

Abstract
Anorexigenic melanocortins decrease food intake by activating MC3/MC4 receptors (MC3/4R); the prevailing view is that the orexigenic neuropeptide agouti-related peptide (AgRP) exerts the opposite action by acting as an antagonist at MC3/MC4 receptors. A total of 370 hypothalamic ventromedial nucleus (VMH) glutamatergic neurons was studied using whole-cell recording in hypothalamic slices from a novel mouse expressing green fluorescent protein (GFP) under control of the vesicular glutamate transporter 2 (vGluT2) promoter. Massive numbers of GFP-expressing VMH dendrites extended out of the core of the nucleus into the surrounding cell-poor shell. VMH dendrites received frequent appositions from AgRP-immunoreactive axons in the shell of the nucleus, but not the core, suggesting that AgRP may influence target VMH neurons. α-MSH, melanotan II (MTII), and selective MC3R or MC4R agonists were all inhibitory, reducing the spontaneous firing rate and hyperpolarizing vGluT2 neurons. The MC3/4R antagonist SHU9119 was excitatory. Unexpectedly, AgRP did not attenuate MTII actions on these neurons; instead, these two compounds showed an additive inhibitory effect. In the absence of synaptic activity, no hyperpolarization or change in input resistance was evoked by either MTII or AgRP, suggesting indirect actions. Consistent with this view, MTII increased the frequency of spontaneous and miniature IPSCs. In contrast, the mechanism of AgRP inhibition was dependent on presynaptic inhibition of EPSCs mediated by Gi/Go-proteins, and was attenuated by pertussis toxin and NF023, inconsistent with mediation by Gs-proteins associated with MC receptors. Together, our data suggest that the mechanism of AgRP actions on these excitatory VMH cells appears to be independent of the actions of melanocortins on MC receptors.
Keywords: NPY, arcuate nucleus, feeding, POMC, vGluT2, glutamate
Introduction
The hypothalamic ventromedial nucleus (VMH) has been suggested as a “satiety center” in the mammalian hypothalamus; lesions of the VMH result in severe obesity (Stellar, 1954; King, 2006), and leptin receptors in the VMH play a key role in resisting obesity (Dhillon et al., 2006). A number of different neuroactive substances have been reported in the VMH, including GABA, glutamate, nitric oxide, enkephalin, and dynorphin (Bhat et al., 1995; Commons et al., 1999; Ziegler et al., 2002; Slamberova et al., 2004; Lin et al., 2006). Glutamate appears to be the primary excitatory neurotransmitter in the medial hypothalamus (van den Pol et al., 1990), and glutamate agonists and antagonists have substantial effects on food intake (Hettes et al., 2003; Lee and Stanley, 2005). However, until now, there has not been any reliable means of selectively studying live glutamate neurons within the hypothalamus.
The central melanocortin system plays a key role in the control of food intake by acting on MC3 and MC4 melanocortin receptors (MC3R and MC4R). It is one of the downstream pathways mediating the regulation of energy homeostasis by leptin and insulin (Schwartz et al., 2000; Benoit et al., 2002). α-MSH released by proopiomelanocortin (POMC) neurons is considered an anorexigenic peptide, which can increase energy expenditure (Poggioli et al., 1986; Brown et al., 1998; Hillebrand et al., 2005). MC3/4R agonists inhibit feeding, whereas the antagonists stimulate feeding (Cowley et al., 1999). POMC and MC4R knock-out mice are obese with hyperphagia and metabolic defects (Huszar et al., 1997; Yaswen et al., 1999); the POMC knock-out phenotype can be reversed by treatment with α-MSH derivatives (Yaswen et al., 1999). In humans, mutations in MC4R have also been linked to obesity (Farooqi et al., 2000; Vaisse et al., 2000). MC3R knock-out mice show increased adiposity and feeding efficiency, suggesting that MC3R may be important in regulating caloric intake and partitioning nutrients into fat (Butler et al., 2000; Chen et al., 2000b).
Agouti-related peptide (AgRP), which colocalizes with NPY in the same set of arcuate nucleus neurons, is another important member of the melanocortin system (Broberger et al., 1998; Hahn et al., 1998; Marks and Cone, 2003). AgRP has been reported to be a specific competitive antagonist of the MC3R and MC4R (Fong et al., 1997; Ollmann et al., 1997; Cone 2005). Intracerebroventricular administration of AgRP stimulates feeding (Rossi et al., 1998), and transgenic overexpression of AgRP causes obesity (Ollmann et al., 1997). Postembryonic ablation of AgRP neurons in mice leads to a lean, hypophagic phenotype (Bewick et al., 2005). Selective ablation of AgRP neurons in adult mice results in an acute reduction of feeding (Gropp et al., 2005; Luquet et al., 2005).
MC3R and MC4R have been reported to be expressed by neurons of the VMH, possibly with greater expression of MC3R (Roselli-Rehfuss et al., 1993; Harrold et al., 1999; Jegou et al., 2000; Kishi et al., 2003, 2005; Liu et al., 2003; Harrold and Williams, 2006). Previous reports have noted a relatively low level of innervation by AgRP (Broberger et al., 1998; Haskell-Luevano et al., 1999) or POMC (Smith and Funder, 1988; Cone, 1999) axons within the VMH; we have addressed this question again using a combination of transgenic mice and immunostaining.
Using transgenic mice expressing green fluorescent protein (GFP) under the control of the vesicular glutamate transporter 2 (vGluT2) promoter to identify excitatory neurons, together with the patch-clamp recording technique, we first examined immunoreactive axonal projections to GFP-positive VMH cells, and then characterized the membrane properties of these excitatory neurons in the VMH, and studied the activity of α-MSH and its analogs, as well as AgRP, on these neurons.
Materials and Methods
Transgenic mice.
We generated transgenic mice using the vGluT2 promoter to drive enhanced GFP expression. In these vGluT2 mice, GFP is selectively expressed in glutamatergic vGluT2-expresssing neurons but not in other cells. The sequence of the vGluT2 promoter construct used to drive GFP expression in these transgenic mice has been described previously (Huang et al., 2006). Briefly, the vGluT2 promoter, 1.8 kb upstream from the vGluT2 sequence, was used to drive GFP expression. Two oligomers, a forward primer with the sequence 5′-ATC TCG AGA CGC ACT CCC CCT GGT TGA TTT AG-3′ and a reverse primer containing 5′-CCG CGG TAC CTC TTG TAA AGA CTG GTG TCC AGC CTT ACC AGA TTTA-3′ corresponding to the region immediately upstream of the mouse vGluT2 coding region were used to generate a PCR fragment from mouse whole-brain genomic DNA template. This sequence was inserted into a plasmid containing the sequence for enhanced codon-optimized GFP.
Immunocytochemistry.
To study AgRP-immunoreactive axons, we used a rabbit anti-AgRP(83–132)-NH2 antibody (Phoenix Pharmaceuticals) described in detail previously (Haskell-Luevano et al., 1999). Adult transgenic mice were given an anesthetic overdose, and perfused transcardially with physiological saline, followed by 4% paraformaldehyde. Ten- to 30-μm-thick sections were cut on a cryostat, and immersed in blocking solution contained 0.1% lysine, 0.1% glycine, 1% bovine serum albumin, and 1% normal goat serum. Sections were then incubated in primary affinity-purified AgRP antiserum overnight at a dilution according to the manufacturer's recommendations, washed, and then immersed in goat anti-rabbit antibody conjugated to Texas Red (1:300). Sections were visualized on an Olympus IX70 microscope. GFP-producing neurons were visualized as green, and AgRP immunoreactivity as red. In addition, goat antisera against α-MSH (1:2000; Millipore Bioscience Research Reagents) and donkey anti-goat conjugated to Alexa 594 (Invitrogen) were used to study the distribution of these axons in and around the VMH using similar methods to those used to visualize AgRP. Photomicrographs were made with a Spot digital camera, and contrast and brightness corrected with Adobe Photoshop 7.0.
Hypothalamic slices.
Two- to 6-week-old mice maintained in a 12 h light/dark cycle, were deeply anesthetized by administering an overdose of sodium pentobarbital (100 mg/kg, i.p.) during the light part of the cycle (10:00 A.M. to 2:00 P.M.). Most slices were from mice 2–3 weeks of age; additional slices were used from adult 6-week-old mice to verify that neuronal responses were similar and independent of age. The brains were then rapidly removed and placed in an ice-cold oxygenated (95% O2, 5% CO2) high-sucrose solution that contained the following (in mm): 220 sucrose, 2.5 KCl, 6 MgCl2, 1 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, 10 glucose, pH 7.4 (when equilibrated with the mixed O2). A tissue block containing VMH was prepared and coronal slices (250–300 μm thick) were cut on a vibratome. The slices were gently moved to an equilibrium chamber filled with gassed, artificial CSF (ACSF) (95% O2, 5% CO2) that contained the following (in mm): 124 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4. After a 1–2 h recovery period, slices were moved to a recording chamber mounted on an Olympus BX51WI upright microscope equipped with video-enhanced, infrared-differential interference contrast (DIC) and fluorescence and perfused with a continuous flow of gassed ACSF. Neurons were viewed using blue excitation light with an Olympus 40× water-immersion lens. Approximately 85% of the cells recorded were from the ventrolateral part of the VMH, and 15% from the dorsomedial part of the VMH. We combined all data because no significant difference in membrane properties or drug responses were found between these two regions of the VMH. The use of mice for these experiments was approved by the Yale University Committee on Animal Use.
Single-cell cytoplasm harvest and reverse transcription-PCR.
Expression of GFP in vGluT2-expressing cells was verified with single-cell PCR. Harvesting of cytoplasmic RNA, reverse transcription (RT), and PCR were performed by modification of the previously described methods (Liss et al., 1999; Kang et al., 2004; Hirrlinger et al., 2005). Briefly, patch pipettes were back-loaded with 5 μl of diethylpyrocarbonate-treated water and were used to harvest the cell cytoplasm. Under visual control using both fluorescent and differential interference contrast imaging, a pipette tip was used to reach GFP-positive cells in the VMH and form a gigaohm seal. The cell contents were carefully aspirated into the pipette by application of gentle negative pressure. The pipettes were then removed and the contents were expelled into a microtube.
The 5 μl volume of diethylpyrocarbonate-treated water containing the single-cell cytoplasm was used as a template in a reverse transcription reaction using the SuperScript III Reverse Transcription kit (Invitrogen) following the manufacturer's instructions. Gene-specific oligos complementary to the mouse β-actin and vGluT2 mRNA sequences were used at a reaction temperature of 53°C and final reaction volume of 20 μl. The resulting single-cell cDNA samples were then treated with 2 U of RNase H (Invitrogen) and incubated at 37°C for 20 min and stored at −20°C until use. All PCR was performed using the Expand High Fidelity PCR kit (Roche Diagnostics). Five microliters of each single-cell cDNA sample was used as template in separate 50 μl PCRs, producing a 523 bp β-actin amplicon and a 279 bp nested vGluT2 amplicon. The β-actin PCR conditions consisted of 35 cycles at an annealing temperature of 56.7°C and an elongation time of 45 s. The nucleotide sequences of the primer pair used in the β-actin reaction were as follows: forward, 5′-GCC AAC CGT GAA AAG ATG AC-3′, and reverse, 5′-CAA CGT CAC ACT TCA TGA TG-3′. The vGluT2 PCR consisted of first a primary PCR from which 1 μl of reaction product was used as template in a second “nested” PCR. The vGluT2 primary PCR conditions consisted of 35 cycles at an annealing temperature of 53.8°C and an elongation time of 45 s. The nucleotide sequences of the primer pair used in the primary vGluT2 reaction were as follows: forward, 5′-CAT TTC AGA TGG CGT TGG CAC-3′, and reverse, 5′-CTT ATA GGT GTA CGC GTC TTG-3′. The vGluT2 nested PCR conditions consisted of 30 cycles at an annealing temperature of 53.6°C and an elongation time of 45 s. The nucleotide sequences of the primer pair used in the nested vGluT2 reaction were as follows: forward, 5′-CTA TCA TTG TTG GTG CAA TGA-3′, and reverse, 5′-AGC CTC CAT TCT CCT GTG AG-3′.
In situ hybridization. In situ hybridization experiments were performed using procedures described by the Allen Institute for Brain Science (www.brain-map.org/pdf/ABADataProductionProcesses.pdf). Digoxigenin-labeled vGluT2 antisense and sense cRNA probes were prepared as described in the Allen Brain Atlas (www.brain-map.org; probe no. RP_050921_01_E03). Briefly, total mRNA was isolated from freshly dissected mouse hypothalamus and reverse-transcribed to produce cDNA using random hexamers. The hypothalamic cDNA was then used as the template for a PCR targeted to bases 2190–2769 of mouse vGluT2 mRNA (GenBank accession no. NM_080853) using the following primer pair: forward, 5′-CCAAATCTTACGGTGCTACCTC-3′, and reverse, 5′-TAGCCATCTTTCCTGTTCCACT-3′. The 580 bp PCR amplification product was then TA-cloned and sequenced, with the resulting plasmid used as template for the generation of both antisense and sense vGluT2 cRNA probes labeled with digoxigenin. Twenty micrometer frozen sections of mouse hypothalamus were hybridized with the cRNA probes, and incubated with sheep antiserum (Fab fragment; Roche Diagnostics) against digoxigenin conjugated to alkaline phosphatase, and labeled with nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-idolyl-phosphate, toluidine salt (Roche Diagnostics).
Patch-clamp recording.
Whole-cell current- and voltage-clamp recordings were performed in 370 cells using pipettes with 4–6 MΩ resistance after filling with pipette solution. Recording pipettes were made of borosilicate glass (World Precision Instruments) using a PP-83 vertical puller (Narishige) and filled with pipette solutions. For most recordings, the composition of the pipette solution was as follows (in mm): 130 KMeSO4 [or KCl for IPSCs and miniature IPSCs (mIPSCs)], 1 MgCl2, 10 HEPES, 1.1 EGTA, 2 Mg-ATP, and 0.5 Na2-GTP, 10 Na2-phosphocreatine, pH 7.3 with KOH. Glutamatergic GFP-expressing neurons in the VMH were initially identified under fluorescence, and then DIC was used to get a seal in these cells. After a gigaohm seal was obtained, a gentle negative pressure was applied to break through to the whole-cell configuration. An EPC10 amplifier and Patchmaster software (HEKA Elektronik) were used for data acquisition. Capacitance was compensated automatically using Patchmaster software. Input resistance was monitored continuously, and only those cells with a stable access resistance (change, <10%) were used for analysis. The recordings were made at 32°C. AgRP, α-MSH, and analogues were applied focally to the recorded neurons via a large 400 um tip diameter flow pipette. Human AgRP was used in most experiments; mouse AgRP was also tested in some cells with similar results. When drugs were not applied, a continuous flow of buffer was applied from the flow pipette.
Pulsefit (HEKA Electronik), Axograph (Molecular Devices) and Igor Pro (WaveMetrics) software were used for analysis. Both miniature and spontaneous postsynaptic currents were detected and measured with an algorithm in Axograph (Bekkers and Stevens, 1995), and only those events with amplitude >5 pA were used, as has been described in detail previously (Gao and van den Pol, 1999). The frequency of action potentials was measured using Axograph as well. In Results, data are expressed as mean ± SE unless otherwise noted. ANOVA with Bonferroni's post hoc test, t test, and Kolmogorov–Smirnoff statistical tests were used. A value of p < 0.05 was considered statistically significant.
To test for Gi/Go-dependent actions of AgRP, slices were preincubated for at least 4 h in either pertussis toxin (PTX) (5 ug/ml) or 8,8′-[carbonylbis(imino-3,1-phenylene)]bis-(1,3,5-naphthalenetrisulfonic acid) (NF023) (10 um) at 37°C as previously described (Beindl et al., 1996; Freissmuth et al., 1996; DeBock et al., 2003; Garic-Stankovic et al., 2005).
Chemicals and reagents.
Bicuculline methiodiode (BIC), dl-2-amino-5-phosphonovaleric acid (AP5), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and picrotoxin were purchased from Sigma-Aldrich; tetrodotoxin (TTX) was obtained from Tocris Bioscience) α-MSH trifluoroacetate salt, (Ac-Nle4,Asp5,d-2-Nal7,Lys10)-α-MSH(4–10)amide (SHU9119), and melanotan II (MTII) were from Bachem. MC-4R agonist [cyclo(β-Ala-His-d-Phe-Arg-Trp-Glu)-NH2] and AgRP(83–132)-NH2 (human, mouse) were from Phoenix Pharmaceuticals. Pertussis toxin, ω-agatoxin IVA, and ω-conotoxin GVIA were also from Sigma-Aldrich, and NF023 was from Calbiochem.
Results
Expression of GFP with vGluT2 promoter in neurons of the VMH
In the present study, we used a transgenic mouse that expressed GFP under the control of the vGluT2 promoter. As shown in Figure 1, A and B, and Figure 2, GFP-expressing neurons and their processes are found throughout the VMH. The shell area around the VMH shows relatively few GFP-positive cells. The expression of GFP in vGluT2-expressing cells was verified with single-cell RT-PCR analysis. All GFP-positive neurons tested in the VMH (n = 30) were vGluT2 mRNA positive (Fig. 1C). Hypothalamic GFP-negative cells (n = 4) and striatal GFP-negative neurons (n = 13) were used as controls; all 17 control neurons were negative on the RT-PCR test. Similarly, a water control replacing template with buffer was vGluT2 mRNA negative. The dense distribution of GFP cells in the VMH area is consistent with the heavy expression of vGluT2 in the VMH (Ziegler et al., 2002; Collin et al., 2003), and supports the view that many VMH neurons use glutamate as an excitatory neurotransmitter (Beart et al., 1988). A rostrocaudal series of micrographs showing GFP expression at different levels of the VMH is seen in Figure 2A–E. In situ hybridization for the vGluT2 mRNA (Fig. 2F) showed a similar distribution of labeled cells in and around the VMH as seen with GFP expression; in areas in which there was no GFP expression such as the striatum, no labeling was found with in situ hybridization. Furthermore, no labeling was found in the VMH with a control sense probe.
Figure 1.
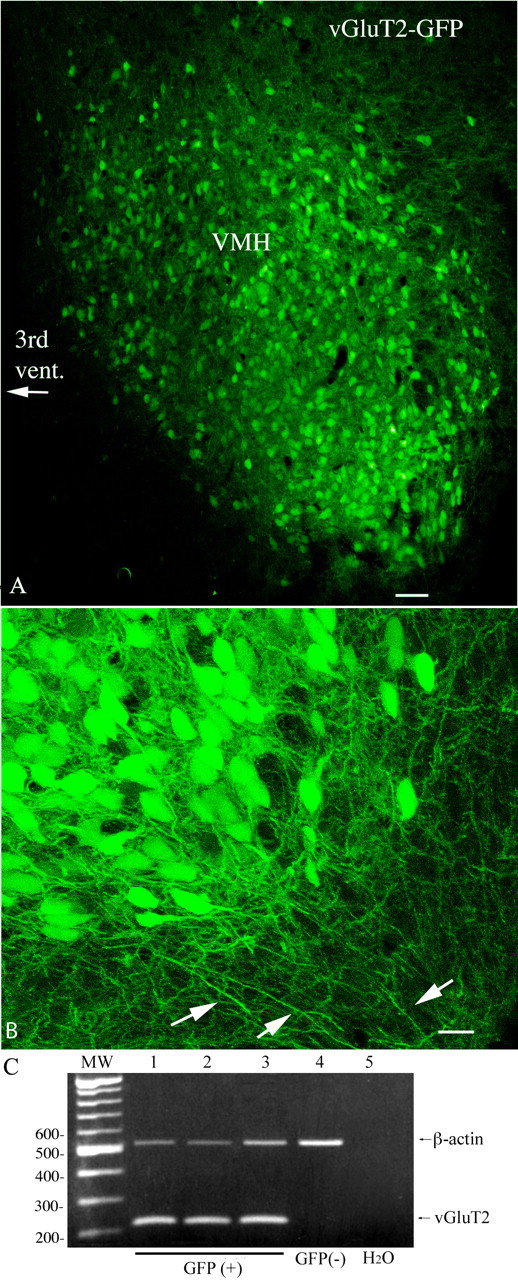
GFP is expressed selectively in vGluT2-containing neurons in VMH. A, GFP-expressing cells in a slice from VMH. Scale bar, 45 μm. B, High-magnification laser confocal image showing the ventrolateral VMH with GFP-expressing processes. Scale bar, 12 μm. C, Photograph of an ethidium bromide-stained gel with single-cell PCR amplicons showing that all GFP-expressing neurons (lanes 1–3) in the VMH are vGluT2 positive, whereas the cell that did not express GFP (lane 4) is vGluT2 negative. A total of 30 GFP-positive cells were harvested from the VMH, and all 30 were positive for vGluT2 mRNA. Lane 5 is the negative PCR control replacing template with water. The molecular weight marker is a successive 100 bp DNA ladder.
Figure 2.
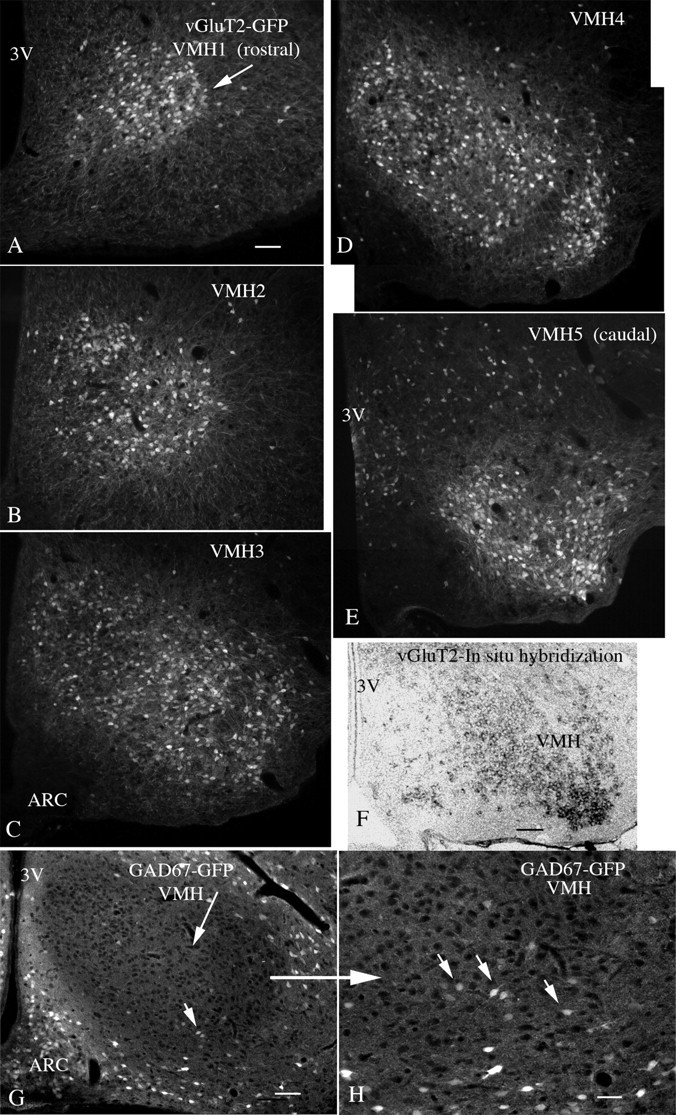
Rostrocaudal expression of GFP in VMH. A–E show GFP expression (white fluorescent cells) in successive rostrocaudal levels of the VMH of the vGluT2-GFP mouse, starting rostrally in A, and proceeding caudally to E (rostral to caudal: VMH1–VMH5). Scale bar, 60 μm. F, In situ hybridization with a probe for vGluT2 mRNA shows expression (dark cells) in the VMH, corresponding to a similar level in E. Scale bar, 60 μm. G shows a VMH section from a transgenic mouse expressing GFP in GABA neurons under control of the GAD67 promoter. The VMH is darker than the surrounding area, but isolated GABA cells can be found. Scale bar, 60 μm. H, Higher magnification in the area of the small arrow in G. Small arrows indicate GABA-GFP neurons. Scale bar, 28 μm. 3V, Third ventricle; ARC, arcuate nucleus.
We also examined sections of the VMH from another transgenic mouse that expressed GFP selectively in GABA neurons under control of the GAD67 promoter (Acuna-Goycolea et al., 2005). A small number of GABA-GFP neurons were detected within the VMH, but a higher number was found around the periphery of the VMH and in the arcuate nucleus (Fig. 2G,H).
Other neurons in the hypothalamus also expressed GFP, consistent with previous descriptions of vGluT2 distribution (Fremeau et al., 2001, 2004; Ziegler et al., 2002). Regions that contained vGluT2 GFP neurons included the dorsomedial nucleus, preoptic area, medial and lateral septum, lateral hypothalamus, arcuate nucleus, and anterior hypothalamus. GFP-positive cells were not found among the GABA neurons of the suprachiasmatic nucleus (SCN), although positive cells were found at the lateral and dorsal borders of the SCN; similarly, only a small number of GFP-positive neurons were found in the arcuate nucleus adjacent to the VMH, consistent with in situ hybridization (Fig. 2F) and with previous reports of vGluT2 expression. Other positive cells were found in the cortex, amygdala, and thalamus (Huang et al., 2006). In contrast, neurons that were known to express vGluT1 and not vGluT2, for instance the CA3 pyramidal cells of the hippocampus (Fremeau et al., 2001), were negative for GFP in this transgenic mouse.
GFP was found in both cell bodies and in dendrites of the VMH. Consistent with previous reports based on Golgi impregnations (Millhouse, 1973a,b), dendrites of GFP-positive neurons consistently extended beyond the borders of the VMH as defined by the cell body density into the cell-poor VMH shell (Figs. 1, 3). In fact, the dendrites of green VMH neurons extended hundreds of micrometers outside the VMH (Fig. 3A), suggesting that synaptic input to these cells is very unlikely to be restricted to those axons entering the VMH core.
Figure 3.
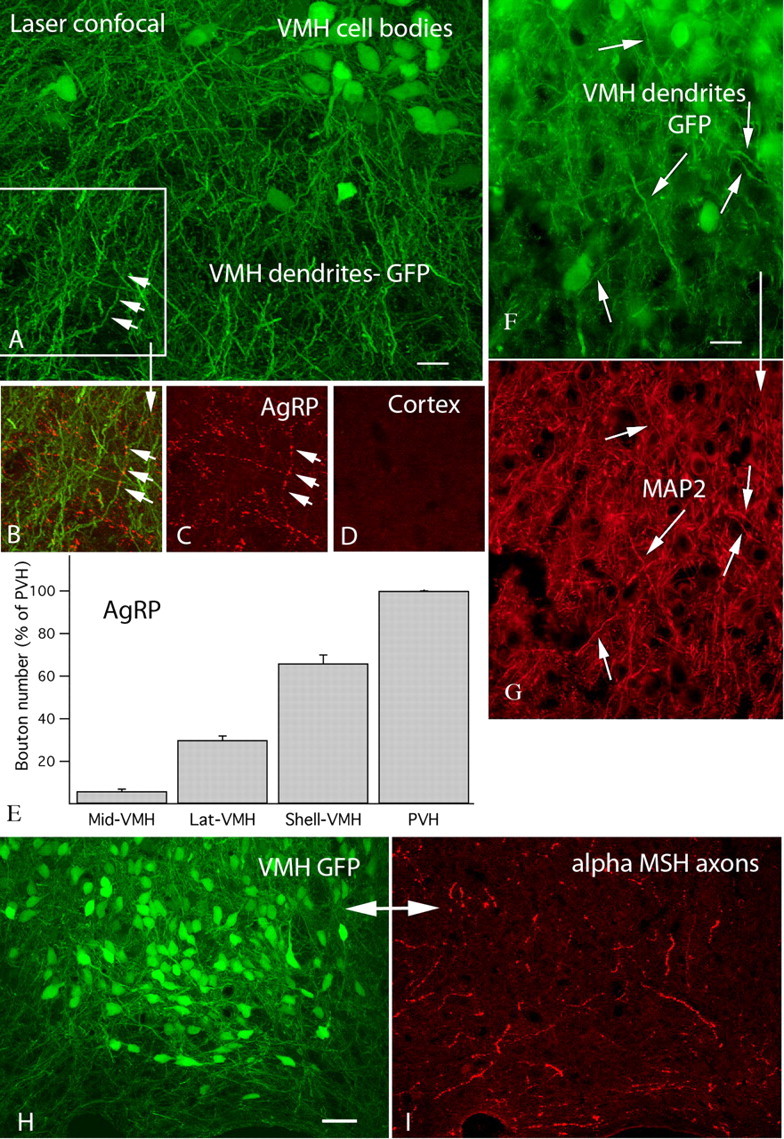
AgRP immunoreactive axons abundant in shell of VMH. A, Laser confocal micrograph of VMH dendrites exiting the ventrolateral region of the nucleus. The dendrite shown by three arrows is also seen in B and C. Scale bar, 16 μm. B, Red AgRP-immunoreactive boutons run along a green VMH dendrite making numerous appositions. C, Only the red AgRP-immunoreactive axons are shown. D, Control section in cortex shows no immunoreactive axons. E, Relative number of AgRP-immunoreactive axonal boutons. Note the higher bouton density in the lateral VMH and shell. Error bars indicate SEM. F, GFP-expressing dendrites in shell of the VMH. Scale bar, 13 μm. G, Immunostaining with the dendritic marker MAP2 labels the same processes, consistent with the view that the processes are dendrites. H, GFP-expressing cells and processes in the ventrolateral VMH. Scale bar, 25 μm. I, In the same field as H, immunoreactive α-MSH axons are found in and around the ventrolateral VMH.
Immunocytochemistry: AgRP-immunoreactive axons innervate VMH dendrites
Previous reports have suggested that the VMH, generally defined as the core of the nucleus, contained only a low number of AgRP axons but that immunoreactive axons surrounded the nucleus (Broberger et al., 1998; Haskell-Luevano et al., 1999). However, we find that VMH neurons send long dendrites outside the nucleus into the shell of the VMH (Fig. 3A), and the VMH shell has a relatively high density of AgRP axons, many of them in apposition to GFP-positive VMH dendrites (Fig. 3 A–C). That the GFP-expressing processes in the shell of the VMH were dendrites was confirmed using antisera against the dendritic marker microtubule-associated protein 2 (MAP2) (Fig. 3F,G). To examine the relative density of AgRP-immunoreactive boutons relative to the VMH, we counted immunoreactive bouton profiles labeled with Texas Red, based on counting immunoreactive boutons in squares 150 μm on a side. In the dorsomedial part of the VMH, we found only a small number of boutons (6.2 ± 1.1 SEM/square). In striking contrast, in the ventrolateral VMH, we found 31.4 ± 2.0. In the area just outside the lateral VMH, but in an area with a very high density of green VMH dendrites, we found 68.4 ± 4.2 boutons (Fig. 3B,C,E). The hypothalamic paraventricular nucleus (PVH), previously described as expressing a high density of AgRP axons had 104 ± 5.6 boutons; the cortex showed few or no AgRP-immunoreactive axons (Fig. 3D). We also examined VMH sections immunostained for α-MSH. The ventrolateral region of the VMH, and the VMH shell containing a high number of VMH dendrites showed substantive axons and boutons immunoreactive for α-MSH (Fig. 3H,I).
Membrane properties of vGluT2 cells in VMH
Using whole-cell recording in hypothalamic slices, we characterized the membrane properties of GFP-expressing cells in VMH. Because the density of AgRP immunoreactive boutons was greater in the ventrolateral VMH, and even higher in the dendritic fields of the ventrolateral VMH, we focused our recordings on this region of the VMH. Of 66 recorded cells, 44 cells fired spontaneously at 1.7 ± 0.3 Hz (0.1–9.3 Hz) at resting membrane potential (−53.0 ± 0.5 mV) and 4 of these 44 cells showed burst firing (Fig. 4A1–A3), whereas 22 other neurons were silent at rest (−63.0 ± 1.7 mV). In six of six silent cells, 10–40 pA current injection evoked action potentials corroborating the neuronal phenotype of the recorded green cells (Fig. 4A4). The average resting membrane potential was −57.0 ± 0.8 mV (−48.3 to −75.5 mV; n = 66), measured in the first 3 min after achieving whole-cell recording, and during the interspike interval. The mean amplitude and duration of afterhyperpolarization measured from the 40 spontaneously firing cells (not including bursting cells) was 9.9 ± 0.4 mV and 201 ± 12 ms, respectively. The firing pattern and resting membrane potential value are similar to that of general VMH neurons reported previously (Priestley, 1992). Slightly more than one-half of the cells (13 of 22) fired continuously after injection of a 3 s square current pulse of 40 or 120 pA, and showed spike frequency adaptation (i.e., a progressive increase in the spike interval was observed over the 3 s stimulation) (Fig. 4B1,B2). Nine cells did not fire continuously after the current injection, and rapidly returned to an inactive state (Fig. 4B3) (Boden and Hill, 1988). The mean input resistance was 591 ± 26 MΩ (range, 320–1280 MΩ; n = 48) calculated from the linear part of the slope of the current–voltage relationship between −60 and 0 mV (Fig. 4C1,C2), which is higher than that of general VMH neurons measured using intracellular recording (Minami et al., 1986; Priestley, 1992). In 13 of 23 cells, outward current injections into cells that were hyperpolarized negative to −70 mV generated a low-threshold spike (LTS) on which one or more fast spikes rode (Fig. 4C1). A similar phenomenon was observed in one type of glucose-responsive neurons in guinea pig VMH (Minami et al., 1986). The LTS was calcium dependent because it persisted in the presence of TTX (0.5 μm) but was blocked by nickel (200 μm) (n = 4) (Fig. 4C3).
Figure 4.

Membrane properties of vGluT2 neurons. A1, Spontaneous firing of a GFP-VMH neuron at resting membrane potential (−52.5 mV). A2, Single action potential from A1. A3, A cell showing spontaneous bursting firing at rest (−55.1 mV). A4, Silent neuron at resting membrane potential of −72.5 mV (top trace), which fired on current injection of 20–40 pA (bottom trace). B1, B2, Voltage responses of a GFP-neuron to 3 s step current injections of 40 and 120 pA. B3, A cell failed to fire continuously during the 3 s current injection of 120 pA. C1, Voltage traces evoked by a step current injection from −100 to 10 pA. C2, Mean current–voltage relationship of 20 GFP-neurons. C3, Traces show that a LTS was evoked when the cell recovered from a hyperpolarization to −75 mV; the LTS persisted in TTX, but was abolished by NiCl2 (200 μm).
In experiments described below, the majority of the cells that were used were spontaneously firing cells, and a minority were nonfiring at rest. Bursting cells were not included in this analysis.
Response to amino acid transmitters
All GFP-neurons recorded in the VMH responded to focal application of the glutamate receptor agonists AMPA and NMDA, and the GABAA receptor agonist muscimol. In current-clamp recording, AMPA (30 μm) evoked a burst of spikes and depolarized the membrane potential by 8.8 ± 1.8 mV (n = 5) (Fig. 5A), and NMDA (50 μm) also increased the spike frequency and induced a 1.7 ± 0.5 mV depolarization (n = 9) (Fig. 5B). The GABAA receptor agonist muscimol (30 μm) attenuated the spike frequency and hyperpolarized the membrane potential by 15.4 ± 2.7 mV (n = 4) (Fig. 5C). With voltage clamp, both spontaneous excitatory and inhibitory synaptic activity was found in these neurons. EPSCs were recorded with KMeSO4 in the pipette solution and BIC in the bath, and could be completely blocked by the glutamate antagonists AP5 (50 μm) and CNQX (10 μm) (Fig. 5D). In the presence of AP5 and CNQX, IPSCs were recorded using KCl pipette solution; IPSCs were completely blocked by 30 μm BIC (Fig. 5E).
Figure 5.
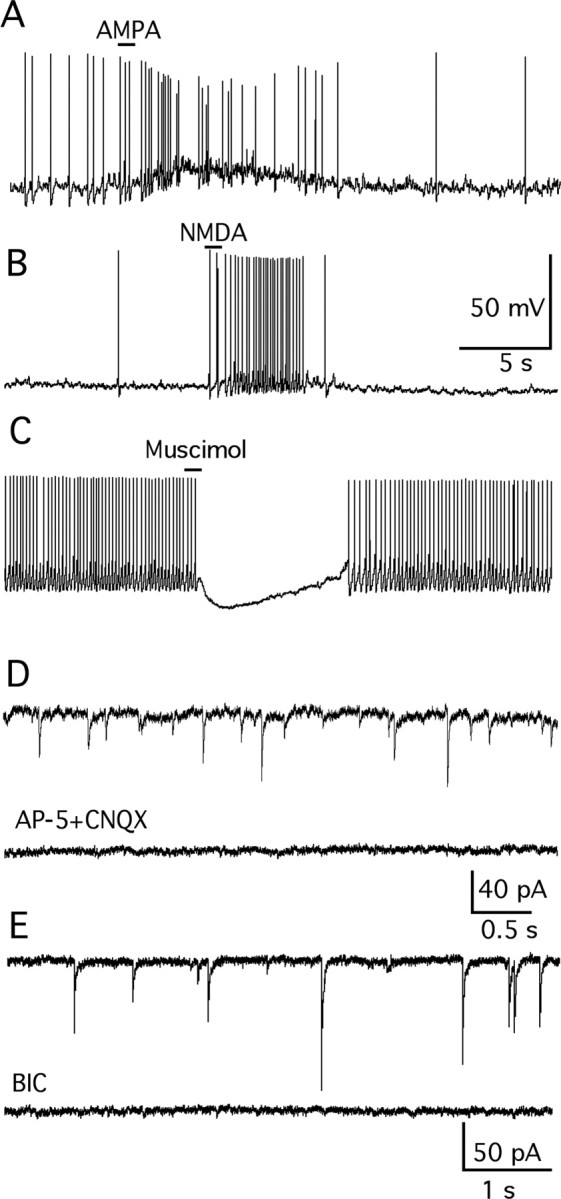
Response of vGluT2 neurons to amino acid neurotransmitters. A, Glutamate receptor agonist AMPA (30 μm) induced a robust increase of spikes and depolarized the membrane potential in current-clamp recording at rest (−54 mV). B, NMDA (50 μm) evoked a burst of spikes and a depolarization in a cell at rest (−51.5 mV). C, GABAA receptor agonist muscimol (30 μm) attenuated the spike frequency and hyperpolarized the membrane potential at rest (−58.8 mV). D, Spontaneous EPSCs recorded with KMeSO4 in the pipette solution and BIC (30 μm) in the bath in voltage clamp (top trace). EPSCs were completely blocked by application of glutamate antagonists AP5 (50 μm) and CNQX (10 μm) (bottom trace). E, Spontaneous IPSCs were recorded in the presence of AP5 and CNQX and using KCl in the pipette solution (top trace). IPSCs were completely blocked by 30 μm BIC (bottom trace).
Application of AP5 (50 μm) and CNQX (10 μm) to block the actions of synaptic glutamate greatly reduced the spike frequency of VMH neurons (by 93.9 ± 3.8%; p < 0.05; t test; n = 5), indicating glutamate synaptic input plays an important role in determining the activity of these cells.
α-MSH and MTII inhibit vGluT2 neurons in VMH
The hypothalamic MC3R and MC4R are important in the regulation of food intake and metabolism (Fan et al., 1997; Chen et al., 2000a,b; Butler and Cone, 2002). These two receptors have been reported in the VMH (Roselli-Rehfuss et al., 1993; Jegou et al., 2000; Kishi et al., 2003, 2005), suggesting that melanocortin peptides might modulate VMH neuronal activity. α-MSH is one of the cleavage products of POMC and an endogenous ligand for melanocortin receptors and appears to play a tonic inhibitory role in feeding and energy storage (Poggioli et al., 1986). The effect of α-MSH on VMH vGluT2 neurons was studied. Of 10 cells tested, 8 cells were inhibited by a 1 min application of α-MSH (0.1–0.5 μm). The membrane potential was hyperpolarized by 2.8 ± 0.6 mV in these cells (p < 0.05; ANOVA), and the spike frequency was decreased by 66.0 ± 8.3% in four spontaneously firing cells (p < 0.05; ANOVA) (Fig. 6A). To ensure that the inhibition was not attributable to changing solutions, a different neuropeptide, hypocretin-2 (200 nm), was applied under current clamp; no consistent effect was observed (spike frequency, 95 ± 23% of control; p > 0.05; n = 4; ANOVA), suggesting that the α-MSH inhibition was a real effect and not attributable to perfusion artifact.
Figure 6.
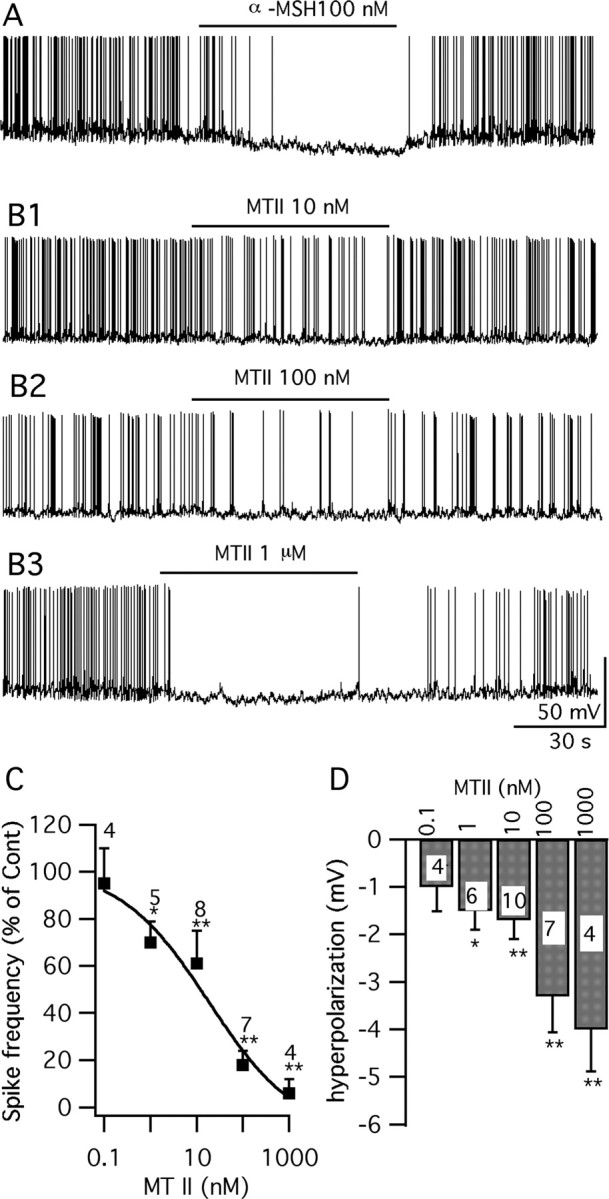
α-MSH and MTII inhibit vGluT2 neurons. A, A trace showing that α-MSH (100 nm) depressed vGluT2 neurons. B1–B3, Traces showing that 10 nm ∼ 1 μm MTII inhibits the firing and hyperpolarizes the membrane potential of GFP-VMH neurons. C, Spike frequency dose-dependent response of MTII. D, Bar graph showing the hyperpolarization of membrane after application of MTII (0.1 nm ∼ 1 μm) (*p < 0.05; **p < 0.01; ANOVA). Error bars indicate SEM. n is shown as a number inside each bar.
MTII, a synthetic analog of α-MSH, is considered a nonspecific melanocortin agonist of MC3R and MC4R, and decreases food intake when injected intracerebroventricularly (Fan et al., 1997; Jonsson et al., 2002). Here, we studied the effect of MTII on GFP-neurons in VMH slices from 2- to 3-week-old mice. Under current-clamp recording conditions, MTII (1 nm to 1 μm) inhibited all 19 cells tested (Fig. 6 B–D). In these cells, MTII evoked a statistically significant hyperpolarization and decreased spike frequency. The reduction of spike frequency was dose-dependent with an IC50 of 18.0 nm (Fig. 6C,D). In parallel experiments, when we used older adult mice, 5–6 weeks of age, to generate brain slices, MTII (100 nm) similarly decreased the firing rate by 64 ± 17% (p < 0.05; n = 5; ANOVA), and hyperpolarized the membrane by 2.8 ± 0.7 mV (p < 0.05; n = 5; ANOVA). Thus, adult mice showed the same response as slightly younger mice. The inhibition of MTII on these neurons was completely reversed after a 1–2 min washout period (Fig. 6B1–B3). Inhibition by MTII is not without precedent and has been suggested in the noradrenergic locus ceruleus neurons (Kawashima et al., 2003).
To study the possible mechanisms of MTII modulation on vGluT2 VMH neurons, glutamate and GABA ionotropic receptor antagonists were used to block the presynaptic input. In the presence of AP5 (50 μm), CNQX (10 μm), and BIC (30 μm), MTII (100 nm) failed to affect the activity of the GFP-expressing VMH neurons (Fig. 7 A–C). No significant change of spike frequency (108 ± 5.0% of control) or membrane potential (−0.3 ± 0.3 mV) was observed after application of MTII 100 nm in this condition (p > 0.05; n = 5; ANOVA) (Fig. 7C). The effect of MTII was further investigated in the presence of TTX (0.5 μm) to block the spontaneous firing along with the glutamate and GABA blockers. No detectable change of the membrane potential (−0.4 ± 0.4 mV; p > 0.05; n = 7; ANOVA) or input resistance (control, 465 ± 66 MΩ; MTII, 445 ± 61 MΩ; p > 0.05; n = 9; t test) was induced by MTII at 0.1–1 μm in GFP-expressing VMH neurons under these conditions (Fig. 7D,E). These results suggest that MTII might modulate the vGluT2 VMH neurons via a presynaptic mechanism.
Figure 7.

No inhibitory activity of MTII on vGluT2 neurons was found when synaptic transmission was blocked. A, Trace showing that the vGluT2 neuron is not inhibited by MTII in the presence of glutamate and GABA receptor antagonists, AP5, CNQX, and BIC, respectively. B, Time course of the response to MTII on spike frequency in the neuron shown in A. C, Bar graph showing that the spike frequency is not decreased by MTII in the presence of AP5, CNQX, and BIC. Error bars indicate SEM. D, Representative trace showing little effect of MTII on membrane potential in the presence of TTX, AP5, CNQX, and BIC. E, MTII did not change the input resistance (n = 9).
We therefore investigated the effect of MTII on spontaneous EPSCs and IPSCs in GFP- VMH neurons. In the presence of AP5 (50 μm) and CNQX (10 μm), IPSCs were recorded at a holding potential of −60 mV using a KCl pipette solution. MTII increased IPSCs in these cells (Fig. 8A1–A3). The frequency of IPSCs was increased by 26.3 ± 9.3% with 1 min application of MTII (100 nm) and then reversed to 111 ± 14.1% of the control after an ACSF wash (p < 0.05; n = 5; ANOVA). This is consistent with a previous report showing that MTII increased IPSCs in hypothalamic paraventricular neurons (Cowley et al., 1999). Next, the effect of MTII on mIPSCs was studied by using TTX (0.5 μm) along with AP5 (50 μm) and CNQX (10 μm) to block the spike-dependent synaptic currents. MTII (100 nm) reversibly increased the mIPSC frequency by 30.7 ± 6.3%, which recovered to 98.0 ± 9.9% of the control (p < 0.01; n = 5; ANOVA) (Fig. 8B1,B2). No significant change in the amplitude of mIPSCs was found after application of MTII (Fig. 8B3). In addition, the decay kinetics of mIPSCs were analyzed. mIPSCs before, during, and after MTII application were averaged and fit to an exponential curve, from which the decay time constant (τ) was obtained. No significant change in the decay kinetics was induced by MTII at 100 nm (τ = 10.4 ± 0.5, 10.8 ± 0.4, and 11.1 ± 0.4 ms for control, MTII, and washout, respectively; p > 0.05; ANOVA; n = 5).
Figure 8.

MTII reversibly increased IPSCs and mIPSCs in GFP-VMH neurons. A1, Representative traces showing the effect of MTII (100 nm) on IPSCs in a typical GFP-VMH cell. A2, Time course of MTII-induced enhancement of IPSCs. A3, Bar graph showing the mean facilitation of MTII on IPSCs (*p < 0.05; ANOVA). Error bars indicate SEM. B1, MTII (100 nm) increased mIPSCs in a typical GFP-VMH neuron in the presence of TTX (0.5 μm). B2, Bar graph showing the mean enhancement of mIPSCs after application of MTII (*p < 0.05; ANOVA). B3, MTII did not change the amplitude of mIPSCs.
Spontaneous EPSCs were recorded using KMeSO4 pipette solution with BIC (30 μm) in the bath. Under these conditions, MTII (100 nm) did not change the frequency of EPSCs (101 ± 9.8% of control; p > 0.05; n = 6; ANOVA). These results suggest that MTII might depress glutamatergic neurons in the VMH by facilitating presynaptic GABA release onto these neurons. Consistent with this view, application of MTII (100 nm) in the presence of BIC (30 μm) had no effect on the firing rate of GFP-VMH neurons (106 ± 11.3% of control; p > 0.05; n = 6; ANOVA). Previous work showed membrane depolarization by MTII in MC4R-expressing cells in the dorsal medial hypothalamus (Liu et al., 2003) and in arcuate nucleus POMC neurons or insulin receptor substrate-2 neurons (Choudhury et al., 2005; Smith et al., 2007). Both the arcuate and dorsomedial nucleus were contained in the same slices as the VMH, and both regions contain numerous GABA neurons (Acuna-Goycolea et al., 2005). Therefore, some of the MTII-increased GABA input to the VMH may arise from these loci.
SHU9119 is a selective antagonist of MC3/4R (Hruby et al., 1995). In six GFP-VMH neurons, after obtaining stable recordings of IPSCs, we pretreated the cell with SHU9119 before applying MTII. After 1 min application of SHU9119 (500 nm), the frequency of IPSCs decreased to 84 ± 5% of control (p < 0.05; ANOVA), suggesting there is a tonic facilitation of GABA release by endogenous MC3/4 receptor agonists. The increase in IPSC frequency normally found with MTII (100 nm) was blocked in the presence of SHU9119 (500 nm) (94 ± 5%; p > 0.05; t test; n = 6). These results suggest that the increase in IPSCs induced by MTII alone is blocked by the pharmacological antagonist of the MC3/4 receptors.
Effects of MC4R and MC3R selective agonists on glutamatergic neurons
The MC4R is thought to play a major role in the regulation of body weight. Targeted deletion of MC4R resulted in hyperphagia and obesity (Huszar et al., 1997), whereas the anorexic effects of MTII were greatly diminished in MC4R knock-out mice (Marsh et al., 1999). MC3R is thought to play a complementary role to MC4R by regulating feeding efficiency and partitioning of nutrients into fat (Chen et al., 2000a). It has also been suggested that MC3/4R show different receptor–ligand interactions (Lee et al., 2001) and may couple to differential downstream signaling systems (Konda et al., 1994). To study whether MC3/4R activation shows different effects in vGluT2 VMH neurons, selective MC4R and MC3R agonists were used. Cyclo(β-Ala-His-d-Phe-Arg-Trp-Glu)-NH2, which shows a 90-fold selectivity over MC3R and >2000-fold selectivity over MC5R (Bednarek et al., 2001) was used as a selective MC4R agonist. Two agonist concentrations were used to identify the MC4R activating effect. One minute application of this MC4R agonist (10 nm and 1 μm) evoked a consistent statistically significant depression of GFP-VMH neurons (Fig. 9A,C,D). d-Trp8-γ-MSH was used as a selective agonist of MC3R given its 40- to 200-fold selectivity over MC4R and MC5R (Grieco et al., 2000; Cowley et al., 2001). In all cells tested, d-Trp8-γ-MSH (10 nm) (1 min) also showed a consistent inhibition of GFP-VMH neurons (Fig. 9 B–D). The inhibitory effect of d-Trp8-γ-MSH is consistent with its effect on increasing miniature IPSC frequency in POMC neurons (Cowley et al., 2001). Our results indicate that, in glutamatergic neurons in the VMH, MC3R and MC4R activation induced similar effects.
Figure 9.
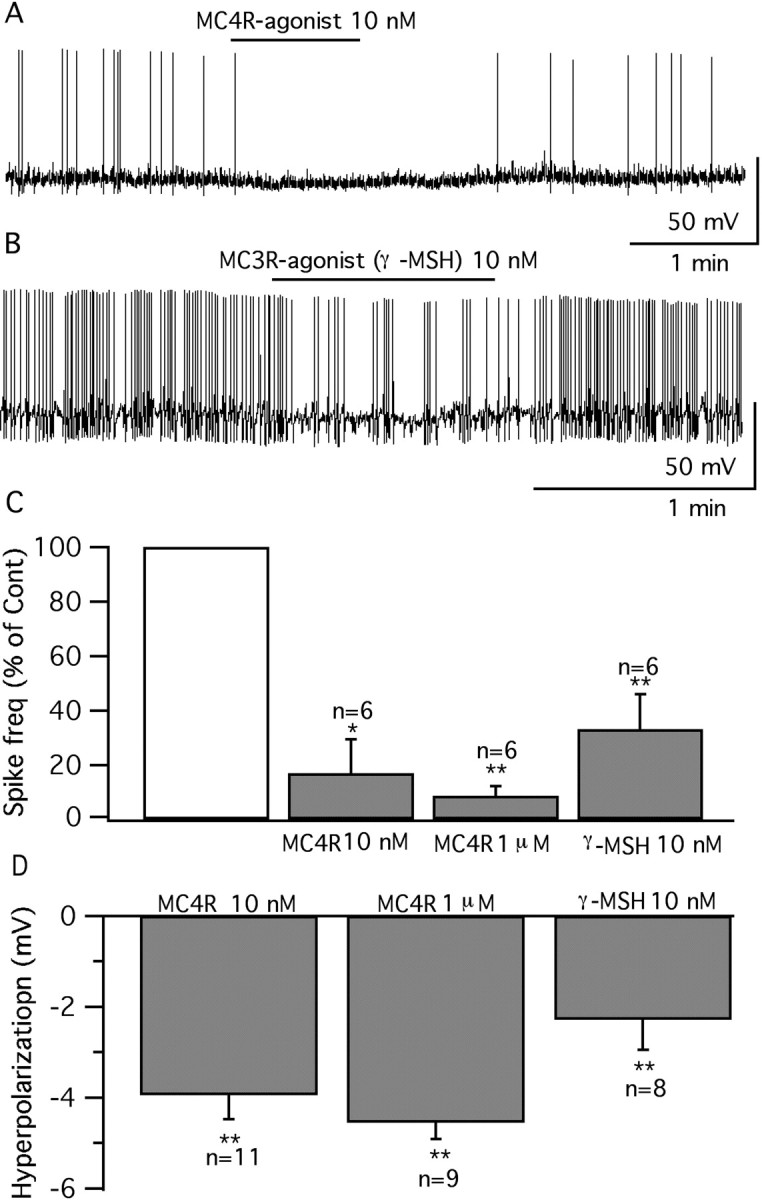
Selective MC4R and MC3R agonists both depress vGluT2 neurons. A, A cell depressed by MC4R agonist (10 nm) under current-clamp recording. B, A cell depressed by MC3R agonist d-Trp8-γ-MSH (10 nm). C, Bar graphs showing the average spike frequency after application of MC4R agonist (10 nm, 1 μm) and γ-MSH (10 nm) (control, 100%). *p < 0.05; **p < 0.01; ANOVA. Error bars indicate SEM. D, Mean hyperpolarization of the membrane potential by MC4R agonist (10 nm and 1 μm) and γ-MSH (10 nm). **p < 0.01; ANOVA.
AgRP depresses glutamatergic neurons in VMH
AgRP, another important member of the melanocortin system, is colocalized with NPY in the same set of arcuate nucleus neurons (Broberger et al., 1998; Marks and Cone, 2003), and these neurons send dense projections to a number of hypothalamic areas, including as we showed previously, the VMH shell. The axonal pathway of the POMC neurons converges in many of the same regions (Broberger et al., 1998; Haskell-Luevano et al., 1999). AgRP has been demonstrated to be a specific competitive antagonist of the MC3/4R (Fong et al., 1997; Ollmann et al., 1997), and intracerebral administration of AgRP stimulates feeding (Rossi et al., 1998). Here, we investigated the effect of AgRP on GFP-expressing glutamatergic neurons in VMH. We used three concentrations of AgRP (1, 10, and 100 nm). Similar to MTII, AgRP depressed glutamatergic VMH neurons dose-dependently (Fig. 10A1–C). The effect of AgRP completely recovered after 1–3 min peptide washout. Similarly, when we used older adult mice of 5–6 weeks, AgRP exerted the same inhibitory effect. AgRP (100 nm) decreased the firing rate in adult slices by 74 ± 13% (p < 0.05; n = 6; ANOVA), and hyperpolarized the membrane by 2.9 ± 0.4 mV (p < 0.05; n = 6; ANOVA). Our results suggest that AgRP, similar to MTII, shows an inhibitory effect in glutamatergic neurons in the VMH.
Figure 10.

Opposite effects of AgRP and SHU9119 on vGluT2 neurons. A1–A3, AgRP (1–100 nm) inhibited the firing and hyperpolarized the membrane of vGluT2 VMH neurons. B, Bar graph showing the dose-dependent depression on average spike frequency by AgRP (1–100 nm) (control, 100%; *p < 0.05; **p < 0.01; ANOVA). Error bars indicate SEM. C, Bar graph showing the hyperpolarization of AgRP (1–100 nm) on GFP neurons. D, Typical trace showing that AgRP (100 nm) does not change the membrane potential with TTX (0.5 μm), AP5 (50 μm), CNQX (10 μm), and BIC (30 μm) in the bath solution. E, Cell that was excited by SHU9119 (SHU) (100 nm). F, Bar graph showing the percentage of spike frequency increased by SHU9119 (SHU) (100 nm) on these cells (*p < 0.05; ANOVA).
The depression of firing after application of AgRP on glutamatergic neurons in the VMH may be attributable to a presynaptic effect, because no hyperpolarization was observed with bath perfusion of TTX (0.5 μm), AP5 (50 μm), CNQX (10 μm), and BIC (30 μm) (−0.2 ± 0.4 mV; p > 0.05; n = 8; ANOVA) (Fig. 10D). Neither was a significant change observed in membrane input resistance (control, 726 ± 126 MΩ; AgRP, 693 ± 124 MΩ; p > 0.05; n = 6; t test).
In contrast to the actions of AgRP, a 1 min application of MC3/4R antagonist, SHU9119 (100 nm), increased the spike frequency in neurons that showed spikes (9 of 11 showed spikes; the other 2 were silent) by 199 ± 61% (p < 0.05; ANOVA) (Fig. 10E,F). SHU9119 shifted the spike threshold in a negative direction by 1.6 ± 0.3 mV (from −44.4 ± 1.3 mV to −46.0 ± 1.3 mV; p < 0.05; t test; n = 9), increasing the probability of action potentials. Consistent with its effect on IPSCs, this result also suggests that glutamatergic neurons in the VMH are under tonic inhibitory modulation by melanocortin receptors. In the presence of TTX, AP5, CNQX, and BIC, SHU9119 (100 nm) did not evoke a significant change in the membrane potential of GFP-VMH neurons (−0.3 ± 0.5 mV; p > 0.05; n = 4; ANOVA), suggesting that it has an indirect, rather than a direct, effect on these neurons.
AgRP does not block the effect of MTII on vGluT2 neurons in VMH
AgRP has been suggested as an endogenous antagonist of the MC3/4R (Fong et al., 1997; Ollmann et al., 1997); we therefore tried applying MTII and human AgRP together on GFP-VMH neurons, to determine whether they can block or attenuate each other's effect. In six cells that were inhibited by both MTII (10 nm) (49.3 ± 10.4%; p < 0.05; ANOVA) and AgRP (10 nm) (63.7 ± 8.1%; p < 0.05; ANOVA), coadministration of these two peptides generated a greater level of inhibition than either alone (MTII plus AgRP inhibition by 74.5 ± 13.3%; p < 0.05; ANOVA) (Fig. 11A1–C). In another seven cells, AgRP (10 nm) was preapplied for 1–1.5 min before adding MTII (10 nm) for 1 min. Again, the effect of AgRP was not reversed or attenuated by MTII. The spike frequency was inhibited by 64 ± 11% (p < 0.01; ANOVA) during the AgRP application, and additional inhibition (by 82 ± 11% vs control) was found after adding MTII (p < 0.01; ANOVA) (Fig. 11D). Coapplication of AgRP and MTII reduced the spike frequency more than AgRP alone (p < 0.05; ANOVA) (Fig. 11E). We also examined the effect of both compounds on inhibitory synaptic activity. Under voltage clamp, preadministration of AgRP (200 nm) for 1–1.5 min, did not block the increase in IPSCs found when MTII (100 nm) was added together with AgRP (126 ± 3.6% compared with AgRP alone; p < 0.05; n = 7; ANOVA). Together, these results suggest that instead of blocking the effect of MTII, as might be expected if AgRP antagonized the MTII action, AgRP showed a similar and additive effect with MTII on these VMH neurons. AgRP therefore does not act as an antagonist of MC3/4R in glutamatergic VMH neurons, but rather has the opposite action. Alternatively, AgRP may act on receptors independent of the known melanocortin system (Marsh et al., 1999; Pritchard and White, 2005).
Figure 11.
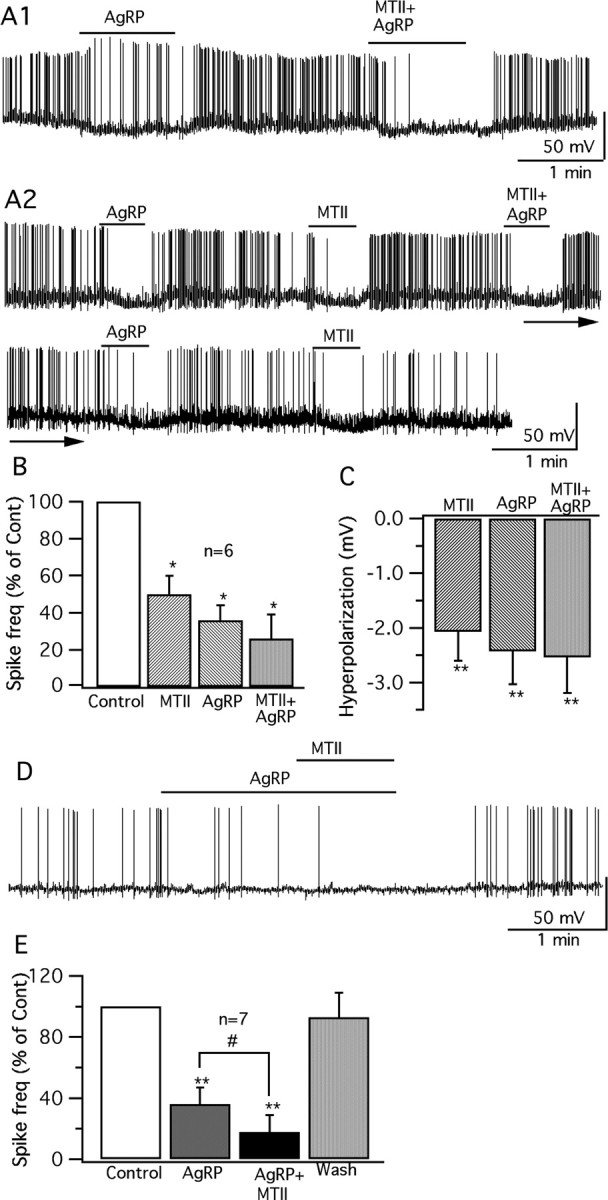
Additive effect of AgRP and MTII on vGluT2 neurons. A1, A vGluT2 cell inhibited by AgRP (10 nm) was inhibited further when both MTII (10 nm) and AgRP (10 nm) were applied. A2, Trace showing that both MTII and AgRP, at 10 nm concentration, show inhibitory effect when applied sequentially on the same cell; coadministration of these two drugs also depresses the cell; and the effects of these two drugs are repeatable. B, Bar graph shows the decrease in spike frequency with MTII (10 nm) and AgRP (10 nm) applied alone and together (*p < 0.05; n = 6; ANOVA). Error bars indicate SEM. C, Bar graph shows the hyperpolarization of the membrane after application of MTII (10 nm) and AgRP (10 nm) alone and together. D, A trace showing that, after pretreatment with AgRP (10 nm), a vGluT2 neuron was still further inhibited after adding MTII (10 nm). E, Bar graph comparing the spike frequency during application of AgRP (10 nm) alone and coapplication with MTII (**p < 0.01; #p < 0.05; n = 7; ANOVA).
In the paragraph above, human AgRP was used; in parallel experiments, we also tested the effect of mouse AgRP on VMH glutamatergic neurons. Mouse AgRP evoked consistent inhibitory effects on these neurons. Mouse AgRP (10 nm) decreased the firing rate by 53 ± 10% (p < 0.01; n = 9; ANOVA), and hyperpolarized the membrane potential by −2.1 ± 0.5 mV (p < 0.05; n = 9; ANOVA). In six cells, mouse AgRP (10 nm), MTII (10 nm), and a combination of these two drugs were applied sequentially in alternate orders on each cell; all cells were inhibited by both MTII (10 nm) (45.2 ± 8.8% reduction in firing rate; p < 0.05; ANOVA) and AgRP (10 nm) (46.7 ± 10.4% reduction in firing rate; p < 0.01; ANOVA). Coadministration of these two peptides generated a greater level of inhibition than either alone (64.2 ± 10.1% reduction in firing rate; p < 0.01; ANOVA). These results suggest that mouse and human AgRP evoked similar inhibitory actions on VMH glutamatergic neurons.
AgRP inhibition in vGlut2 neurons in VMH is Gi/o-protein dependent
Because no direct effect of AgRP on vGlut2 neurons was found, we studied the effects of AgRP on glutamatergic and GABAergic synaptic input to the GFP-expressing VMH neuron. Under voltage-clamp recording, spontaneous and miniature postsynaptic currents were recorded as described previously. AgRP (100 nm) decreased the frequency of EPSCs by 36.6 ± 4.3%, with a recovery to 85.5 ± 5.0% in 1–3 min (p < 0.01; ANOVA; n = 9) (Fig. 12 A–C). Because AgRP did not act via MC3/4 receptors, which are coupled to Gs- and/or Gq-proteins (Konda et al., 1994; Lee et al., 2001; Srinivasan et al., 2003), we investigated the possibility that AgRP might act through the Gi/o-protein pathway. The Gi/o-protein antagonists PTX and NF023 were used (Beindl et al., 1996; Freissmuth et al., 1996). Brain slices were preincubated in PTX or NF023 as described previously (DeBock et al., 2003; Garic-Stankovic et al., 2005) before using for whole-cell recording. AgRP (100 nm) failed to decrease the frequency of EPSCs significantly in either PTX-pretreated slices (84 ± 6% of control; p > 0.05; ANOVA; n = 6) or NF023-pretreated slices (94 ± 12% of control; p > 0.05; ANOVA; n = 7) (Fig. 12C), suggesting a dependence on Gi/Go-protein mediation.
Figure 12.

AgRP reduces excitatory synaptic currents by presynaptic Gi/o G-protein mediation. A, Traces showing that AgRP (100 nm) reversibly decreased the frequency of spontaneous EPSCs, recorded with BIC (30 μm) in the bath. B, Time course of AgRP (100 nm) actions on EPSCs. C, Bar graphs show the mean frequency of EPSCs before, during, and after application of AgRP (100 nm) in normal conditions and after the slices were pretreated with PTX or NF023. The effects of AgRP were significantly attenuated when slices were pretreated with PTX or NF023; the slight reduction in EPSC frequency induced by AgRP after treatment with PTX or NF023 was not significantly different (NS) from the pre-AgRP control frequency. The statistical indication placed within each set of bars indicates a within-group ANOVA comparison (i.e., AgRP caused a statistically significant decrease under control conditions). The horizontal lines above the bars (*p < 0.05; t test) indicate that AgRP under control conditions induced a statistically greater decrease in EPSC frequency than did AgRP in the presence of PTX or NF023. Error bars indicate SEM. D, Bar graphs showing the mean frequency of mEPSCs was depressed by AgRP in normal conditions (p < 0.05; n = 6; ANOVA), whereas no depression was observed after the slices were pretreated with NF023. NF023 caused a statistically significant attenuation in the reduction of EPSC frequency (**p < 0.01; t test). E, Whereas AgRP did attenuate the frequency of mEPSCs under normal conditions, it did not change the amplitude (p > 0.05; n = 6; t test).
AgRP decreased the frequency of mEPSCs by 32 ± 7% (p < 0.05; ANOVA; n = 6) without changing the amplitude (control, 9.3 ± 1.6 pA; AgRP, 8.6 ± 1.6 pA; p > 0.05; n = 6; t test) (Fig. 12D,E). In NF023-pretreated slices, AgRP failed to change the frequency (103 ± 8% of control) (Fig. 12D) and amplitude of mEPSCs (12.4 ± 0.9 pA for AgRP vs 12.0 ± 0.7 pA for control; p > 0.05; ANOVA; n = 5). Our results suggest that AgRP attenuates presynaptic glutamate release, at least, partially via the Gi/o G-protein pathway.
Calcium channels, especially N- and P/Q-type channels, are involved in neurotransmitter release from presynaptic terminals (Turner et al., 1993), and they are also downstream targets of the Gi/o-protein signal pathway. To determine whether calcium channels are involved in the effect of AgRP on vGluT2 cells in the VMH, Cd2+ and specific calcium blockers were used. In the presence of Cd2+ (200 um) in the bath solution, AgRP (100 nm) slightly decreased the frequency of EPSCs by 26 ± 16% (not significant; p > 0.05; ANOVA; n = 6). In the presence of ω-conotoxin GVIA (1 μm) and ω-agatoxin IVA (200 nm) in the bath solution to block the N- and P/Q-type calcium channels, the frequency of EPSCs was decreased by 20 ± 11% by AgRP (100 nm) (not statistically significant; p > 0.05; ANOVA; n = 4). These results suggest that, although AgRP may attenuate calcium currents to a modest degree, it is not a major factor.
We also tested the effect of AgRP on inhibitory synaptic activity. AgRP (100 nm) did not change the frequency of IPSCs (98.1 ± 15.5%; p > 0.05; n = 8; ANOVA). Neither the frequency (89.7 ± 8.2% of control; p < 0.05; n = 4; ANOVA) nor the amplitude (control, 48.7 ± 7.7 pA; AgRP, 48.4 ± 6.2 pA; p > 0.05; n = 4; t test) of mIPSCs was changed by AgRP. The above results suggest that AgRP may depress the activity of vGluT2 VMH neurons by decreasing presynaptic glutamate release.
In the experiments above, we found no clear evidence for direct actions of either AgRP or melanocortin agonists on VMH neurons. To ensure that this was not attributable to dialyzing some factor in the cytoplasm of the recorded cell and thereby blocking a direct effect, we also used cell-attached recording, which would not alter the intracellular milieu. These experiments were done in the presence of GABA and the glutamate receptor antagonists BIC, AP5, and CNQX to block indirect synaptic actions of the peptides. Application of AgRP or MTII had no statistically significant effect on the frequency of action currents studied with cell-attached recording (110 ± 29 and 116 ± 22% of control, respectively; n = 7; p > 0.05).
NPY inhibits vGluT2 neurons in VMH
NPY is colocalized with AgRP in the same arcuate nucleus neurons; similar to AgRP, NPY (1 μm) consistently inhibited vGluT2 neurons in VMH, decreasing the spike frequency by 66 ± 14% (p < 0.01; n = 6; ANOVA) and hyperpolarizing the membrane by 3.7 ± 1.1 mV (p < 0.01; n = 6; ANOVA) (Fig. 13A,D). The hyperpolarization of NPY was blocked by TTX (0.5 μm), AP5 (50 μm), CNQX (10 μm), and BIC (30 μm) (−0.6 ± 0.4 mV; p > 0.05; n = 6; ANOVA) (Fig. 13B). The input resistance was not changed by 1 μm NPY under these conditions [566 ± 64 MΩ (control) vs 569 ± 65 MΩ (NPY); p > 0.05; n = 5; ANOVA]. These results suggest an indirect effect of NPY on these neurons. Under voltage-clamp recording, in the presence of BIC (30 μm) in the bath, NPY (1 μm) decreased the frequency of spontaneous EPSCs by 46 ± 11% (p < 0.05; n = 6; ANOVA) (Fig. 13C,E), suggesting that NPY depressed the vGluT2 VMH neurons by inhibiting glutamate release from presynaptic excitatory neurons, similar to the effect of NPY on dopamine-responsive VMH neurons (Davidowa et al., 2002) and hypocretin neurons in the lateral hypothalamus (Fu et al., 2004), and similar to the inhibitory effect of AgRP on EPSCs we describe above.
Figure 13.
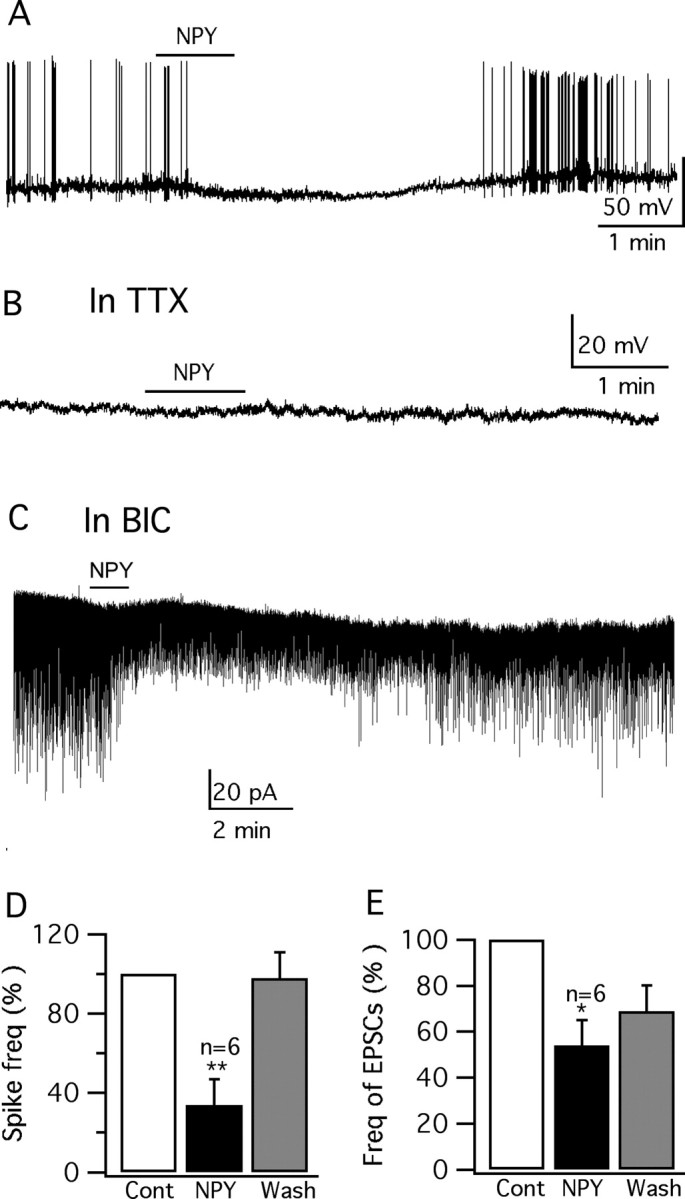
NPY inhibits vGluT2-GFP neurons. A, Trace showing that application of NPY (1 μm) inhibits the firing and hyperpolarizes the GFP-VMH cell. B, In the presence of TTX (0.5 μm) in the bath solution, no hyperpolarization is induced by NPY (1 μm). C, In the presence of BIC (30 μm), NPY (1 μm) attenuates the spontaneous EPSCs. D, E, Bar graphs showing the decrease of spike frequency and EPSCs mediated by NPY (1 μm) (*p < 0.05; **p < 0.01; ANOVA). Error bars indicate SEM.
Discussion
Because a number of neuroactive substances have been reported in the VMH, suggesting a cellular heterogeneity, we generated a transgenic mouse to detect a single type of cell, the glutamatergic neuron expressing a GFP reporter gene under control of the vGluT2 promoter (Ziegler et al., 2002; Fremeau et al., 2004). Selective expression of GFP in vGluT2-expressing VMH cells was confirmed with single-cell RT-PCR. Using whole-cell voltage-clamp and current-clamp recording, we investigated the activity of melanocortin receptor agonists and antagonists on this subset of VMH neurons. MC3/4R agonists, α-MSH, MTII, as well as selective MC4R and MC3R agonists, all depressed these neurons. Surprisingly, AgRP, a possible MCR antagonist, had the same effect as the agonists and also inhibited glutamatergic cells. In contrast, the synthesized MC3/4R antagonist SHU9119 excited these cells. Most of the activity of these compounds in the VMH appeared to be attributable to mechanisms related to modulation of synaptic input. Little direct effect of α-MSH, MTII, AgRP, or SHU9119 was detected on these neurons.
Arcuate nucleus axons target dendrites in VMH shell
Although MC receptors have been described in the VMH (Roselli-Rehfuss et al., 1993; Harrold et al., 1999; Jegou et al., 2000; Kishi et al., 2003, 2005; Liu et al., 2003; Harrold and Williams, 2006), previous reports have suggested that AgRP and α-MSH containing axons do not show a strong innervation of the VMH (Smith and Funder, 1988; Broberger et al., 1998; Cone, 1999; Haskell-Luevano et al., 1999), raising the relevant question whether the VMH normally receives afferent information from the arcuate POMC or AgRP neurons. Of potential importance in this regard, when we examined the dendrites of the vGluT2 neurons of the VMH, we found that they consistently extended out of the body of the VMH and formed a dense shell surrounding the VMH. Relatively high densities of AgRP boutons were found in the VMH shell, and red immunoreactive boutons were commonly apposed to green vGluT2-expressing dendrites in the cell-poor shell region. Similarly, immunoreactive α-MSH axons, scarce within the dorsomedial VMH, were more common in the other parts of the VMH and its shell. That much of the input to the VMH innervates the shell rather than the body of the nucleus has received little attention in recent years, but was the subject of several classic studies based on Golgi silver chromate impregnations. In Golgi studies, all neurons within the VMH were found to have dendrites that extended out of the nucleus, and many of the long afferent projections to the VMH selectively innervated the shell and not the core (Millhouse, 1973a,b).
Melanocortins depress VMH glutamate neurons
VMH neurons were inhibited by MTII, an MC3/4R agonist. This inhibition is probably indirect, because neither spike frequency, membrane potential, nor input resistance was changed by MTII when synaptic transmission was blocked. MTII significantly increased the frequency of spontaneous IPSCs and mIPSCs without changing the amplitude, suggesting that MTII may facilitate GABA release presynaptically. The increased GABAergic tone is a primary mechanism whereby MTII inhibits these excitatory cells. This mechanism of MTII inhibition in the VMH is similar to that found in the PVH (Cowley et al., 1999). MC receptors couple via Gs-proteins to activate adenylyl cyclase (cAMP) (Cone et al., 1996), which may underlie the MTII-mediated increase in IPSCs and mIPSCs.
Our results suggest that selective MC3R and MC4R activation shows very similar inhibitory effects on glutamatergic neurons. Both MC4R and MC3R agonists showed consistent inhibitory actions in all cells tested. MC3R and MC4R show different receptor–ligand interaction (Lee et al., 2001) and may couple to different downstream signaling systems (Konda et al., 1994).
SHU9119, the synthesized MC3/4R antagonist, showed excitatory actions on the cells tested, consistent with its antagonist actions on MC3/4R. SHU9119 did not appear to act on the VMH cells directly, because no effect was detected with TTX in the bath.
AgRP also depresses VMH glutamate neurons and does not antagonize MCR agonists
AgRP consistently inhibited glutamatergic VMH neurons, even at a very low concentration of 1 nm. Similar inhibitory actions were evoked by NPY, coreleased by neurons that contain AgRP (Broberger et al., 1998). We did not find excitatory actions of AgRP as reported in the dorsomedial VMH and PVH in rats (Davidowa et al., 2003; Li and Davidowa, 2004). Experiments with single-unit recording of unidentified cells in the VMH have suggested complex excitatory and inhibitory actions of MC3/4R agonists (Li and Davidowa, 2004). This diversity of response may be attributable to the complex phenotypic heterogeneity of neurons in most regions of the hypothalamus. In contrast, our data suggest that, by selectively recording from one type of cell, in our case the presumptive GFP-expressing glutamatergic neurons of the VMH, the neuronal responsiveness to AgRP and MC3/4R agonists is substantially more homogeneous.
AgRP appeared to modulate excitatory VMH neurons by a presynaptic mechanism, because no change in membrane potential or input resistance was obtained in the presence of TTX together with glutamate and GABA receptor blockers. In contrast to melanocortins that increased presynaptic GABA activity, AgRP depressed these cells by decreasing presynaptic glutamate release, reducing the frequency of EPSCs and mEPSCs with no change in mEPSC amplitude.
AgRP has been suggested to act by a mechanism of antagonizing the MC3/4R. In contrast to this view, when MTII and AgRP were applied sequentially to the same cell, they both exhibited similar inhibitory effects. Furthermore, coadministration of these two drugs to the same cell induced an additive inhibition. These results suggest that, in our study, AgRP may not act as an antagonist to MC3/4R. That the MC3/4R in the VMH can be antagonized is shown by control experiments with the synthesized antagonist SHU9119. Alternately, AgRP may act on other receptors independent of MC receptors; this possibility has been raised previously (Marsh et al., 1999; Nijenhuis et al., 2001; Pritchard and White, 2005). We addressed the question as to the mechanism of action for AgRP and found it to be based on a presynaptic inhibition of excitatory synaptic input dependent on Gi/Go-protein mediation. The source of the AgRP-sensitive glutamatergic input to the VMH has not been determined, but one possibility is these axons arise as collaterals from excitatory VMH neurons, or from the nearby dorsomedial nucleus.
It has been suggested that AgRP can in some circumstances act as an inverse agonist (Haskell-Luevano and Monck, 2001; Chai et al., 2003; Smith et al., 2007), in this case an agent that would act at the same receptor as MC agonists, but have the opposite effect. However, our data cannot be explained by inverse agonism, because AgRP did not have an effect opposite to that of MC3/4R agonists; furthermore, the combined application of an inverse agonist together with the agonist should cancel the effect of the agonist, but in our experiments, the two agents instead showed an additive effect. These data do not argue against the prevailing view that AgRP can act to antagonize the MC3/4R in other systems, but rather that the actions on excitatory cells of the VMH do not fit with a simple model of antagonism of the MC3/4 R on the cell bodies or on presynaptic terminals innervating the glutamate neurons of the VMH.
Relevance to energy homeostasis and hypothalamic function
The VMH is an important part of the CNS regulatory system in the control of food intake and body weight (Schwartz et al., 2000) and has been suggested as a “satiety” center in the mammalian hypothalamus (Brobeck, 1946; Elmquist et al., 1999). Lesions of the VMH produce hyperphagia and obesity (Keesey and Powley, 1986). The POMC and AgRP/NPY systems are sensitive to circulating signals of energy homeostasis including leptin, ghrelin, and insulin (Elias et al., 1999; Schwartz et al., 2000; Benoit et al., 2002; Chen et al., 2004), and have been suggested as driving forces in the regulation of energy homeostasis (Huszar et al., 1997; Cone, 1999). In the context of the VMH operating to reduce food intake, we show that AgRP/NPY neurons project to the shell of the VMH, and the inhibition of excitatory VMH neurons by both AgRP and NPY is consistent with the putative orexigenic role of these neurons. Because the VMH projects to the POMC neurons (Sternson et al., 2005), a possible model might suggest the AgRP/NPY inhibition of excitatory neurons in the VMH may secondarily serve to reduce excitation of the POMC cells, thereby further reducing the anorexigenic tone during times of lowered energy stores.
Our results indicate that α-MSH, the synthesized α-MSH analog MTII, and the selective MC4R and MC3R agonists all showed inhibitory actions on glutamatergic VMH neurons. This is not consistent with their anorexigenic activity, because inhibition of the VMH may be expected to increase food intake. It is possible that the VMH may not be a key nucleus for melanocortin modulation of feeding under normally fed baseline conditions. MC3/4R may not be restricted to a role in food intake. A number of experiments suggest that MC3/4R may also play a central role in anxiety, depression, reproduction, stress, pain sensation, blood pressure, heart rate, and drug addiction (Gonzalez et al., 1996; Bellasio et al., 2003; Chaki and Okuyama, 2005; Chaki et al., 2005; Cone, 2005). The inhibitory actions of MC3/4R agonists in the VMH may play a role in these other states. Consistent with this view, melanocortin injections into the VMH have been reported to increase anxiety and aggression (Gee et al., 1983; Chaki and Okuyama, 2005); in parallel, animals with VMH lesions are often quite aggressive (Stellar, 1954; Keesey and Powley, 1986). Additional functional studies of melanocortins and AgRP in the VMH should further clarify the roles of these neuropeptides in this region. Finally, because AgRP and melanocortins appear to act on different presynaptic axons within the VMH, the relative effect of each peptide will in part be dependent on the level of activity of each set of presynaptic neurons, which may vary in different physiological states.
Footnotes
This work was supported by National Institutes of Health Grants NS34887, NS48476, and NS41454. We thank Dr. P. Ghosh, J. Davis, Y. Yang, and V. Rogulin for help.
References
- Acuna-Goycolea C, Tamamaki N, Yanagawa Y, Obata K, van den Pol AN. Mechanisms of neuropeptide Y, peptide YY, and pancreatic polypeptide inhibition of identified green fluorescent protein-expressing GABA neurons in the hypothalamic neuroendocrine arcuate nucleus. J Neurosci. 2005;25:7406–7419. doi: 10.1523/JNEUROSCI.1008-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beart PM, Nicolopoulos LS, West DC, Headley PM. An excitatory amino acid projection from ventromedial hypothalamus to periaqueductal gray in the rat: autoradiographic and electrophysiological evidence. Neurosci Lett. 1988;85:205–211. doi: 10.1016/0304-3940(88)90352-7. [DOI] [PubMed] [Google Scholar]
- Bednarek MA, MacNeil T, Tang R, Kalyani RN, Van der Ploeg LH, Weinberg DH. Potent and selective peptide agonists of alpha-melanotropin action at human melanocortin receptor 4: their synthesis and biological evaluation in vitro. Biochem Biophys Res Commun. 2001;286:641–645. doi: 10.1006/bbrc.2001.5444. [DOI] [PubMed] [Google Scholar]
- Beindl W, Mitterauer T, Hohenegger M, Ijzerman AP, Nanoff C, Freissmuth M. Inhibition of receptor/G protein coupling by suramin analogues. Mol Pharmacol. 1996;50:415–423. [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Quantal analysis of EPSCs recorded from small numbers of synapses in hippocampal cultures. J Neurophysiol. 1995;73:1145–1156. doi: 10.1152/jn.1995.73.3.1145. [DOI] [PubMed] [Google Scholar]
- Bellasio S, Nicolussi E, Bertorelli R, Reggiani A. Melanocortin receptor agonists and antagonists modulate nociceptive sensitivity in the mouse formalin test. Eur J Pharmacol. 2003;482:127–132. doi: 10.1016/j.ejphar.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Benoit SC, Air EL, Coolen LM, Strauss R, Jackman A, Clegg DJ, Seeley RJ, Woods SC. The catabolic action of insulin in the brain is mediated by melanocortins. J Neurosci. 2002;22:9048–9052. doi: 10.1523/JNEUROSCI.22-20-09048.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewick GA, Gardiner JV, Dhillo WS, Kent AS, White NE, Webster Z, Ghatei MA, Bloom SR. Post-embryonic ablation of AgRP neurons in mice leads to a lean, hypophagic phenotype. FASEB J. 2005;19:1680–1682. doi: 10.1096/fj.04-3434fje. [DOI] [PubMed] [Google Scholar]
- Bhat GK, Mahesh VB, Lamar CA, Ping L, Aguan K, Brann DW. Histochemical localization of nitric oxide neurons in the hypothalamus: association with gonadotropin-releasing hormone neurons and co-localization with N-methyl-d-aspartate receptors. Neuroendocrinology. 1995;62:187–197. doi: 10.1159/000127004. [DOI] [PubMed] [Google Scholar]
- Boden P, Hill RG. Effects of cholecystokinin and related peptides on neuronal activity in the ventromedial nucleus of the rat hypothalamus. Br J Pharmacol. 1988;94:246–252. doi: 10.1111/j.1476-5381.1988.tb11521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brobeck JR. Mechanism of the development of obesity in animals with hypothalamic lesions. Physiol Rev. 1946;26:541–559. doi: 10.1152/physrev.1946.26.4.541. [DOI] [PubMed] [Google Scholar]
- Broberger C, Johansen J, Johansson C, Schalling M, Hokfelt T. The neuropeptide Y/agouti gene-related protein (AgRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci USA. 1998;95:15043–15048. doi: 10.1073/pnas.95.25.15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KS, Gentry RM, Rowland NE. Central injection in rats of alpha-melanocyte-stimulating hormone analog: effects on food intake and brain Fos. Regul Pept. 1998;78:89–94. doi: 10.1016/s0167-0115(98)00127-x. [DOI] [PubMed] [Google Scholar]
- Butler AA, Cone RD. The melanocortin receptors: lessons from knockout models. Neuropeptides. 2002;36:77–84. doi: 10.1054/npep.2002.0890. [DOI] [PubMed] [Google Scholar]
- Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- Chai BX, Neubig RR, Millhauser GL, Thompson DA, Jackson PJ, Barsh GS, Dickinson CJ, Li JY, Lai YM, Gantz I. Inverse agonist activity of agouti and agouti-related protein. Peptides. 2003;24:603–609. doi: 10.1016/s0196-9781(03)00104-9. [DOI] [PubMed] [Google Scholar]
- Chaki S, Okuyama S. Involvement of melanocortin-4 receptor in anxiety and depression. Peptides. 2005;26:1952–1964. doi: 10.1016/j.peptides.2004.11.029. [DOI] [PubMed] [Google Scholar]
- Chaki S, Oshida Y, Ogawa S, Funakoshi T, Shimazaki T, Okubo T, Nakazato A, Okuyama S. MCL0042: a nonpeptidic MC4 receptor antagonist and serotonin reuptake inhibitor with anxiolytic- and antidepressant-like activity. Pharmacol Biochem Behav. 2005;82:621–626. doi: 10.1016/j.pbb.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Chen AS, Metzger JM, Trumbauer ME, Guan XM, Yu H, Frazier EG, Marsh DJ, Forrest MJ, Gopal-Truter S, Fisher J, Camacho RE, Strack AM, Mellin TN, MacIntyre DE, Chen HY, Van der Ploeg LH, et al. Role of the melanocortin-4 receptor in metabolic rate and food intake in mice. Transgenic Res. 2000a;9:145–154. doi: 10.1023/a:1008983615045. [DOI] [PubMed] [Google Scholar]
- Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 2000b;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- Chen HY, Trumbauer ME, Chen AS, Weingarth DT, Adams JR, Frazier EG, Shen Z, Marsh DJ, Feighner SD, Guan XM, Ye Z, Nargund RP, Smith RG, Van der Ploeg LH, Howard AD, MacNeil DJ, Qian S. Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and agouti-related protein. Endocrinology. 2004;145:2607–2612. doi: 10.1210/en.2003-1596. [DOI] [PubMed] [Google Scholar]
- Choudhury AI, Heffron H, Smith MA, Al-Qassab H, Xu AW, Selman C, Simmgen M, Clements M, Claret M, Maccoll G, Bedford DC, Hisadome K, Diakonov I, Moosajee V, Bell JD, Speakman JR, Batterham RL, Barsh GS, Ashford ML, Withers DJ. The role of insulin receptor substrate 2 in hypothalamic and beta cell function. J Clin Invest. 2005;115:940–950. doi: 10.1172/JCI24445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin M, Backberg M, Ovesjo ML, Fisone G, Edwards RH, Fujiyama F, Meister B. Plasma membrane and vesicular glutamate transporter mRNAs/proteins in hypothalamic neurons that regulate body weight. Eur J Neurosci. 2003;18:1265–1278. doi: 10.1046/j.1460-9568.2003.02840.x. [DOI] [PubMed] [Google Scholar]
- Commons KG, Kow LM, Milner TA, Pfaff DW. In the ventromedial nucleus of the rat hypothalamus, GABA-immunolabeled neurons are abundant and are innervated by both enkephalin- and GABA-immunolabeled axon terminals. Brain Res. 1999;816:58–67. doi: 10.1016/s0006-8993(98)01084-1. [DOI] [PubMed] [Google Scholar]
- Cone RD. The central melanocortin system and energy homeostasis. Trends Endocrinol Metab. 1999;10:211–216. doi: 10.1016/s1043-2760(99)00153-8. [DOI] [PubMed] [Google Scholar]
- Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- Cone RD, Lu D, Koppula S, Vage DI, Klungland H, Boston B, Chen W, Orth DN, Pouton C, Kesterson RA. The melanocortin receptors: agonists, antagonists, and the hormonal control of pigmentation. Recent Prog Horm Res. 1996;51:287–317. [PubMed] [Google Scholar]
- Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AgRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. 1999;24:155–163. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Davidowa H, Li Y, Plagemann A. Differential response to NPY of PVH and dopamine-responsive VMH neurons in overweight rats. NeuroReport. 2002;13:1523–1527. doi: 10.1097/00001756-200208270-00007. [DOI] [PubMed] [Google Scholar]
- Davidowa H, Li Y, Plagemann A. Altered responses to orexigenic (AGRP, MCH) and anorexigenic (alpha-MSH, CART) neuropeptides of paraventricular hypothalamic neurons in early postnatally overfed rats. Eur J Neurosci. 2003;18:613–621. doi: 10.1046/j.1460-9568.2003.02789.x. [DOI] [PubMed] [Google Scholar]
- DeBock F, Kurz J, Azad SC, Parsons CG, Hapfelmeier G, Zieglgänsberger W, Rammes G. Alpha2-adrenoreceptor activation inhibits LTP and LTD in the basolateral amygdala: involvement of Gi/o-protein-mediated modulation of Ca2+-channels and inwardly rectifying K+-channels in LTD. Eur J Neurosci. 2003;17:1411–1424. doi: 10.1046/j.1460-9568.2003.02544.x. [DOI] [PubMed] [Google Scholar]
- Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, Coppari R, Balthasar N, Cowley MA, Chua S, Jr, Elmquist JK, Lowell BB. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Yeo GS, Keogh JM, Aminian S, Jebb SA, Butler G, Cheetham T, O'Rahilly S. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000;106:271–279. doi: 10.1172/JCI9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong TM, Mao C, MacNeil T, Kalyani R, Smith T, Weinberg D, Tota MR, Van der Ploeg LH. ART (protein product of agouti-related transcript) as an antagonist of MC-3 and MC-4 receptors. Biochem Biophys Res Commun. 1997;237:629–631. doi: 10.1006/bbrc.1997.7200. [DOI] [PubMed] [Google Scholar]
- Freissmuth M, Boehm S, Beindl W, Nickel P, Ijzerman AP, Hohenegger M, Nanoff C. Suramin analogues as subtype-selective G protein inhibitors. Mol Pharmacol. 1996;49:602–611. [PubMed] [Google Scholar]
- Fremeau RT, Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Fu LY, Acuna-Goycolea C, van den Pol AN. Neuropeptide Y inhibits hypocretin/orexin neurons by multiple presynaptic and postsynaptic mechanisms: tonic depression of the hypothalamic arousal system. J Neurosci. 2004;24:8741–8751. doi: 10.1523/JNEUROSCI.2268-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XB, van den Pol AN. Neurotrophin-3 potentiates excitatory GABAergic synaptic transmission in cultured developing hypothalamic neurones of the rat. J Physiol (Lond) 1999;518:81–95. doi: 10.1111/j.1469-7793.1999.0081r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garic-Stankovic A, Hernandez MR, Chiang PJ, Debelak-Kragtorp KA, Flentke GR, Armant DR, Smith SM. Ethanol triggers neural crest apoptosis through the selective activation of a pertussis toxin-sensitive G protein and a phospholipase C beta-dependent Ca2+ transient. Alcohol Clin Exp Res. 2005;29:1237–1246. doi: 10.1097/01.alc.0000172460.05756.d9. [DOI] [PubMed] [Google Scholar]
- Gee CE, Chen CL, Roberts JL, Thompson R, Watson SJ. Identification of proopiomelanocortin neurones in rat hypothalamus by in situ cDNA-mRNA hybridization. Nature. 1983;306:374–376. doi: 10.1038/306374a0. [DOI] [PubMed] [Google Scholar]
- Gonzalez MI, Vaziri S, Wilson CA. Behavioral effects of alpha-MSH and MCH after central administration in the female rat. Peptides. 1996;17:171–177. doi: 10.1016/0196-9781(95)02092-6. [DOI] [PubMed] [Google Scholar]
- Grieco P, Balse PM, Weinberg D, MacNeil T, Hruby VJ. d-Amino acid scan of gamma-melanocyte-stimulating hormone: importance of Trp(8) on human MC3 receptor selectivity. J Med Chem. 2000;43:4998–5002. doi: 10.1021/jm000211e. [DOI] [PubMed] [Google Scholar]
- Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, Bruning JC. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8:1289–1291. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of AgRP and NPY in fasting-activated hypothalamic neurons. Nat Neurosci. 1998;1:271–272. doi: 10.1038/1082. [DOI] [PubMed] [Google Scholar]
- Harrold JA, Williams G. Melanocortin-4 receptors, beta-MSH and leptin: key elements in the satiety pathway. Peptides. 2006;27:365–371. doi: 10.1016/j.peptides.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Harrold JA, Widdowson PS, Williams G. Altered energy balance causes selective changes in melanocortin-4 (MC4-R), but not melanocortin-3 (MC3-R), receptors in specific hypothalamic regions: further evidence that activation of MC4-R is a physiological inhibitor of feeding. Diabetes. 1999;48:267–271. doi: 10.2337/diabetes.48.2.267. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C, Monck EK. Agouti-related protein functions as an inverse agonist at a constitutively active brain melanocortin-4 receptor. Regul Pept. 2001;99:1–7. doi: 10.1016/s0167-0115(01)00234-8. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C, Chen P, Li C, Chang K, Smith MS, Cameron JL, Cone RD. Characterization of the neuroanatomical distribution of agouti-related protein immunoreactivity in the rhesus monkey and the rat. Endocrinology. 1999;140:1408–1415. doi: 10.1210/endo.140.3.6544. [DOI] [PubMed] [Google Scholar]
- Hettes SR, Gonzaga J, Heyming TW, Perez S, Wolfsohn S, Stanley BG. Dual roles in feeding for AMPA/kainate receptors: receptor activation or inactivation within distinct hypothalamic regions elicits feeding behavior. Brain Res. 2003;992:167–178. doi: 10.1016/j.brainres.2003.08.032. [DOI] [PubMed] [Google Scholar]
- Hillebrand JJ, Kas MJ, Adan RA. α-MSH enhances activity-based anorexia. Peptides. 2005;26:1690–1696. doi: 10.1016/j.peptides.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Hirrlinger J, Moeller H, Kirchhoff F, Dringen R. Expression of multidrug resistance proteins (Mrps) in astrocytes of the mouse brain: a single cell RT-PCR study. Neurochem Res. 2005;30:1237–1244. doi: 10.1007/s11064-005-8795-y. [DOI] [PubMed] [Google Scholar]
- Hruby VJ, Lu D, Sharma SD, Castrucci AL, Kesterson RA, al-Obeidi FA, Hadley ME, Cone RD. Cyclic lactam alpha-melanotropin analogues of Ac-Nle4-cyclo[Asp5, d-Phe7, Lys10] alpha-melanocyte-stimulating hormone-(4–10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J Med Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- Huang H, Ghosh P, van den Pol AN. Prefrontal cortex-projecting glutamatergic thalamic paraventricular nucleus-excited by hypocretin: a feedforward circuit that may enhance cognitive arousal. J Neurophysiol. 2006;95:1656–1668. doi: 10.1152/jn.00927.2005. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Jegou S, Boutelet I, Vaudry H. Melanocortin-3 receptor mRNA expression in pro-opiomelanocortin neurones of the rat arcuate nucleus. J Neuroendocrinol. 2000;12:501–505. doi: 10.1046/j.1365-2826.2000.00477.x. [DOI] [PubMed] [Google Scholar]
- Jonsson L, Skarphedinsson JO, Skuladottir GV, Watanobe H, Schioth HB. Food conversion is transiently affected during 4-week chronic administration of melanocortin agonist and antagonist in rats. J Endocrinol. 2002;173:517–523. doi: 10.1677/joe.0.1730517. [DOI] [PubMed] [Google Scholar]
- Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes. 2004;53:549–559. doi: 10.2337/diabetes.53.3.549. [DOI] [PubMed] [Google Scholar]
- Kawashima N, Chaki S, Okuyama S. Electrophysiological effects of melanocortin receptor ligands on neuronal activities of monoaminergic neurons in rats. Neurosci Lett. 2003;353:119–122. doi: 10.1016/j.neulet.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Keesey RE, Powley TL. The regulation of body weight. Annu Rev Psychol. 1986;37:109–133. doi: 10.1146/annurev.ps.37.020186.000545. [DOI] [PubMed] [Google Scholar]
- King BM. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol Behav. 2006;87:221–244. doi: 10.1016/j.physbeh.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol. 2003;457:213–235. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- Kishi T, Aschkenasi CJ, Choi BJ, Lopez ME, Lee CE, Liu H, Hollenberg AN, Friedman JM, Elmquist JK. Neuropeptide Y Y1 receptor mRNA in rodent brain: distribution and colocalization with melanocortin-4 receptor. J Comp Neurol. 2005;482:217–243. doi: 10.1002/cne.20432. [DOI] [PubMed] [Google Scholar]
- Konda Y, Gantz I, DelValle J, Shimoto Y, Miwa H, Yamada T. Interaction of dual intracellular signaling pathways activated by the melanocortin-3 receptor. J Biol Chem. 1994;269:13162–13166. [PubMed] [Google Scholar]
- Lee EJ, Lee SH, Jung JW, Lee W, Kim BJ, Park KW, Lim SK, Yoon CJ, Baik JH. Differential regulation of cAMP-mediated gene transcription and ligand selectivity by MC3R and MC4R melanocortin receptors. Eur J Biochem. 2001;268:582–591. doi: 10.1046/j.1432-1327.2001.01900.x. [DOI] [PubMed] [Google Scholar]
- Lee SW, Stanley BG. NMDA receptors mediate feeding elicited by neuropeptide Y in the lateral and perifornical hypothalamus. Brain Res. 2005;1063:1–8. doi: 10.1016/j.brainres.2005.09.045. [DOI] [PubMed] [Google Scholar]
- Li YZ, Davidowa H. Food deprivation decreases responsiveness of ventromedial hypothalamic neurons to melanocortins. J Neurosci Res. 2004;77:596–602. doi: 10.1002/jnr.20192. [DOI] [PubMed] [Google Scholar]
- Lin S, Boey D, Lee N, Schwarzer C, Sainsbury A, Herzog H. Distribution of prodynorphin mRNA and its interaction with the NPY system in the mouse brain. Neuropeptides. 2006;40:115–123. doi: 10.1016/j.npep.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Liss B, Bruns R, Roeper J. Alternative sulfonylurea receptor expression defines metabolic sensitivity of K-ATP channels in dopaminergic midbrain neurons. EMBO J. 1999;18:833–846. doi: 10.1093/emboj/18.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Kishi T, Roseberry AG, Cai X, Lee CE, Montez JM, Friedman JM, Elmquist JK. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J Neurosci. 2003;23:7143–7154. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- Marks DL, Cone RD. The role of the melanocortin-3 receptor in cachexia. Ann NY Acad Sci. 2003;994:258–266. doi: 10.1111/j.1749-6632.2003.tb03188.x. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Miura GI, Yagaloff KA, Schwartz MW, Barsh GS, Palmiter RD. Effects of neuropeptide Y deficiency on hypothalamic agouti-related protein expression and responsiveness to melanocortin analogues. Brain Res. 1999;848:66–77. doi: 10.1016/s0006-8993(99)01962-9. [DOI] [PubMed] [Google Scholar]
- Millhouse OE. The organization of the ventromedial hypothalamic nucleus. Brain Res. 1973a;55:71–87. [PubMed] [Google Scholar]
- Millhouse OE. Certain ventromedial hypothalamic afferents. Brain Res. 1973b;55:89–105. doi: 10.1016/0006-8993(73)90490-3. [DOI] [PubMed] [Google Scholar]
- Minami T, Oomura Y, Sugimori M. Electrophysiological properties and glucose responsiveness of guinea-pig ventromedial hypothalamic neurones in vitro. J Physiol (Lond) 1986;380:127–143. doi: 10.1113/jphysiol.1986.sp016276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijenhuis WA, Oosterom J, Adan RA. AgRP(83–132) acts as an inverse agonist on the human-melanocortin-4 receptor. Mol Endocrinol. 2001;15:164–171. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- Poggioli R, Vergoni AV, Bertolini A. ACTH-(1–24) and alpha-MSH antagonize feeding behavior stimulated by kappa opiate agonists. Peptides. 1986;7:843–848. doi: 10.1016/0196-9781(86)90104-x. [DOI] [PubMed] [Google Scholar]
- Priestley T. The effect of baclofen and somatostatin on neuronal activity in the rat ventromedial hypothalamic nucleus in vitro. Neuropharmacology. 1992;31:103–109. doi: 10.1016/0028-3908(92)90018-k. [DOI] [PubMed] [Google Scholar]
- Pritchard LE, White A. Agouti-related protein: more than a melanocortin-4 receptor antagonist? Peptides. 2005;26:1759–1770. doi: 10.1016/j.peptides.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD. Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc Natl Acad Sci USA. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Kim MS, Morgan DG, Small CJ, Edwards CM, Sunter D, Abusnana S, Goldstone AP, Russell SH, Stanley SA, Smith DM, Yagaloff K, Ghatei MA, Bloom SR. A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology. 1998;139:4428–4431. doi: 10.1210/endo.139.10.6332. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- Slamberova R, Hnatczuk OC, Vathy I. Expression of proopiomelanocortin and proenkephalin mRNA in sexually dimorphic brain regions are altered in adult male and female rats treated prenatally with morphine. J Pept Res. 2004;63:399–408. doi: 10.1111/j.1399-3011.2004.00134.x. [DOI] [PubMed] [Google Scholar]
- Smith AI, Funder JW. Proopiomelanocortin processing in the pituitary, central nervous system, and peripheral tissues. Endocr Rev. 1988;9:159–179. doi: 10.1210/edrv-9-1-159. [DOI] [PubMed] [Google Scholar]
- Smith MA, Hisadome K, Al-Qassab H, Heffron H, Withers DJ, Ashford ML. Melanocortins and agouti-related protein modulate the excitability of two arcuate nucleus neuron populations by alteration of resting potassium conductances. J Physiol (Lond) 2007;578:425–438. doi: 10.1113/jphysiol.2006.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Vaisse C, Conklin BR. Engineering the melanocortin-4 receptor to control G(s) signaling in vivo. Ann NY Acad Sci. 2003;994:225–232. doi: 10.1111/j.1749-6632.2003.tb03184.x. [DOI] [PubMed] [Google Scholar]
- Stellar E. The physiology of motivation. Psychol Rev. 1954;61:5–22. doi: 10.1037/h0060347. [DOI] [PubMed] [Google Scholar]
- Sternson SM, Shepherd GM, Friedman JM. Topographic mapping of VMH → arcuate nucleus microcircuits and their reorganization by fasting. Nat Neurosci. 2005;8:1356–1363. doi: 10.1038/nn1550. [DOI] [PubMed] [Google Scholar]
- Turner TJ, Adams ME, Dunlap K. Multiple Ca2+ channel types coexist to regulate synaptosomal neurotransmitter release. Proc Natl Acad Sci USA. 1993;90:9518–9522. doi: 10.1073/pnas.90.20.9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106:253–262. doi: 10.1172/JCI9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Wuarin JP, Dudek FE. Glutamate, the dominant excitatory transmitter in neuroendocrine regulation. Science. 1990;250:1276–1278. doi: 10.1126/science.1978759. [DOI] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- Ziegler DR, Cullinan WE, Herman JP. Distribution of vesicular glutamate transporter mRNA in rat hypothalamus. J Comp Neurol. 2002;448:217–229. doi: 10.1002/cne.10257. [DOI] [PubMed] [Google Scholar]