding sham samples (Fig. 1b). Calibration bar: 50 μm. (h) Neuronal expression of AQP-1 (red) was confirmed using the neuronal-specific marker NueN that selectively stains neuronal nuclei and cell bodies (green). Calibration bar: 25 μm. (i) Hypertrophic, reactive GFAP-positive astrocytes (green, ii) that formed SCI-induced scar in the dorsal column (marked with oval in Fig. 1d) expressed the highest AQP-1 levels (red; i) versus all other astrocytes. Calibration bar: 25 μm. A magnified astrocyte (iv; marked with arrows) was co-labeled with GFAP (green), AQP-1(red), and DAPI (blue), and showed high expression levels of AQP-1 in both the cell body and processes.
Spinal cord injury induced persistent up-regulation of AQP-1 throughout the spinal cords, in ependymal cells (Fig. 1c; marked with star; n = 21), dorsal horn fibers (Fig. 1c and e, marked with arrow), neuronal cell bodies (Fig. 1c, f, and g), and reactive scar-forming astrocytes (Fig. 1h) 5 months after SCI at T9. AQP-1 cellular localization was the same in injured spinal cords analyzed 7 days or 3 months after SCI (n = 3 per group; data not shown). In this, and all other experiments we focused on chronic changes in AQP-1 because: (i) histopathological and molecular changes in chronically injured spinal cords have not been extensively studied, compared with acutely injured spinal cords and (ii) AQP-1 may have a role in pathological outcomes that occur in the chronic post-injury phase (such as development of neuropathic pain or syringomyelia).
Aquaporin-1 was not found in OX-42-positive resting or reactive microglia; or in CC1-positive oligodendrocytes, in either uninjured or injured spinal cords (n = 3 per cell marker/per group; not shown). AQP-1 was also not expressed in the spinal cord blood vessels labeled with rat endothelial cell antigen (RECA)-1, although epidural blood vessels were positive for AQP-1 (not shown), in agreement with the finding that AQP-1 is expressed in endothelial cells outside CNS (Verkman 2006).
Although the overall number of TUJ1-labeled axons decreased in chronically injured spinal cords (T9; 5 months after SCI), including dorsal horn TUJ1-positive fibers (Fig. 1d; n = 3), AQP-1 expression was increased, suggesting that elevated AQP-1 immunolabeling resulted from the up-regulation of AQP-1 in surviving dorsal horn fibers, but more so from the increased area occupied by AQP-1 expressing fibers below laminae I and II, that is normally not occupied by sensory fibers that robustly express AQP-1. Note the length of the white lines in Fig. 1a and c depicting twofold increases in the depth of AQP-1 immunolabeling in the dorsal horns after SCI. This result suggests that elevated AQP-1 results from excessive axonal sprouting occurring among sensory fibers in the dorsal horns after SCI. AQP-1 immunoreactivity increased not only in the dorsal horn fibers, but also in other axons (see white matter in Fig. 1c).
Figure 1e depicted co-localization of TUJ1 and AQP-1 in the dorsal root and dorsal root entry zone marked with the square in Fig. 1c. This image showed AQP-1 expression in sensory fibers of all diameters, including large ones (∼20 μm; marked with the arrow), but more so in the small diameter fibers (< 5 μm) occupying the superficial laminae of the dorsal horn (marked with double arrow). Some of those AQP-1-positive small diameter fibers were nociceptive fibers expressing Substance P (not shown), as previously reported by Oshio et al. 2005 and thoroughly studied by Shields et al. (2007). In contrast, AQP-1 expression was weak, almost undetectable in the large-diameter fibers, in agreement with Shields et al. (2007).
Aquaporin-1 levels were also increased in surviving spinal neurons (Fig. 1f; n = 3); motoneuronal cell bodies contained considerably more AQP-1 than uninjured spinal motoneurons (Fig. 1b). Neuronal expression of AQP-1 was confirmed with the neuronal marker NeuN (Fig. 1g; n = 3).
Spinal cord injury also increased AQP-1 levels in reactive, hypertophic scar-forming GFAP-positive astrocytes within the dorsal column surrounding the lesion site (Fig. 1h; marked with oval in Fig. 1c; n = 3). Contrary to the uninjured spinal cords, AQP-1 was expressed not only in the astrocytic cell body, but also in GFAP-positive processes [Fig. 1(h1)].
These observations are consistent with white matter astrocytes expressing more AQP-1 than astrocytes in the gray matter, both before and after SCI. However, the markedly higher AQP-1 levels were present in the astroglial cells surrounding the lesion site.
SCI persistently increases AQP-1 protein levels
We used western blot analysis to quantitatively measure AQP-1 levels over time after SCI. As shown in Fig. 2a, the AQP-1 antibody recognized only one band of ∼24 kDa, which appeared only in the membrane-enriched fractions, not in the cytosolic or nuclear fractions, suggesting that all AQP-1 proteins reside in membranes, where they are functional both in sham-treated (S; Fig. 2a) and SCI samples (I; Fig. 2a). The SCI-induced increases in AQP-1 began soon after SCI (approximately fivefold increase at 3 days; Fig. 2b), and persisted for at least 11 months (approximately sixfold increase compared with AQP-1 sham levels at 11 months after sham treatment Fig. 2b). In summary, SCI-induced increases in AQP-1 levels compared with sham levels at each time point were robust and significant (p < 0.01), and resulted from increased AQP-1 levels in spinal neurons/fibers and reactive astrocytes (Fig. 1).
Fig. 2.
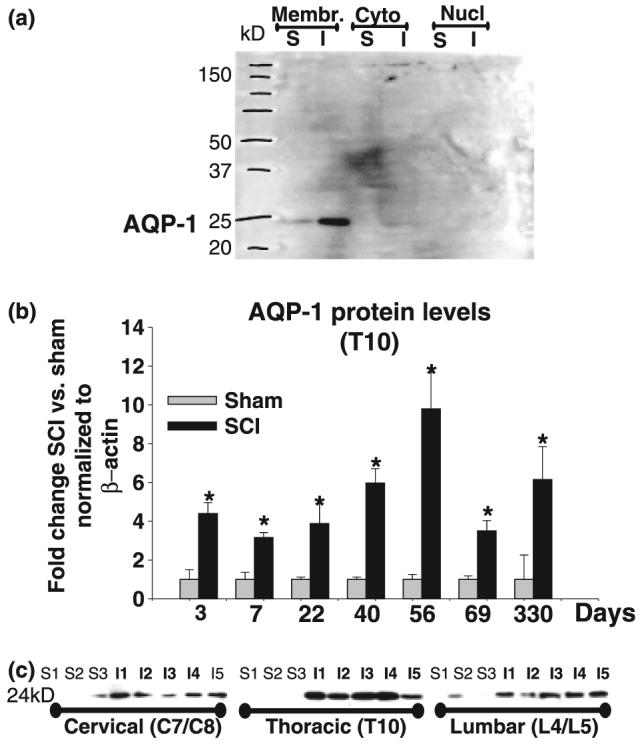
AQP-1 western blots. (a) AQP-1 western blot analysis of different subcellular compartments. AQP-1 western blot obtained using polyclonal anti-AQP-1 Ab showed only one band of ∼24 kDa. Here, we show representative AQP-1 western blot analysis in different subcellular compartments: membrane-enriched fraction (Membr); cytoplasmic (Cyto), and nuclear (Nucl). We analyzed one sham (S) and one SCI sample (I), obtained by pooling three segments: T9, T10 (site of injury) and T11, 35 days after sham treatment or SCI. The AQP-1 band appeared only in the membrane-enriched fraction, and not in the cytoplasmic or nuclear fractions. The intensity of AQP-1 band was markedly increased in injured spinal cord. (b) Time course of AQP-1expression changes after SCI. Quantitative analyses of immunoblotted AQP-1 expression (western blots) in uninjured and injured spinal cords (at the site of injury, T10) at different time points after SCI: 3 days (n = 3 for sham; n = 4 for SCI and n = 2 for naïve; not shown), 7 days (n = 4 for sham, n = 5 for SCI and n = 4 for naïve, not shown); 22 days (n = 4 for both sham and SCI); 40 days (n = 3 for sham and n = 7 for SCI); 56 days (n = 6 for sham and n = 4 for SCI); and 69 days (n = 5 for sham, n = 5 for SCI and n = 2 naïve; not shown); presented as equidistant time points on the X-axis. The Y-axis represents the relative intensity of the 24 kDa AQP-4 band normalized to β-actin, and then to sham values (set to 1). AQP-1 protein levels showed significant and chronic up-regulation in months after SCI (Bonferroni multiple comparisons tests; *p < 0.05). (c) SCI-induced AQP-1 changes in cervical and lumbar segments. Representative AQP-1 western blots showing the AQP-1 band (∼24 kDa) in three sham samples (S1–S3) and five SCI samples (I1–I5) 69 days after SCI in three spinal regions: cervical (pooled C7 and C8); site of injury (T10) and lumbar (pooled L4 and L5).
There was also a significant increase in AQP-1 not only at the site of injury (T10), but also at cervical (C7/C8; Fig. 2c) and lumbar regions (L4/L5; Fig. 2c), suggesting widespread changes in AQP-1 throughout injured spinal cords. Although the changes were most robust at the site of injury (T10; approximately four- to eightfold increase over time), the AQP-1 protein levels changes detected in cervical and lumbar segments were approximately threefold higher than in sham-treated rats, 69 days after SCI. We performed the same analysis 56 days after SCI, with similar changes in both lumbar and cervical segments (n = 6 per group, not shown). However, those changes did not occur concomitantly with AQP-1 increases at the site of injury. We did not find AQP-1 changes in lumbar or cervical segments 3 days after SCI (not shown), although, SCI induced fourfold increases at the site of injury (Fig. 2a), which suggests a temporal spread of AQP-1 changes, from the time of injury.
Aquaporin-1 changes in C7/C8 and L4/L5, suggest that those increases may contribute to the pathological processes underlying the development of hypersensitivity in forelimbs (corresponding to C7/C8) or impairment of the locomotor recovery (L4/L5). Sham-treatment did not significantly affect AQP-1-protein levels compared with naïve rats at different time points, in thoracic, lumbar, or cervical segments (not shown).
Melatonin, but not hypertonicity affects AQP-1 expression
Although the mechanisms that control synthesis of AQP-1 in uninjured or injured spinal cord cells are not known, they have been characterized in other non-CNS AQP-1-expressing cells. For example, it has been shown that anisotonicity and oxidative stress can up-regulate AQP-1 in kidney or blood vessels, respectively (Umenishi et al. 2004, 2005; Echevarria et al. 2007).
Hypertonicity stimulates AQP-1 expression in renal cells (Umenishi et al. 2004, 2005) via a hypertonicity-responsive element in the AQP-1 gene (Umenishi and Schrier 2003). Therefore, we hypothesized that anisotonicity affects AQP-1 expression in spinal cords and that disturbed osmoregulation in injured spinal cords (Frisbie 2007) may be the upstream signal that maintains the persistent elevation of AQP-1. To test this hypothesis we administered a hypertonic solution (6% NaCl) into the i.th. space of uninjured spinal cords over 3 weeks using osmotic minipumps. We chose to administer the hypertonic solution for a prolonged time (3 weeks) to: (i) assure extracellular hypertonicity in all compartments of the spinal cords; (ii) avoid detecting compensatory changes that occur during transient anisotonicity in the brain; and (iii) simulate possible permanent anisotonicity in injured spinal cords, and test the hypothesis that this putative anisotonicity may be the signal that maintains persistent elevation of AQP-1, which may affect behavioral outcomes that occur only in chronically injured spinal cords, such as neuropathic pain (Nesic et al. 2005).
We chose 6% NaCl as a hypertonic solution based on an earlier study that showed that acutely (i.v.) administered 7.5% of NaCl was safe and beneficial in SCI (Legos et al. 2001; and Sumas et al. 2001). As we administered hypertonic solution intrathecally and for prolonged time, we decided to decrease hypertonicity to 6% NaCl. To the best of our knowledge, prolonged extracellular hypertonicity in the spinal cords has not been previously tested. There were no adverse behavioral effects associated with the 6% NaCl treatment of uninjured rats.
Spinal cords were extracted after 3 weeks, proteins from T10 segments isolated and western blots for AQP-1 and -4 performed. The AQP-1 levels were only slightly and insignificantly affected (Fig. 3a). However, 6% NaCl solution treatment did affect expression of another spinal cord aquaporin-AQP-4, which increased significantly (Fig. 3b). AQP-4, present in spinal cord astrocytes, markedly and persistently increased after SCI (Nesic et al. 2006), much like AQP-1 (Fig. 2). This result confirmed that 6% NaCl was sufficiently hypertonic to induce changes in AQP expression and that AQP-1 may not be primarily involved in regulating isotonicity in uninjured spinal cords, as in other tissues, but likely has other functions, as we proposed in the discussion.
Fig. 3.

Melatonin, but not hypertonicity affects AQP-1 expression. (a) Effect of hypertonicity on AQP-1 in uninjured rats. Two groups of sham-treated rats (n = 5 per group) were implanted with intrathecal catheters connected to osmotic minipumps. One group received isotonic solution (0.9% NaCl) continuously (2.5 μL/h) over 3 weeks; the other received hypertonic solution (6% NaCl) continuously (2.5 μL/h) over 3 weeks. Spinal cords were extracted after 3 weeks; proteins extracted from T10, and AQP-1 western blots performed. Western blot data are presented below the bar graph. S1–S5: sham rats receiving isotonic solution; H1–H5: sham rats receiving hypertonic solution. Quantitative analysis of the western blot data (bar graphs) showed that hypertonic solution did not significantly increase AQP-1 levels. Only one sample (H3) had higher AQP-1 levels that may have resulted from an accidental injury during intrathecal catheterization, and not from the differences in the sample loading (see β-actin). Because of that outlier, average AQP-1 levels seem higher in rats treated with hypertonic solution, although the difference was not statistically significant. However, AQP-1 levels in all other uninjured samples (n = 4) treated with hypertonic solution were indistinguishable from samples treated with isotonic solution (n = 5). AQP-1 protein levels were first normalized to β-actin (shown below the AQP-1 western blot data). All data were then normalized to AQP-1 levels in sham rats receiving isotonic solution (set to 1). (b) Effect of hypertonicity on AQP-4 in uninjured rats. In the same samples used in (a), we separately measured the protein levels of AQP-4 using western blots (∼30 kDa). All five samples from sham rats treated with hypertonic solution (H1–H5) had higher AQP-4 protein levels than uninjured samples treated with isotonic solution (S1–S5). The bar graph shows a statistically significant increase in AQP-4 protein levels induced by hypertonicity (*p < 0.05). (c) Hif-1α is increased in chronically injured spinal cords. SCI induced significant up-regulation of nuclear Hif-1α at T10, both at 14 and 42 days after SCI. Here, we present Hif-1α western blots of five sham samples and seven SCI samples 14 days after SCI, and three sham samples and six SCI samples 42 days after SCI. (d) The effect of the antioxidant melatonin on AQP-1 expression 14 days after SCI. Representative western blot data showing AQP-1 levels in T10 of: (1) sham-treated rats (n = 5; S1–S5) that received intraperitoneal injections of vehicle daily for 14 days; (2) SCI rats that received vehicle n = 5(I1–I5); and (3) SCI rats that received melatonin (10 mg/kg; n = 5; M1–M5). Rats were killed 2 weeks after SCI, proteins extracted from T10 and AQP-1 western blots performed for AQP-1 and β-actin. (e) Quantitative analysis of western blot data presented as bar graphs show that SCI induced 5.3-fold increases in AQP-1-protein levels 14 days after SCI (*p < 0.05), in agreement with our data presented in Fig. 2. Melatonin treatment significantly reduced AQP-1 levels at day 14 post-contusion by ∼60% (#p < 0.05). (f) The effect of the antioxidant melatonin on AQP-1 expression 35 days after SCI. In a similar experimental paradigm, one group of sham rats (n = 4) was treated with vehicle daily for 35 days, one group of SCI rats was treated with vehicle (n = 5), and one group of SCI rats was treated with melatonin (n = 4) for 35 days. SCI-induced fourfold significant increases in AQP-1 levels in T10, 35 days after SCI were significantly reduced (#p < 0.05) in melatonin treated SCI rats by 41.5%. (g) SCI-induced protein nitrosylation decreases in melatonin-treated SCI rats. Western blot analysis of nitrosylated proteins in nine samples, in the same experimental paradigm described under (d). Here, we present a representative but unidentified protein band of ∼25 kDa in three sham samples (treated with vehicle), three SCI samples (treated with vehicle), and three SCI samples treated with melatonin (SCI + M). The intensity of that band significantly increased in injured spinal cords (T10; 35 days after SCI; p < 0.05), but was significantly decreased (p < 0.01) in melatonin-treated SCI rats.
It has also been shown that hypoxia regulates AQP-1 expression in endothelial cells (Echevarria et al. 2007). Hypoxic conditions, which are characterized by increased protein levels of the transcription factor Hif-1α (Semenza 2000) have been reported in acutely injured spinal cords (Xiaowei et al. 2006), but not in chronically injured ones. We found a significant increase in nuclear Hif-1α in subchronic (1.6-fold at 14 days) and chronic (1.5-fold at 42 days) post-injury phases at the site of injury (T10; Fig. 3c). These observations confirmed the presence of chronic hypoxia in injured spinal cords and supported the hypothesis that hypoxia may be the mechanism triggering and maintaining increased AQP-1 protein levels after SCI (Fig. 2).
To test this hypothesis, we administered melatonin, a well-characterized antioxidant (Reiter et al. 2001) i.p. daily for 14 or 35 days (1 mL i.p.; 10 mg/kg). We decided to use melatonin as an antioxidant because it is not toxic; it is safe even in humans (Weishaupt et al., 2006); an important characteristic for a drug that had to be administered for several weeks. Furthermore, melatonin can be administered i.p. as it readily crosses blood–brain barrier (Reiter et al. 2004). Intraperitoneal injections are more reliable for long-term administrations, as drug instability and clogging of the catheters can pose a problem when osmotic minipumps are used for i.th. or direct injections. The concentration of melatonin (10 mg/kg body weight) was chosen based on the report by Kaur et al. (2006), showing beneficial effects of melatonin treatment after ischemic brain injury and decreased ischemia-induced AQP-4 levels. Melatonin was dissolved in 100% ethanol and diluted with sterile saline (0.9% NaCl) such that the final concentration of ethanol was 5%. Accordingly, the vehicle solution used in experiments was sterile saline containing 5% ethanol.
As shown in Fig. 3d and e, SCI induced significant 5.3-fold increases in AQP-1 protein levels compared with sham levels at T10 (*p < 0.05), while melatonin treatment significantly decreased AQP-1 protein levels by 61.5% (#p < 0.05). Similar results were obtained after 5 weeks of melatonin treatment (Fig. 3f).
We confirmed that i.p. administration of melatonin at the dose used had the expected effect in diminishing radicals in melatonin-treated SCI rats by measuring nitrosylation levels of proteins in three groups of rats: three sham rats treated with vehicle, three SCI rats treated with vehicle, and three SCI rats treated with melatonin. Melatonin has been shown to decrease nitric oxide synthesis and resultant protein nitrosylation of the tyrosine residues (3-nitrotyrosine, Conti et al. 2007). SCI induced marked nitrosylation of several unidentified proteins in T10, 14 days after SCI. In Fig. 3g, we presented one unidentified nitrosylated protein of about 25 kDa. Melatonin significantly decreased nitrosylation levels of this, and several other unidentified proteins (∼50 and ∼75 kDa). However, not all proteins nitrosylated after SCI were sensitive to melatonin treatment (not shown).
Melatonin decreases pain and AQP-1 in the dorsal horns after SCI
Increased sensitivity to mechanical stimuli that are normally innocuous (e.g. mechanical allodynia) is one of the devastating manifestations of the neuropathic pain that majority of SCI patients develop after injury (Tasker and Dostrovsky 1989). Mechanical allodynia is also well established in the rat model of contusion SCI (Hulsebosch et al. 2000) that was used in all our experiments. Increased sensitivity to mechanical stimuli in the trunk region in vehicle-treated SCI rats was significantly diminished in melatonin-treated SCI rats, as shown in Fig. 4a. We showed mechanical allodynia in the dermatomes (trunk region) corresponding to spinal regions around the site of injury, where we showed the effect of melatonin on AQP-1 protein levels (Fig. 3d–f). The average threshold in the vehicle-treated SCI group was ∼80% lower than the average threshold in the same group of rats measured before the injury. However, the corresponding threshold value for the SCI group treated with melatonin decreased by only ∼30%, not statistically different from the average pre-SCI threshold. The difference between the average thresholds in the vehicle- and melatonin-treated SCI groups 35 days after SCI was statistically significant (p = 0.013).
Fig. 4.

Melatonin decreases pain and AQP-1 in the dorsal horns after SCI. (a) Mechanical allodynia in the trunk region (girdling). We used Von Frey filaments to determine pain threshold values (in grams, presented on Y-axis as average values ± SD). The pain thresholds in all rats before SCI were between 40 and 50 g. Rats were divided into three groups: (1) rats that will be injured, and will receive vehicle (V) via i.p injection for 35 days, (2) rats that will be injured and will receive i.p. melatonin (M) for 35 days, (3) and rats that will undergo sham treatment and will receive i.p. vehicle for 35 days (not shown). The threshold values for sham rats (n = 13) 35 days after sham-treatment were unchanged compared with their baseline values before undergoing sham treatment (not shown). The average threshold for vehicle-treated SCI rats (SCI + V; n = 9) before SCI was 49.78 ± 19 g, but significantly decreased to 8.25 ± 0.7 g 35 days after SCI. However, the average threshold for melatonin-treated SCI rats (SCI + M; n = 10) decreased from 51.15 ± 32.84 before SCI to 32.4 ± 24 g 35 days after SCI. Therefore, melatonin-treated SCI rats have significantly higher pain threshold values than vehicle-treated SCI rats (p = 0.013). (b) Locomotor recovery. BBB scores were measured over a 35 days period in two groups of SCI rats: rats that received vehicle for 35 days (I + V; n = 9) and rats that received melatonin for 35 days (I + M; n = 10). The experimental paradigm used in this experiment is explained in Fig 3d. BBB scores were presented as average values ± SD in both vehicle and melatonin-treated SCI rats, and were undistinguishable. (c) AQP-1 immunolabeling in dorsal horns of T9 segments 35 days after SCI, in two groups of rats: SCI rats that received vehicle for 35 days (SCI + V; n = 3) and SCI rats that received melatonin for 35 days (SCI + M; n = 3). Here we show a representative image of AQP-1 immunoreactivity (IR) in the left panel, and semi-quantitative analysis of AQP-1 IR in the dorsal horns, in the bar graph below. For semiquantitative analysis of AQP-1 in the dorsal horns, we normalized AQP-1 intensity to the area of the dorsal horn taken for each measurement in each of five sections per rat. Not only did the average intensity of the AQP-1 signals markedly decrease in melatonin-treated rats (bar graph), but melatonin also reduced the depth of AQP-1 labeling in the dorsal horns of SCI rats (see white line in the images above). The images used for the analysis were taken in segment T9 from three rats per group, 35 days after SCI.
Locomotor recovery measured using the BBB scale (Basso et al. 1995) showed that BBB scores were indistinguishable between vehicle- and melatonin-treated SCI rats (Fig. 4b). This result may suggest that melatonin did not decrease overall injury or lesion size, in contrast to the previous reports (Genovese et al. 2005; Gul et al. 2005).
Analysis of the AQP-1 labeling in the dorsal horn revealed that the intensity of AQP-1 immunolabeling was also diminished in the dorsal horns of melatonin-treated SCI-rats (Fig. 4c), suggesting that decreased AQP-1 in dorsal horns of SCI rats treated with melatonin may be causally linked to the reduced pain in melatonin-treated SCI rats, as shown in Fig. 4a. Furthermore, decreased AQP-1 immunolabeling resulted primarily from the reduced area occupied by AQP-1-expressing fibers; e.g. the depth of the AQP-1 immunoreactivity of AQP-1-positive sensory fibers in the dorsal horn was decreased (see the white line in the representative AQP-1 immunolabeling), indicating that melatonin reduced excessive, aberrant sprouting of sensory AQP-1-positive fibers in dorsal horn regions below laminae I and II, and thus diminished mechanical allodynia.
Discussion
Aquaporin-1 protein levels are significantly and persistently elevated for up to 11 months after SCI in sensory axons, neurons, astrocytes, and ependymal cells, despite considerable loss of AQP-1-expressing neurons and axons at the site of injury. Multicellular localization and widespread up-regulation of AQP-1 suggest its possible contribution to different neuropathological outcomes after SCI.
To identify signals that can influence AQP-1 expression, we tested the effect of extracellular hypertonicity, given the presence of a hypertonicity-responsive element in the AQP-1 promoter (Umenishi and Schrier 2003). However, even long-lasting, continuous hypertonicity did not affect AQP-1 levels, although it significantly increased AQP-4 levels (not reported before). Increased AQP-4 is likely an astrocytic compensatory mechanism that counterbalances extracellular hypertonicity, for example astrocytes take up more water from the blood and redistribute it into the extracellular space. Because of the AQP-4 changes, and probably other osmoadaptive processes, rats exposed to long-term hypertonicity did not show any pathological behavioral changes (not shown). If astrocytic AQP-4 was responsible for osmoadaptive changes and for maintaining spinal cord isotonicity, what then is the function of AQP-1? If not anisotonicity, what are the signals that underlie persistent AQP-1 up-regulation after SCI?
We identified oxidative stress as one SCI-triggered process that contributes to AQP-1 up-regulation, as the antioxidant melatonin reduced AQP-1 increases. Although melatonin may have an immunosuppressive role (Ekmekcioglu 2006), its best documented role is decreasing reactive oxygen species levels (Allegra et al. 2003; Tan et al. 2003; Rodriguez et al. 2004; Reiter et al. 2005). The effect of melatonin on AQP-1 has not been previously investigated.
Hypoxic conditions characterized by increased Hif-1α levels exist not only soon after SCI (Xiaowei et al. 2006), but also in chronically injured spinal cords, as we report here. Thus, chronic hypoxia may contribute to persistent increases in AQP-1 after SCI. Hypoxia stimulates AQP-1 expression in endothelial cells (Echevarria et al. 2007), where AQP-1 plays a role in angiogenesis and migration by affecting cytoskeletal rearrangement and lamelipodia protrusion (Verkman 2006). We believe that AQP-1 in the spinal cord may play a role in astrocytic migration or in the axonal elongation via a similar mechanism involving the intake of water through AQP-1 channels that precedes axonal elongation.
As shown in Fig. 5, we found that the pattern of AQP-1 expression in the dorsal horns was similar to that of GAP-43, important in axonal growth during embryogenesis, in synaptic remodeling and plasticity, and in regeneration after injury (Kruger et al. 1993; Carulli et al. 2004). GAP-43 resides in the growth cones of growing neurites, where it interacts with F-actin-associated adhesion molecules and/or extracellular matrix complexes to promote neurite extension (He et al. 1997; Shen et al. 2002). We found that GAP-43 co-localizes with AQP-1 in both uninjured and injured spinal cords (Fig. 5a and b). As with AQP-1, the highest GAP-43 labeling was found within unmyelinated, small-diameter nerve fibers in the superficial laminae of the dorsal horns, in agreement with Nacimiento et al. (1993). This indicates a high degree of plasticity of sensory axons; e.g. they are capable of sprouting and structural reorganization that appear to be necessary for normal processing of sensory information. Although correlative, our findings (Fig. 5) may indicate a possible role for AQP-1 in axonal remodeling and plasticity, necessary for normal sensory processing.
Fig. 5.
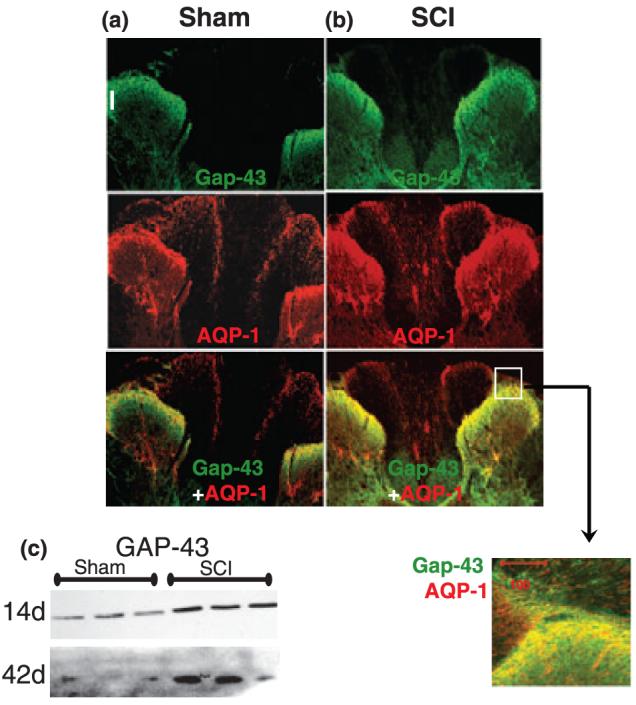
Co-localization of AQP-1 and GAP-43 in uninjured (a) and injured spinal cords (b). We analyzed GAP-43 and AQP-1 immunolabeling in T9 sections of sham and SCI spinal cords, 1 and 5 months after sham treatment or SCI (n = 3 per group). We used a GAP-43 antibody that recognizes both non-phosphorylated and phosphorylated (activated) GAP-43. As with AQP-1, GAP-43 was most abundant in the superficial laminae of the dorsal horn. Analogous to AQP-1, GAP-43 was increased after SCI throughout the spinal cords, including the GAP-43 labeling in the dorsal horns of SCI rats. AQP-1 and GAP-43 co-localized within the same afferent sensory fibers, as presented in the high-magnification image of the spinal region labeled with the white square. (c) SCI-induced increases in GAP-43 were confirmed with GAP-43 western blots 14 or 42 days after sham-treatment or SCI in T10 segments. We analyzed three sham and three SCI samples.
As dorsal horn fibers normally have high GAP-43 expression levels (Fig. 5a), it is not surprising that SCI-stimulated GAP-43 increases (Fig. 5c) result in non-specific and excessive sprouting in nociceptive fibers, which has been proposed as a mechanism of neuropathic pain after SCI (Macias et al., 2006), or underlying autonomic dysreflexia (Cameron et al. 2006). Our results demonstrate that because of excessive sprouting, AQP-1-positive fibers, which normally occupy only laminae I and II, penetrate deeper into the injured dorsal horn. Our experiments also show that melatonin treatment of SCI rats reduces: (i) AQP-1 levels; (ii) appearance of AQP-1-positive fibers below laminae I and II; and (iii) mechanical allodynia, thus indicating a possible causal relationship between AQP-1 with excessive axonal sprouting and subsequent chronic pain.
A possible consequence of increased AQP-1 levels in all spinal axons and neurons is the well-established neuronal/axonal swelling in injured spinal cords (Nashmi and Fehlings 2001). It is likely that the mechanism of SCI-induced neuronal/axonal swelling has not previously been defined, as neuronal AQPs have not been identified in spinal cords until now. Thus, hypoxic conditions may contribute to chronic accumulation of water within neurons and cytotoxic edema that can account for the increased water content in chronically injured spinal cords, that we previously reported (Nesic et al. 2006).
Sustained up-regulation of AQP-1 in ependymal cells may result in the over-production of CSF and formation of CSF-filled cysts after SCI. Formation and enlargement of cysts (syringomyelia) is a serious and untreatable complication after SCI (Klekamp and Samii, 2002; Evans et al. 2002). It has already been shown that AQP-1 has a role in the formation of CSF-filled cysts (Basaldella et al. 2007; Longatti et al., 2006).
We found markedly increased AQP-1 levels in reactive astrocytes, surrounding the lesion site. AQP-1 expression has been reported in reactive astrocytes in several neuropathological conditions (Rodríguez et al. 2006; Suzuki et al. 2006; Chen et al. 2006; Barrachina et al., 2007), although its role is still unclear. McCoy and Sontheimer (2007) have recently suggested that AQP-1 may play a role in the migration of malignant astrocytes. Consistent with this is our finding that only reactive astrocytes around the lesion site showed high AQP-1 expression levels, while reactive astrocytes at a distance from the lesion rim expressed markedly less AQP-1. Therefore, given that hypoxia stimulates astrocytic migration (Zhang et al. 1999; Xu et al. 2007) it is possible that hypoxic conditions after SCI trigger AQP-1 synthesis in astrocytes, as an attempt of injured spinal cords to facilitate astrocytic migration to the lesion site. However, the role of AQP-1 in the migration of astrocytes after SCI remains to be investigated. Considering the lack of safe and specific AQP-1 inhibitors (Yang et al. 2006), in the future we plan to use AQP-1 small interfering RNA (Splinter et al. 2003; Boassa et al. 2006) to directly test a causal relationship between AQP-1 and astrocyte migration after SCI in rats.
In summary, constant hypoxic conditions in chronically injured spinal cords persistently elevate AQP-1 in neurons, ependymal cells, astrocytes, and sensory fibers, and so may contribute to different pathological conditions after SCI, such as neuronal/axonal swelling, over-production of CSF and formation of cysts, or excessive axonal sprouting underlying the development of neuropathic pain and autonomic dysreflexia. Therefore, it is necessary to further characterize AQP-1 functions in SCI and identify safe interventions that specifically target AQP-1 functions.
Acknowledgements
This study was supported by Mission Connect, a project of TIRR Foundation, and NIH HDO039833. I would like to thank Drs Wayne Bolen and Joerg Rosgen for helpful discussion about osmoregulation, and Dr David Konkel for critically editing the manuscript.
Abbreviations used:
- AQP
aquaporin
- BBB
Basso, Beattie, and Bresnahan Scale
- DTT
dithiothreitol
- GAP-43
growth-associated protein 43
- Hif-1α
hypoxia inducible factor-1α
- i.th.
intrathecal
- SCI
spinal cord injury
- TUJ1
class III β-tubulin
References
- Allegra M, Reiter RJ, Tan DX, Gentile C, Tesoriere L, Livrea MA. The chemistry of melatonin's interaction with reactive species. J. Pineal Res. 2003;34:1–10. doi: 10.1034/j.1600-079x.2003.02112.x. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Lindland H, Zelenin S, Roberg BA, Gundersen BB, Petersen P, Rinvik E, Torgner IA, Ottersen OP. Brain mitochondria contain aquaporin water channels: evidence for the expression of a short AQP9 isoform in the inner mitochondrial membrane. FASEB J. 2005;19:1459–1467. doi: 10.1096/fj.04-3515com. [DOI] [PubMed] [Google Scholar]
- Basaldella L, Orvieto E, Dei Tos AP, Della Barbera M, Valente M, Longatti P. Causes of arachnoid cyst development and expansion. Neurosurg. Focus. 2007;22:00. doi: 10.3171/foc.2007.22.2.4. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J. Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Boassa D, Stamer WD, Yool AJ. Ion channel function of aquaporin-1 natively expressed in choroid plexus. J. Neurosci. 2006;26:7811–7819. doi: 10.1523/JNEUROSCI.0525-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calamita G, Ferri D, Gena P, Liquori GE, Cavalier A, Thomas D, Svelto M. The inner mitochondrial membrane has aquaporin-8 water channels and is highly permeable to water. J. Biol. Chem. 2005;280:17149–17153. doi: 10.1074/jbc.C400595200. [DOI] [PubMed] [Google Scholar]
- Cameron AA, Smith GM, Randall DC, Brown DR, Rabchevsky AG. Genetic manipulation of intraspinal plasticity after spinal cord injury alters the severity of autonomic dysreflexia. J. Neurosci. 2006;26:2923–2932. doi: 10.1523/JNEUROSCI.4390-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carulli D, Buffo A, Strata P. Reparative mechanisms in the cerebellar cortex. Prog. Neurobiol. 2004;72:373–398. doi: 10.1016/j.pneurobio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Chen Y, Tachibana O, Oda M, Xu R, Hamada J, Yamashita J, Hashimoto N, Takahashi JA. Increased expression of aquaporin 1 in human hemangioblastomas and its correlation with cyst formation. J. Neurooncol. 2006;80:219–225. doi: 10.1007/s11060-005-9057-1. [DOI] [PubMed] [Google Scholar]
- Conti A, Miscusi M, Cardali S, Germano A, Suzuki H, Cuzzocrea S, Tomasello F. Nitric oxide in the injured spinal cord: synthases cross-talk, oxidative stress and inflammation. Brain Res. Rev. 2007;54:205–218. doi: 10.1016/j.brainresrev.2007.01.013. Review. [DOI] [PubMed] [Google Scholar]
- Crown ED, Ye Z, Johnson KM, Xu GY, McAdoo DJ, Westlund KN, Hulsebosch CE. Upregulation of the phosphorylated form of CREB in spinothalamic tract cells following spinal cord injury: relation to central neuropathic pain. Neurosci. Lett. 2005;384:139–144. doi: 10.1016/j.neulet.2005.04.066. [DOI] [PubMed] [Google Scholar]
- Crown ED, Ye Z, Johnson KM, Xu GY, McAdoo DJ, Hulsebosch CE. Increases in the activated forms of ERK 1/2, p38 MAPK, and CREB are correlated with the expression of at-level mechanical allodynia following spinal cord injury. Exp. Neurol. 2006;199:397–407. doi: 10.1016/j.expneurol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Echevarria M, Munoz-Cabello AM, Sanchez-Silva R, Toledo-Aral JJ, Lopez-Barneo J. Development of cytosolic hypoxia and HIF stabilization are facilitated by aquaporin 1 expression. J. Biol. Chem. 2007;12:282(41):30207–30215. doi: 10.1074/jbc.M702639200. [DOI] [PubMed] [Google Scholar]
- Ekmekcioglu C. Melatonin receptors in humans: biological role and clinical relevance. Biomed. Pharmacother. 2006;60:97–108. doi: 10.1016/j.biopha.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Evans A, Stoodley N, Halpin S. Magnetic resonance imaging of intraspinal cystic lesions: a pictorial review. Curr. Probl. Diagn. Radiol. 2002;31:79–94. doi: 10.1067/cdr.2002.125402. [DOI] [PubMed] [Google Scholar]
- Frisbie JH. Salt wasting, hypotension, polydipsia, and hyponatremia and the level of spinal cord injury. Spinal Cord. 2007;45:563–568. doi: 10.1038/sj.sc.3101984. [DOI] [PubMed] [Google Scholar]
- Gao H, He C, Fang X, Hou X, Feng X, Yang H, Zhao X, Ma T. Localization of aquaporin-1 water channel in glial cells of the human peripheral nervous system. Glia. 2006;53:783–787. doi: 10.1002/glia.20336. [DOI] [PubMed] [Google Scholar]
- Genovese T, Mazzon E, Muia C, Bramanti P, De Sarro A, Cuzzocrea S. Attenuation in the evolution of experimental spinal cord trauma by treatment with melatonin. J. Pineal Res. 2005;38:198–208. doi: 10.1111/j.1600-079X.2004.00194.x. [DOI] [PubMed] [Google Scholar]
- Gul S, Celik SE, Kalayci M, Tasyurekli M, Cokar N, Bilge T. Dose-dependent neuroprotective effects of melatonin on experimental spinal cord injury in rats. Surg. Neurol. 2005;64:355–361. doi: 10.1016/j.surneu.2005.03.036. [DOI] [PubMed] [Google Scholar]
- He Q, Dent EW, Meiri KF. Modulation of actin filament behavior by GAP-43 (neuromodulin) is dependent on the phosphorylation status of serine 41, the protein kinase C site. J. Neurosci. 1997;17:3515–3524. doi: 10.1523/JNEUROSCI.17-10-03515.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Strittmatter SM. Regulating axon growth within the postnatal central nervous system. Semin. Perinatol. 2004;28:371–378. doi: 10.1053/j.semperi.2004.10.001. Review. [DOI] [PubMed] [Google Scholar]
- Hulsebosch CE, Xu GY, Perez-Polo JR, Westlund KN, Taylor CP, McAdoo DJ. Rodent model of chronic central pain after spinal cord contusion injury and effects of gabapentin. J. Neurotrauma. 2000;17:1205–1217. doi: 10.1089/neu.2000.17.1205. [DOI] [PubMed] [Google Scholar]
- Kaur C, Sivakumar V, Zhang Y, Ling EA. Hypoxia-induced astrocytic reaction and increased vascular permeability in the rat cerebellum. Glia. 2006;54:826–839. doi: 10.1002/glia.20420. [DOI] [PubMed] [Google Scholar]
- Kim JG, Son YJ, Yun CH, Kim YI, Nam-Goong IS, Park JH, Park SK, Ojeda SR, D'Elia AV, Damante G, Lee BJ. Thyroid transcription factor-1 facilitates cerebrospinal fluid formation by regulating aquaporin-1 synthesis in the brain. J. Biol. Chem. 2007;282:14923–14931. doi: 10.1074/jbc.M701411200. [DOI] [PubMed] [Google Scholar]
- Klekamp J, Batzdorf U, Samii M, Bothe HW. Treatment of syringomyelia associated with arachnoid scarring caused by arachnoiditis or trauma. J. Neurosurg. 1997;86:233–240. doi: 10.3171/jns.1997.86.2.0233. [DOI] [PubMed] [Google Scholar]
- Kruger L, Bendotti C, Rivolta R, Samanin R. Distribution of GAP-43 mRNA in the adult rat brain. J. Comp. Neurol. 1993;333:417–434. doi: 10.1002/cne.903330308. [DOI] [PubMed] [Google Scholar]
- Legos JJ, Gritman KR, Tuma RF, Young WF. Coadministration of methylprednisolone with hypertonic saline solution improves overall neurological function and survival rates in a chronic model of spinal cord injury. Neurosurgery. 2001;49:1427–1433. doi: 10.1097/00006123-200112000-00022. [DOI] [PubMed] [Google Scholar]
- Liang Y, Yan C, Nylander KD, Schor NF. Early events in Bcl-2-enhanced apoptosis. Apoptosis. 2003;8:609–616. doi: 10.1023/A:1026135509274. [DOI] [PubMed] [Google Scholar]
- Longatti P, Basaldella L, Orvieto E, Tos AP, Martinuzzi A. Aquaporin 1 expression in cystic hemangioblastomas. Neurosci. Lett. 2006;392:178–180. doi: 10.1016/j.neulet.2005.09.083. [DOI] [PubMed] [Google Scholar]
- Macias MY, Syring MB, Pizzi MA, Crowe MJ, Alexanian AR, Kurpad SN. Pain with no gain: allodynia following neural stem cell transplantation in spinal cord injury. Exp. Neurol. 2006;201:335–348. doi: 10.1016/j.expneurol.2006.04.035. [DOI] [PubMed] [Google Scholar]
- McCoy E, Sontheimer H. Expression and function of water channels (aquaporins) in migrating malignant astrocytes. Glia. 2007;55:1034–1043. doi: 10.1002/glia.20524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacimiento W, Töpper R, Fischer A, Oestreicher AB, Nacimiento AC, Gispen WH, Noth J, Kreutzberg GW. Immunocytochemistry of B-50 (GAP-43) in the spinal cord and in dorsal root ganglia of the adult cat. J. Neurocytol. 1993;22:413–424. doi: 10.1007/BF01181562. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Fehlings MG. Changes in axonal physiology and morphology after chronic compressive injury of the rat thoracic spinal cord. Neuroscience. 2001;104:235–251. doi: 10.1016/s0306-4522(01)00009-4. [DOI] [PubMed] [Google Scholar]
- Nesic O, Lee J, Johnson KM, et al. Transcriptional profiling of spinal cord injury-induced central neuropathic pain. J. Neurochem. 2005;95(4):998–1014. doi: 10.1111/j.1471-4159.2005.03462.x. [DOI] [PubMed] [Google Scholar]
- Nesic O, Lee J, Ye Z, Unabia GC, Rafati D, Hulsebosch CE, Perez-Polo JR. Acute and chronic changes in aquaporin 4 expression after spinal cord injury. Neuroscience. 2006;143:779–792. doi: 10.1016/j.neuroscience.2006.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshio K, Song Y, Verkman AS, Manley GT. Aquaporin-1 deletion reduces osmotic water permeability and cerebrospinal fluid production. Acta Neurochir. Suppl. 2003;86:525–528. doi: 10.1007/978-3-7091-0651-8_107. [DOI] [PubMed] [Google Scholar]
- Oshio K, Watanabe H, Song Y, Verkman AS, Manley GT. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel aquaporin-1. FASEB J. 2005;19:76–78. doi: 10.1096/fj.04-1711fje. [DOI] [PubMed] [Google Scholar]
- Pérez E, Barrachina M, Rodríguez A, Torrejón-Escribano B, Boada M, Hernández I, Sánchez M, Ferrer I. Aquaporin expression in the cerebral cortex is increased at early stages of Alzheimer disease. Brain Res. 2007;1128:164–174. doi: 10.1016/j.brainres.2006.09.109. [DOI] [PubMed] [Google Scholar]
- Reiter RJ, Tan DX, Manchester LC, Qi W. Biochemical reactivity of melatonin with reactive oxygen and nitrogen species: a review of the evidence. Cell Biochem. Biophys. 2001;34:237–256. doi: 10.1385/CBB:34:2:237. [DOI] [PubMed] [Google Scholar]
- Reiter RJ, Tan DX, Gitto E, et al. Pharmacological utility of melatonin in reducing oxidative cellular and molecular damage. Pol. J. Pharmacol. 2004;56:159–170. [PubMed] [Google Scholar]
- Reiter RJ, Tan DX, Maldonado MD. Melatonin as an antioxidant: physiology versus pharmacology. J. Pineal Res. 2005;39:215–216. doi: 10.1111/j.1600-079X.2005.00261.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez C, Mayo JC, Sainz RM, Antolín I, Herrera F, Martin V, Reiter RJ. Regulation of antioxidant enzymes: a significant role for melatonin. J. Pineal Res. 2004;36:1–9. doi: 10.1046/j.1600-079x.2003.00092.x. [DOI] [PubMed] [Google Scholar]
- Rodríguez A, Pérez-Gracia E, Espinosa JC, Pumarola M, Torres JM, Ferrer I. Increased expression of water channel aquaporin 1 and aquaporin 4 in Creutzfeldt-Jakob disease and in bovine spongiform encephalopathy-infected bovine-PrP transgenic mice. Acta Neuropathol. (Berl.) 2006;112:573–585. doi: 10.1007/s00401-006-0117-1. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000;88:1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- Shen Y, Mani S, Donovan SL, Schwob JE, Meiri KF. Growth-associated protein-43 is required for commissural axon guidance in the developing vertebrate nervous system. J. Neurosci. 2002;22:239–247. doi: 10.1523/JNEUROSCI.22-01-00239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields SD, Mazario J, Skinner K, Basbaum AI. Anatomical and functional analysis of aquaporin 1, a water channel in primary afferent neurons. Pain. 2007;131:8–20. doi: 10.1016/j.pain.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Speake T, Freeman LJ, Brown PD. Expression of aquaporin 1 and aquaporin 4 water channels in rat choroid plexus. Biochim. Biophys. Acta. 2003;1609:80–86. doi: 10.1016/s0005-2736(02)00658-2. [DOI] [PubMed] [Google Scholar]
- Splinter PL, Masyuk AI, LaRusso NF. Specific inhibition of AQP1 water channels in isolated rat intrahepatic bile duct units by small interfering RNAs. J. Biol. Chem. 2003;278:6268–6274. doi: 10.1074/jbc.M212079200. [DOI] [PubMed] [Google Scholar]
- Sumas ME, Legos JJ, Nathan D, Lamperti AA, Tuma RF, Young WF. vTonicity of resuscitative fluids influences outcome after spinal cord injury. Neurosurgery. 2001;48:167–172. doi: 10.1097/00006123-200101000-00029. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Okuda M, Asai J, Nagashima G, Itokawa H, Matsunaga A, Fujimoto T, Suzuki T. Astrocytes co-express aquaporin-1, -4, and vascular endothelial growth factor in brain edema tissue associated with brain contusion. Acta Neurochir. Suppl. 2006;96:398–401. doi: 10.1007/3-211-30714-1_82. [DOI] [PubMed] [Google Scholar]
- Tan DX, Manchester LC, Hardeland R, Lopez-Burillo S, Mayo JC, Sainz RM, et al. Melatonin: a hormone, a tissue factor, an autocoid, a paracoid, and an antioxidant vitamin. J. Pineal Res. 2003;34:75–78. doi: 10.1034/j.1600-079x.2003.02111.x. [DOI] [PubMed] [Google Scholar]
- Tasker RR, Dostrovsky JO. Deafferentation and central pain. In: Wall PD, Melzack R, editors. Textbook of Pain. 2nd ed Churchill Livingstone; New York: 1989. pp. 154–180. [Google Scholar]
- Umenishi F, Schrier RW. Identification and characterization of a novel hypertonicity-responsive element in the human aquaporin-1 gene. Biochem. Biophys. Res. Commun. 2002;292:771–775. doi: 10.1006/bbrc.2002.6709. [DOI] [PubMed] [Google Scholar]
- Umenishi F, Schrier RW. Hypertonicity-induced aquaporin-1 (AQP-1) expression is mediated by the activation of MAPK pathways and Hypertonicity-responsive element in the AQP-1 gene. J. Biol. Chem. 2003;278:15765–15770. doi: 10.1074/jbc.M209980200. [DOI] [PubMed] [Google Scholar]
- Umenishi F, Narikiyo T, Schrier RW. Hypertonic induction of aquaporin-1 water channel independent of transcellular osmotic gradient. Biochem. Biophys. Res. Commun. 2004;325:595–599. doi: 10.1016/j.bbrc.2004.10.076. [DOI] [PubMed] [Google Scholar]
- Umenishi F, Yoshihara S, Narikiyo T, Schrier RW. Modulation of hypertonicity-induced aquaporin-1 by sodium chloride, urea, betaine, and heat shock in murine renal medullary cells. J. Am. Soc. Nephrol. 2005;16:600–607. doi: 10.1681/ASN.2004030241. (Epub 2005 Jan 12) [DOI] [PubMed] [Google Scholar]
- Verkman AS. Aquaporins in endothelia. Kidney Int. 2006;69:1120–1123. doi: 10.1038/sj.ki.5000226. Review. [DOI] [PubMed] [Google Scholar]
- Weishaupt JH, Bartels C, Polking E, et al. Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J. Pineal Res. 2006;1:313–323. doi: 10.1111/j.1600-079X.2006.00377.x. [DOI] [PubMed] [Google Scholar]
- Xiaowei H, Ninghui Z, Wei X, Yiping T, Linfeng X. The experimental study of hypoxia-inducible factor-1alpha and its target genes in spinal cord injury. Spinal Cord. 2006;44:35–43. doi: 10.1038/sj.sc.3101813. [DOI] [PubMed] [Google Scholar]
- Xu Q, Wang S, Jiang X, et al. Hypoxia-induced astrocytes promote the migration of neural progenitor cells via vascular endothelial factor, stem cell factor, stromal-derived factor-1alpha and monocyte chemoattractant protein-1 upregulation in vitro. Clin. Exp. Pharmacol. Physiol. 2007;34:624–631. doi: 10.1111/j.1440-1681.2007.04619.x. [DOI] [PubMed] [Google Scholar]
- Yang B, Kim JK, Verkman AS. Comparative efficacy of HgCl2 with candidate aquaporin-1 inhibitors DMSO, gold, TEA+ and acetazolamide. FEBS Lett. 2006;580:6679–6684. doi: 10.1016/j.febslet.2006.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Porat RM, Alon T, Keshet E, Stone J. Tissue oxygen levels control astrocyte movement and differentiation in developing retina. Brain Res. Dev. Brain Res. 1999;118:135–145. doi: 10.1016/s0165-3806(99)00140-6. [DOI] [PubMed] [Google Scholar]
- Zhang D, Vetrivel L, Verkman AS. Aquaporin deletion in mice reduces intraocular pressure and aqueous fluid production. J. Gen. Physiol. 2002;119:561–569. doi: 10.1085/jgp.20028597. [DOI] [PMC free article] [PubMed] [Google Scholar]