

Abstract
An analysis of the x-ray structure of homodimeric avian farnesyl diphosphate synthase (geranyltransferase, EC 2.5.1.10) coupled with information about conserved amino acids obtained from a sequence alignment of 35 isoprenyl diphosphate synthases that synthesize farnesyl (C15), geranylgeranyl (C20), and higher chain length isoprenoid diphosphates suggested that the side chains of residues corresponding to F112 and F113 in the avian enzyme were important for determining the ultimate length of the hydrocarbon chains. This hypothesis was supported by site-directed mutagenesis to transform wild-type avian farnesyl diphosphate synthase (FPS) into synthases capable of producing geranylgeranyl diphosphate (F112A), geranylfarnesyl (C25) diphosphate (F113S), and longer chain prenyl diphosphates (F112A/F113S). An x-ray analysis of the structure of the F112A/F113S mutant in the apo state and with allylic substrates bound produced the strongest evidence that these mutations caused the observed change in product specificity by directly altering the size of the binding pocket for the growing isoprenoid chain in the active site of the enzyme. The proposed binding pocket in the apo mutant structure was increased in depth by 5.8 Å as compared with that for the wild-type enzyme. Allylic diphosphates were observed in the holo structures, bound through magnesium ions to the aspartates of the first of two conserved aspartate-rich sequences (D117–D121), with the hydrocarbon tails of all the ligands growing down the hydrophobic pocket toward the mutation site. A model was constructed to show how the growth of a long chain prenyl product may proceed by creation of a hydrophobic passageway from the FPS active site to the outside surface of the enzyme.
The isoprenoid biosynthetic pathway is built around a family of diphosphate esters of linear alcohols that contain increasing numbers of isoprene units. Beginning with the C5 molecule dimethylallyl diphosphate (DMAPP), a series of C10 (geranyl diphosphate, GPP), C15 (farnesyl diphosphate, FPP), C20 (geranylgeranyl diphosphate, GGPP), and higher molecular weight isoprenoid diphosphates are formed by the 1′–4 addition of isopentenyl diphosphate (IPP) to the growing chain. These compounds are the substrates for biosynthesis of all isoprenoid metabolites, including monoterpenes, sesquiterpenes, diterpenes, sterols, carotenoids, ubiquinones, dolichols, and prenylated proteins. Thus far, over 23,000 individual isoprenoid compounds have been identified. They form the most chemically diverse family of molecules found in nature and are widely distributed among archaea, bacteria, and eukareya.
The 1′–4 condensation reactions are catalyzed by a family of prenyltransferases, the isoprenyl diphosphate synthases, that are highly selective for the chain lengths and double bond stereochemistries of both substrates and products. For example, yeast contains at least four distinct enzymes for chain elongation. FPP synthase (FPS) converts DMAPP to FPP for synthesis of sterols, farnesylated proteins, heme a, and higher chain length isoprenoid diphosphates (1). A newly discovered yeast GGPP synthase converts FPP to GGPP for synthesis of geranylgeranylated proteins (2). A hexaprenyl diphosphate synthase synthesizes the C30 isoprenoid chain found in yeast ubiquinones (3), and an as-yet uncharacterized eicosaprenyl diphosphate synthase is required to convert FPP to the C100 precursor of the dolichols needed for glycoprotein biosynthesis. Although different in detail, similar scenarios are found in other organisms.
Avian FPS catalyzes the sequential chain elongations of DMAPP to GPP and GPP to FPP. It is the only prenyltransferase for which an x-ray structure has been obtained (4). The enzyme is a homodimer, and the subunits each contain a single site for the C5 to C15 elongation. The putative catalytic site consists of a large central cavity formed by a bundle of 10 α-helices. Two aspartate-rich sequences (DDXXD) that are highly conserved among the isoprenyl diphosphate synthases, and are essential for enzymatic activity, are located on opposite walls of this cavity, with the aspartate side-chains ≈12 Å apart and facing each other. A solvent accessible surface constructed from FPS revealed two hydrophobic depressions in the wall of the active site, the deeper of the two being located with its opening adjacent to the first DDXXD sequence. The dimensions of the deeper depression suggested that it is sufficiently spacious to serve as a pocket for binding GPP, the allylic substrate for the last of the two condensations with IPP during synthesis of FPP, by accommodating its hydrophobic C10 isoprenoid chain. With some movement of the protein side chains, FPP will also fit into this pocket. When forced, avian FPS will catalyze addition of a third IPP to synthesize GGPP from FPP, although this activity is not important under normal physiological conditions (5).
The aromatic ring of F113 forms the “floor” of the putative allylic substrate binding pocket in avian FPS and is buttressed by an offset face-to-face stacking with the aromatic ring of F112. A survey of amino acid sequences from 35 isoprenyl diphosphate synthases,‖ representing a wide variety of organisms, reveals some general patterns. The eukaryotic FPSs have Phe or Tyr residues at the positions corresponding to F112 and F113 in the avian enzyme, while their eubacterial and archaebacterial counterparts have F112 and S113 or T113 substitutions. In contrast, GGPP and higher chain length synthases have amino acids with smaller, more flexible side chains at these positions, typically A112, S113 or M112, S113 motifs. Model studies based on the structure of avian FPS indicated that the active site hydrophobic pocket could be enlarged to readily accommodate a farnesyl residue by replacing the phenylalanines with small amino acids. Three mutants of avian FPS (F112A, F113S, and F112A/F113S) were constructed to ascertain if the selectivity for synthesis of FPP, GGPP, and perhaps longer chain length isoprenoid diphosphates was indeed dictated by the size of the large hydrophobic pocket. We now report biochemical and structural studies of the mutants that provide new insights about how isoprenyl diphosphate synthases control chain length during the 1′–4 condensation reaction.
MATERIALS AND METHODS
Preparation of Mutant FPS.
The avian FPS gene was mutated in pBluescript II SK+ (8). The primers used to construct the F112A, F113S, and F112A/F113S mutants were 5′-CACCAGGAAGGCGGCCTGGAAGAGCTCGATGCAC-3′, 5′-CAGCCACCAGGGAGAAAGCTTGGAACAAC-3′, and 5′-CATCAGCCACCACGCTAGCGGCCTGGAACAAC-3′, respectively. The base changes are shown in boldface type, and underlined regions indicate restriction sites (SacI for F112A, HindIII for F113S, and NheI for F112A/F113S) introduced as silent mutations to facilitate screening. NdeI–partial XhoI digests gave 1.1-kb fragments containing the mutant FPS genes. The resulting fragments were isolated, and ligated into the NdeI/SalI sites of the Escherichia coli expression vector pTTQ18N. These plasmids were then used to transform E. coli XA90 cells. Mutant and wt FPS were overproduced by the procedures described for the yeast enzyme (9), except for the use of isopropyl β-d-thiogalactoside for induction, and purified to homogeneity as described (10). The activities of wilt-type (wt) FPS, the F112A mutant, and the F113S mutant were determined by an acid lability assay (11). Activity of the F112A/F113S mutant was determined by a butanol extraction method (12). Short chain prenyl diphosphate products were analyzed directly using reversed-phase HPLC (13). Long chain products produced by the F112A/F113S mutant were first converted to alcohols by acid phosphatase (14), and then separated by reversed-phase HPLC. The stereochemistry of the geranylgeraniol was assigned by coinjection with a synthetic sample on normal phase HPLC.
Crystallization.
The crystallization conditions that were used to solve the structure of avian recombinant FPS (4) were changed to prevent tartrate from interfering with substrate binding by chelating magnesium in the solutions. Calorimetric binding and kinetic inhibition studies showed that tartrate-Hepes solutions inhibited binding of substrates to the enzyme, while normal binding was observed in PEG-endo-cis-bicyclo[2.2.1]heptene dicarboxylic acid (BHDA) solutions (unpublished results). A sparse scan screen (15) of 100 test solutions produced crystals in PEG-NaCl solutions that had the same bipyrimidal shape as those grown from tartrate. The optimum conditions for producing both wt and F112A/F113S mutant FPS crystals were determined to be 12% PEG-6000, 1.3 M NaCl, 30 mM BHDA, and 5 mM magnesium (pH 7.0), and a final FPS concentration of 16–20 mg/ml.
Data Collection and Refinement.
Crystal structures of mutant and wt FPS containing allylic diphosphates, and a substrate analog (16), geranylmethyl phosphonophosphate (GCPP), were obtained by soaking the crystals in solutions containing the ligands. Crystals were soaked in DMAPP, GPP, and GCPP at 400 μM ligand and 5 mM magnesium in mother liquor. FPP and GGPP were soaked into the crystals by incremental addition to a final concentration of 200 μM followed by addition of magnesium to a concentration of 500 μM. The different procedure for FPP and GGPP was necessary because of solubility and crystal cracking problems experienced with these compounds. All crystallographic data were produced with a RU-200 (Rigaku, Tokyo) generator, using monochromatic copper Kα-radiation, and collected with a Siemens X1000A multiwire area detector (Madison, WI). Data were reduced using the Siemens sadie and saint (17) packages. Native FPS coordinates were refined against the mutant structure factors, first using the xplor (18) package. The refined coordinates were then further refined using the tnt package (19). The resulting difference electron density (Fo − Fc) maps were then used to place ligand and water molecules in the structure and to manually reposition atoms of the protein, using the tom (20) program. Magnesium atoms were fit into spherical 3-σ difference electron density whose centers were within 2.0 Å of allylic substrate diphosphate oxygens. The process of adding waters, model building, and refinement with tnt was repeated until no 3-σ or greater difference electron density was observed.
RESULTS AND DISCUSSION
Mutations in F112 and F113 Alter the Chain Length Selectivity of Avian FPS.
Each of the recombinant mutants catalyzed addition of IPP to FPP at rates substantially faster than the low basal level observed for the avian enzyme. Incubation of the F112A mutant with [1-14C]IPP and FPP gave E,E,E-GGPP. A similar incubation with the F113S mutant yielded two diphosphates, one with a retention time identical to GGPP and one more hydrophobic than GGPP. The alcohol obtained from hydrolysis of the less hydrophobic material was E,E,E-geranylgeraniol. A high-resolution mass spectrum of the more hydrophobic product did not contain a molecular ion, but instead had a strong peak at m/z 341.3233. This exact mass corresponds to a C25H41 fragment, consistent with loss of water from geranylfarnesol. Analysis of the alcohols obtained from the F112A/F113S mutant product mixture after treatment with phosphatase, followed by reversed-phase chromatography, gave a mixture of radioactive compounds whose retention times corresponded to isoprenoid alcohols in the C20–C70 range. The most abundant components varied depending on the reaction conditions but were typically in the C35–C40 range, as shown in Fig. 1. A recent study (21) described how through random mutagenesis of a bacterial FPS, four mutants with GGPP synthase activity were obtained. The two most active had a Y → H mutation at the position equivalent to F112 in FPS.
Figure 1.
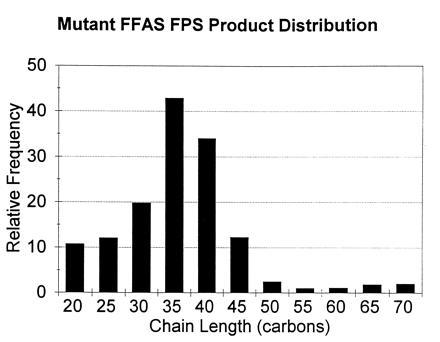
Mutant F112A/F113S FPS product distribution shows relative frequency of production, on a molar basis, of different chain length isoprenes. Each observed cpm for [14C]IPP incorporated into the product was normalized by dividing by the number of IPPs incorporated into the product, and then by the lowest value of the series, giving a value of one for the lowest value of the distribution.
Steady-State Kinetic Parameters for wt FPS and the F112A, F113S, and F112A/F113S Mutants.
Steady-state kinetic constants provided important insights about the shift in the distribution of products to higher chain lengths as the hydrophobic pocket in FPS was progressively enlarged in the F112A, F113S, and F112A:F113S mutants. As shown in Table 1, a 130-fold decrease in kcat and 30-fold increases in KMIPP and KMFPP over the respective values for GPP with wt protein combined to terminate chain elongation cleanly at FPP.
Table 1.
Kinetic constants for wt and mutant FPS
| KMGPP, μM | KMIPP, μM | kcat, s−1 | KMFPP, μM | KMIPP, μM | kcat, s−1 | KMGGPP, μM | KMIPP, μM | kcat, s−1 | |
|---|---|---|---|---|---|---|---|---|---|
| Wild Type | 0.3 | 0.6 | 1.6 | 9* | 20* | 0.012* | — | — | — |
| F112A | 0.1 | 1.0 | 1.2 | 0.2 | 1.0 | 0.51 | — | — | — |
| F113S | 0.2 | 0.6 | 0.68 | 0.7 | 0.4 | 0.59 | 1.0 | 0.7 | 0.09 |
| F112A/F113S | 0.03 | 0.04 | 0.09 | 0.04 | 0.08 | 0.18 | 0.2 | 0.04 | 0.07 |
The Michaelis constants were determined from plots of initial velocities versus substrate concentration by nonlinear curve fitting using grafit (Sigma). KM values for the allylic substrates are apparent values determined at 4 μM IPP. KM values for IPP were determined at saturating concentrations of the allylic substrate.
Ref. 10.
Chain length selectivity was extended by one isoprene residue when the buttressing aromatic ring in F112 was replaced by a methyl group in the F112A mutant. The KM values for the C10 to C15 and the C15 to C20 steps were similar to those for C10 to C15 elongation in wt enzyme, and kcat was only 2-fold lower. Chain elongation terminated cleanly at GGPP. Replacement of the phenylalanine at the floor of the hydrophobic pocket by serine in the F113S mutant gave an enzyme whose catalytic constants for the C10 to C15 and C15 to C20 steps were similar to the F112A mutant. However, the F113S protein also catalyzed synthesis of a C25 product with a kcat for C20 to C25 elongation only 18-fold less than the C10 to C15 step for wt FPS. Now, the chain terminated cleanly after the C20 to C25 step.
Replacement of the aromatic rings in both phenylalanine residues by smaller substituents in the F112A/F113S mutant altered the ability of the enzyme to regulate the chain length of its products to the point where the protein lost its ability to control the number of molecules of IPP added to the growing allylic chain. The kcat values for prenyltransfer initiated with GPP, FPP, and GGPP decreased by 4- to 13-fold relative to kcat for GPP with the wt enzyme, while the Michaelis constants for IPP and the allylic substrates for the F112A/F113S decreased. The mechanism for binding substrates by wt FPS is ordered, with the allylic substrate binding first (22). According to this mechanism, the equations for KMIPP and KMAllylic both contain k4, the rate constant for release of product, in their numerator:
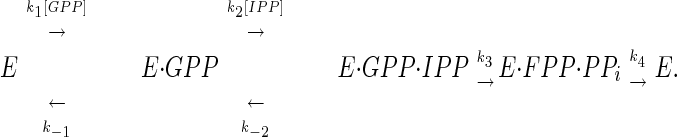 |
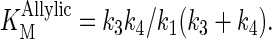 |
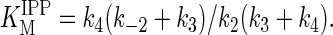 |
Release of FPP from the wt enzyme-product complex was rate limiting when the enzyme was operating at steady state, and k3 > k4 (22). Under these conditions, KMAllylic ≈ k4/k1 and KMIPP ≈ k4(k-2 + k3)/k2k3. If k4 decreases substantially in the F112A/F113S mutant as the hydrocarbon chain grows beyond five isoprene units, the KM values for both substrates should also decrease, assuming that the rate constants for addition of allylic substrate (k1), addition of IPP (k2), and catalysis (k3) do not change substantially. The kinetic constants for the double mutant are consistent with a scenario where the enzyme catalyzes addition of IPP to the growing allylic chain by a processive mechanism resulting from a substantial decrease in the rate constant for dissociation of the longer chain isoprenoid products. However, a step-wise mechanism where newly formed product bound tightly and competed with the allylic substrate for IPP, even at low conversions, would also explain the results.
Overall Mutant FPS Structure.
The F112A/F113S mutant of FPS crystallized from PEG-NaCl solutions in the tetragonal space group, I4122, with unit cell dimensions of a = b = 88.5 Å, c = 275.0 Å, and α = β = γ = 90°, containing one subunit of the dimer per asymmetric unit. These crystals were isomorphous with wt crystals, with or without bound ligands. The crystals produced from PEG-NaCl solutions were also isomorphous with those grown in tartrate, and used in the initial determination of the wt FPS crystal structure (4). The apo structure and the structures containing bound GPP and FPP were refined to 2.5-Å resolution, while the DMAPP containing structure was refined to 2.4 Å (Table 2).
Table 2.
F112A/F113S mutant structure refinement statistics
| Structure | Resolution, Å | Completeness, % | R-factor, % |
|---|---|---|---|
| F112A/F113S-apo | 2.5 | 88 | 20.2 |
| F112A/F113S-DMAPP | 2.4 | 87 | 20.2 |
| F112A/F113S-GPP | 2.4 | 89 | 19.2 |
| F112A/F113S-FPP | 2.4 | 86 | 20.1 |
The structure of the mutant enzyme, with or without bound ligands, is essentially the same as the wt protein, with the exception of the two mutated side chains. A superposition of the backbone atoms of the F112A/F113S structure on the wt FPS structure, using insight (23), gave an rms difference of only 0.58 Å between the two structures. Similarly, a superposition of the F112A/F113S-FPP structure backbone atoms on the apo F112A/F113S-FPS structure gave a rms difference of 0.51 Å between the two structures. Most of the difference occurred in two fairly disordered loops containing residues 190–200 and 262–280.
The large central cavity formed by the 10 core α-helices (Fig. 2) contains the putative active site of each FPS subunit. The surface of this cavity contains all five regions of conserved amino acid sequence found in the alignment of 13 members of the isoprenyl diphosphate synthase family (6). The two conserved DDXXD sequences thought to contain residues that bind the diphosphate moieties (24) are located on opposite walls of the cavity ≈12 Å apart. The roof of the cavity is formed by two long flexible loops that are formed from residues 122–137 and 264–282. These loops contain conserved basic residues R126 and K271, respectively. A van der Waals surface of the FPS active site showed that the first DDXXD sequence was near a large hydrophobic pocket (Fig. 2) that contained the mutated F112 and F113 residues. This pocket is proposed to bind the growing hydrocarbon tail of the isoprenyl product. Avian FPS is active as a homodimer (10). Because only one subunit of the protein was found in the asymmetric unit in the tetragonal crystal, it was proposed (4) that the subunits of the dimer were related by a crystallographic 2-fold axis. This configuration results in a dimer interface that is highly hydrophobic, causing 6 aromatic and 17 aliphatic residue side chains to be buried. The dimer configuration places the single active site of each subunit at the top of the dimer, as viewed in Fig. 2, and facing away from each other so that they each open outward toward the bulk solvent.
Figure 2.
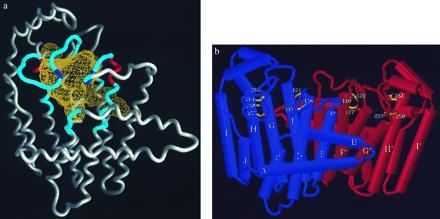
(a) Structure of mutant F112A/F113S FPS showing the FPS monomer with active site cavity volume in yellow mesh. The mutation site is at the bottom of the long protrusion at the lower right of the cavity. The five conserved sequences that line the cavity are shown in blue. The conserved DDXXD sequences involved in substrate binding are shown in red (D117–D121 on right, and D257–D261 on left). The conserved basic residues associated with these two sequences (R126–R127 and K271) are shown in purple. (b) FPS dimer with α-helices shown as tubes. The purple monomer’s active site is facing forward and shows the aspartates of the two DDXXD sequences on opposite walls of the cavity. The active site of the orange monomer is facing away from the viewer.
The Mutated Residues Increase the Active Site Hydrophobic Pocket Depth.
Based on an analysis of amino acid sequences for FPSs, GGPP synthases, and long chain synthases, along with the x-ray structure of wt FPS, it was surmised that F112 and F113 at the floor of the hydrophobic pocket were important residues for controlling chain length in wt avian FPS. In addition, F112A and F113S mutations in the avian enzyme would increase the depth of the pocket and allow mutant FPSs to produce longer chain isoprenes in a similar manner as wt GGPP and long chain synthases. For example, the GGPP synthase from Methanobacterium thermoautotrophicum contains phenylalanine and serine residues corresponding to F112 and F113, respectively, in avian FPS. The crystal structure of apo F112A/F113S FPS showed that the smaller side chains of the mutated residues increased the hydrophobic pocket depth by 5.8 Å. This increase in the depth of the cavity agrees with the 5.2-Å increase in chain length going from FPP (C15) to GGPP (C20). The total depth of the pocket is 17.1 Å. The diameter of the pocket varies from ≈7.9 Å at the opening where the first conserved DDXXD sequence is located to 6.2 Å at the bottom of the pocket. These dimensions snugly accommodate the prenyl tail of GGPP when the diphosphate head is bound to the first DDXXD sequence, as would be the case following the condensation of IPP with FPP. The pocket is approximately horizontal (in the orientation presented in Fig. 2) from the opening to about 8.9 Å in depth, where it makes a bend to continue down vertically 8.2 Å into the interior of the enzyme. The surface of the extended part of the pocket in the mutant F112A/F113S remains hydrophobic. One side of the surface is formed from residues F109, A112, S113, S146, E150, T181, E182, and Y218, while the other side is formed from residues I143, N144, F147, and L148 from the other subunit of the dimer.
Allylic Compounds Bind at the First DDXXD Sequence.
X-ray structures obtained by soaking crystals in solutions containing DMAPP, GPP, or FPP showed continuous 3-σ difference (Fo − Fc) electron density at the putative allylic substrate binding site, near the first aspartate-rich sequence (D117–D121) and continuing into the adjoining hydrophobic pocket, while there was no difference electron density observed at the second DDXXD (D257–D261) sequence (the putative homoallylic binding site). The diphosphate groups of the allylic compounds bound to the aspartate carboxyl oxygens (see Materials and Methods) through magnesium bridges as originally predicted (24). The F112A/F113S structure with bound DMAPP, refined to 2.4-Å resolution, clearly showed two magnesium atoms involved in binding the diphosphate group to the aspartates. The distance between the refined position of each magnesium and its nearest diphosphate oxygen ligand was 1.9 Å, and each magnesium was coordinated to 4 or 5 oxygens. This observation is consistent with a previous study (25) which showed that two Mn2+ atoms and one GPP or FPP molecule bound per monomer of FPS. The magnesium that produced consistently stronger difference electron density was within 3.0 Å, and therefore interacting with the bridging diphosphate oxygen, a carboxyl oxygen of D117, a carboxyl oxygen of D121, and a nonbridging oxygen in the internal (P1) phosphate. The remaining coordinating atoms, presumably waters, were not resolved. The resulting coordination was approximately octahedral. The other magnesium was interacting with one of the same nonbridging P1 oxygen, the second D117 carboxyl oxygen, a carboxyl oxygen of D121, one of the D188 carboxyl oxygens, a resolved water molecule, and presumably another water that was not resolved in the structure. This coordination was not in an octahedral configuration. The two magnesiums were 4.1 Å apart in the novel enzyme-bound diphosphate structure.
The F112A/F113S structures containing bound GPP and FPP showed magnesium atoms in approximately the same locations and coordinated similarly as observed in the DMAPP structure. This distance between the two magnesiums was 3.8 Å in the GPP structure and 4.7 Å in the FPP structure. However, there were some differences among the three structures. In the GPP structure (Fig. 3), a resolved water was coordinated to the first magnesium, and this metal was coordinated to a nonbridging P2 oxygen rather than one from P1. In addition, the Fo − Fc electron density for the magnesium site was weaker in the GPP and FPP structures than in the DMAPP structure. The difference may have been produced as the result of the poorer diffracting crystals. Alternately, the second magnesium site in the GPP and FPP structures may actually be occupied by a water molecule. A superposition of the DMAPP, GPP, and FPP-F112A/F113S structures on the apo structure showed that the first magnesium atoms in the DMAPP and GPP structures were within 0.5 Å of each other, while the first magnesium from the FPP structure was displaced 1.1 and 1.6 Å from these two, respectively. A similar result was obtained for the second magnesium in the three structures. The DMAPP and GPP sites were 0.2 Å from each other, whereas the FPP site was 0.9 and 0.8 Å from the former two, respectively. This alignment also showed that in all the liganded structures the distance between the carboxyl groups of Asp-117 and Asp-121 decreased. The change was from 4.5 to 3.6 Å in going from the apo to the FPP structure.
Figure 3.
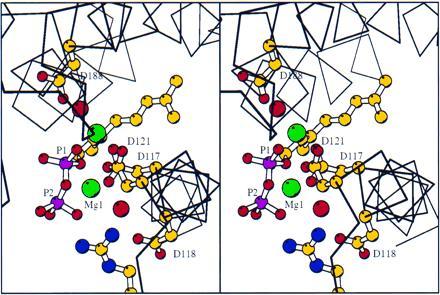
Stereoview of GPP binding in the active site of mutant F112A/F113S-FPS. The GPP is shown with phosphorus atoms magenta. The nitrogens of R126 are colored blue. Magnesiums are shown in green, and waters are shown as red spheres slightly larger than the carboxylate oxygens of the aspartates.
Two additional interactions between highly conserved amino acids and the allylic diphosphate may be important for catalysis. The first is between the nonbridging oxygens of P2 and the guanidino nitrogens of R126. This interaction may participate in binding the allylic diphosphate during catalysis. Additionally, it could function by exerting an electrostatic force that would aid in lysis of the diphosphate, and/or help pull it out of the active site afterward. Similarly, the conserved R127, located next to R126, could participate by pulling the diphosphate further out toward bulk solvent. The R126–diphosphate interaction may help explain why monophosphate esters of isoprenes are not substrates of FPS. The second interaction, between K214 and P1 of the allylic diphosphate, could aid catalysis by electrostatically stabilizing developing negative charge on the oxygen attached to C1, or by donating an ɛ-amino proton to the P1 oxygen as the diphosphate leaves. A deprotonated K214 would be in position to provide dipolar stabilization to the carbocation intermediates in the reaction, and perhaps to assist with removal of the C2 proton of IPP. A yeast mutant (26) was described that had a K → E mutation in the residue equivalent to K124 in avian FPS. Cell-free extracts from the mutant strain had reduced FPS activity and an increase in the amount of GPP that accumulated relative to FPP when chain elongation was initiated with IPP and DMAPP.
The orientations of the DMAPP, GPP, and FPP hydrocarbon tails in the F112A/F113S crystal structures provide the strongest evidence for the important role of the mutated phenylalanine residues in chain length regulation. The tails of all three compounds were oriented in the same configuration, growing down the hydrophobic pocket toward the mutation site. The F112A/F113S structure containing bound FPP (Fig. 4) shows the distal end of the isoprene tail located at precisely the site where F112 and F113 were located in the wt enzyme. When these side chains from the wt structure were overlaid on the F112A/F113S-FPP structure it was evident that chain growth beyond 15 carbons would be strongly inhibited in the wt enzyme by steric hindrance from the phenyl rings.
Figure 4.
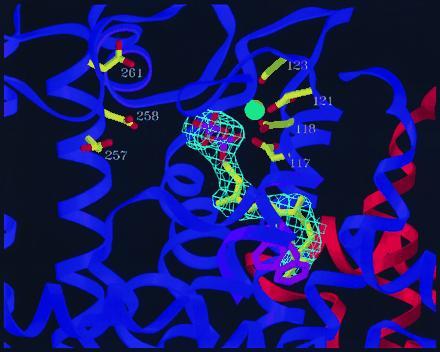
Mutant F112A/F113S-FPS dimer showing orientation of bound FPP in the active site. The ligand is shown bound to the purple subunit, while the other (orange) subunit makes up part of the back wall of the binding pocket. The diphosphate group of FPP is bound to the aspartates of the first conserved DDXXD sequence (D117–D121) through a bridge of two magnesiums (one shown). Residues 109–115 of the wt structure (α-helix ribbon in light purple, sidechains of F112 and F113 in pink) are superimposed on the F112A/F113S structure. The prenyl tail of the ligand ends at precisely where the phenyl rings of residues F112 and F113 would be in the wt structure, thus blocking further chain elongation. Electron density (2Fo − Fc) observed for the ligand is shown in light blue.
During chain elongation catalyzed by avian FPS, the double bond in IPP is alkylated by C1 of the allylic substrate in a stereospecific reaction (27). The alkylation is from the si face of the double bond and proceeds with inversion of configuration at C1 of the allylic substrate (Fig. 5). The pro-R hydrogen is then removed from C2 of IPP with concomitant formation of an E-double bond in the allylic product (27). The allylic substrates in the cocrystal structures are oriented so the back side of C1 faces outward toward the putative DDXXD binding site for IPP, in a manner consistent with inversion at that carbon. Because attempts to obtain interpretable difference electron density for IPP in maps prepared from cocrystals of IPP-GCPP-F112A/F113S were unsuccessful, IPP was modeled into diffuse electron density observed at its putative binding site. The resulting structure placed C4 of IPP within 2.0 Å of C1 of GCPP. The structure is not sufficiently refined to specify interactions between the diphosphate moiety in IPP and the second DDXXD residues or to define the base responsible for stereospecific deprotonation of C2 in IPP.
Figure 5.
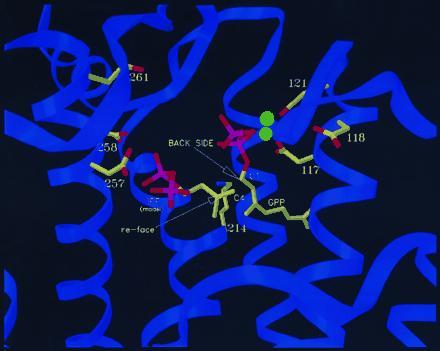
Mutant F112A/F113S-FPS-GPP structure with IPP modeled into proposed conformation. The configuration of bound GPP is consistent with the observed stereochemistry of the reaction. The proposed conformation of bound IPP places it in an arrangement such that the si-face of its double bond can attack GPP-C1 to produce an E-double bond. The ɛ-amino group of K214 is in proximity of the allylic diphosphate and IPP.
A Model for Long Chain Isoprene Growth in F112A/F113S Mutants.
Attempts to obtain crystal structures of F112A/F113S with products greater that 15 carbons in length were unsuccessful due to the decreasing solubility of the longer chain isoprenoid diphosphates in the presence of magnesium. As an alternative, molecular modeling was used to produce hypothetical structures of F112A/F113S containing all E-octaprenyl diphosphate (OPP). It was surmised that mutant FPS may form isoprene products longer than 20 carbons by undergoing conformational changes that deepen the binding pocket. This may be accomplished by displacing of the side chains of the residues that form the new floor of the hydrophobic pocket. For example, by rotating the side chains of just two residues at the bottom of the binding pocket (Y154 and E150), a passageway is opened through the dimer interface into the bottom of the adjoining subunit’s binding pocket. These rotations do not generate collisions with the side chains of adjacent residues. Rotations of the side chains of eight more residues (L157, K158, R162, Y167, L171, E172, F174, L175) open a new passageway to the outside surface of the enzyme. Again, these side chains can be rotated without interference from the side chains of neighboring residues. The resulting passageway was lined almost entirely by hydrophobic residues. OPP was modeled into the passage, with its diphosphate group placed in the same position as observed in the F112A/F113S-FPP structure (Fig. 6). The allylic diphosphate spanned the distance from the first aspartate binding site, into the binding pocket, and through the newly opened passageway to the outside surface of the enzyme. The resulting structure was minimized, using discover (23), to an rms energy derivative of 4.2 kcal·mol−1·Å−1.
Figure 6.
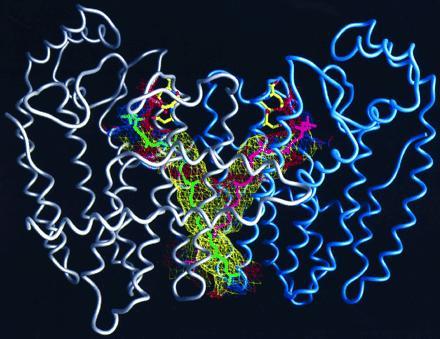
Structure of mutant F112A/F113S-FPS dimer with two C-40 isoprenes, shown in green and magenta, modeled into proposed binding channels. The first subunit of the dimer is shown with a gray α-carbon worm, and the second is in blue. The diphosphate heads of the two isoprenes are shown binding to the aspartates of the first conserved DDXXD sequences of each subunit in the same configuration observed for DMAPP, GPP, and FPP. The walls of the channels that contain the growing hydrophobic tails of the prenyl products are shown in mesh. The mesh is colored according to the hydrophobicity of the protein atoms that it is composed of, with yellow being hydrophobic, red acidic, and blue basic.
An analysis of the x-ray structure of wt avian FPS suggested that the ultimate length of the polyisoprenoid chain obtained during successive condensations of the growing allylic substrate with IPP is governed by the size of a hydrophobic pocket in the interior of the enzyme. Substitution of the benzyl groups in F112 and F113 with smaller side chains gave FPS mutants that synthesized C20 (F112A), C25 (F113S), and longer (F112A/F113S, F112A/F113S) isoprenoid products. X-ray structures were obtained for the apo form of the F112A/F113S mutant along with those for the protein ligated with DMAPP, GPP, and FPP. The structures with bound allylic substrates DMAPP, GPP, and FPP showed the hydrocarbon chain growing into the hydrophobic pocket as predicted during an analysis of the wt FPS structure and confirms the role of the pocket in substrate binding and regulation of chain length. The F112A/F113S structures also revealed that the diphosphate moiety in the allylic substrate was bound to the side chains of D117 and D121 in the first conserved aspartate-rich region through a magnesium bridge. A second magnesium was located in the DMAPP form of the enzyme that also formed a bridge between the carboxylates of D117 and D121 and the diphosphate moiety. An interaction between oxygens of the P2 phosphorus and R126 appears to help anchor the diphosphate, and may also aid in catalysis. The position of the ɛ-amino group of K214 suggests that it may function in catalysis by activation of the diphosphate leaving group, and perhaps subsequent stabilization of the carbocation intermediates and deprotonation of C2 of IPP. These studies have provided the clearest picture thus far for how an isoprenyl diphosphate synthase catalyzes prenyl transfer and regulates the length of the polyisoprenoid chain in its product.
Acknowledgments
We thank Susan Baughman for purification of the F112A/F113S mutant for crystallization studies. P.J.P. thanks the National Institutes of Health for a National Research Service Award (GM16732). This work was supported by National Institutes of Health Grants GM07260 (L.C.T.), GM45859 (J.C.S.), and GM21328 (C.D.P.).
Footnotes
Abbreviations: DMAPP, dimethylallyl diphosphate; FPP, farnesyl diphosphate; FPS, farnesyl diphosphate synthase; GPP, geranyl diphosphate; GCPP, geranylmethyl phosphonophosphate; GGPP, geranylgeranyl diphosphate; IPP, isopentenyl diphosphate; wt, wild type.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, Chemistry Department, Brookhaven National Laboratory, Upton, NY 11973 [reference 1UBV (apo), 1UBW (GPP), 1UBX (FPP), 1UBY (DMAPP)].
Over 30 additional prenyltransferase sequences have been deposited in data bases since the study with 13 sequences (6) was published. The sequences were identified by performing a blast search (7) of the nonredundant protein data bases through the National Center for Biotechnology Information using FPP and GGPP synthase sequences.
References
- 1.Anderson M S, Yarger J G, Burck C L, Poulter C D. J Biol Chem. 1989;264:19176–19184. [PubMed] [Google Scholar]
- 2.Jiang Y, Proteau P, Poulter C D, Ferro-Novick S J. J Biol Chem. 1995;270:21793–21799. doi: 10.1074/jbc.270.37.21793. [DOI] [PubMed] [Google Scholar]
- 3.Ashby M N, Edwards P A. J Biol Chem. 1990;265:13157–13164. [PubMed] [Google Scholar]
- 4.Tarshis L C, Yan M, Poulter C D, Sacchettini J C. Biochemistry. 1994;33:10871–10877. doi: 10.1021/bi00202a004. [DOI] [PubMed] [Google Scholar]
- 5.Reed B C, Rilling H C. Biochemistry. 1975;14:50–54. doi: 10.1021/bi00672a009. [DOI] [PubMed] [Google Scholar]
- 6.Chen A, Kroon P, Poulter C D. Protein Sci. 1994;3:600–607. doi: 10.1002/pro.5560030408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 8.Kunkel T A, Robert J D, Zakour R A. Methods Enzymol. 1986;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 9.Song L, Poulter C D. Proc Natl Acad Sci USA. 1994;91:3044–3048. doi: 10.1073/pnas.91.8.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reed B C, Rilling H C. Biochemistry. 1976;15:3739–3744. doi: 10.1021/bi00662a015. [DOI] [PubMed] [Google Scholar]
- 11.Bartlett D L, King C-H R, Poulter C D. Methods Enzymol. 1985;110:171–184. doi: 10.1016/s0076-6879(85)10073-x. [DOI] [PubMed] [Google Scholar]
- 12.Ohnuma S, Koyama T, Ogura K. J Biol Chem. 1991;266:23706–23713. [PubMed] [Google Scholar]
- 13.Zhang D, Poulter C D. Anal Biochem. 1993;213:356–361. doi: 10.1006/abio.1993.1432. [DOI] [PubMed] [Google Scholar]
- 14.Fujii H, Koyama T, Ogura K. Biochim Biophys Acta. 1982;712:716–718. [PubMed] [Google Scholar]
- 15.Jancarik J, Kim S H. J Appl Crystallogr. 1991;24:409–411. [Google Scholar]
- 16.Corey E J, Volante R P. J Am Chem Soc. 1976;85:1291–1293. [Google Scholar]
- 17.Siemens Industrial Automation (1995) saint Software Reference Manual (Siemens, Madison, WI), Part 269–014200.
- 18.Brunger A T, Kuriyan J, Karplus M. Science. 1987;235:458–460. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- 19.Tronrud, D. E. (1994) tnt 5E Refinement Package (Oregon State Board of Higher Education, Eugene, OR).
- 20.Jones T A. Methods Enzymol. 1985;115:157–171. doi: 10.1016/0076-6879(85)15014-7. [DOI] [PubMed] [Google Scholar]
- 21.Ohnuma S, Nakazawa T, Hemmi H, Hallberg A M, Koyama T, Ogura K, Nishino T. J Biol Chem. 1996;271:10087–10095. doi: 10.1074/jbc.271.17.10087. [DOI] [PubMed] [Google Scholar]
- 22.Laskovics F M, Poulter C D. Biochemistry. 1981;20:1893–1901. doi: 10.1021/bi00510a027. [DOI] [PubMed] [Google Scholar]
- 23.Molecular Simulations (1995) insight/discover (Molecular Simulations, San Diego), Version 95.0.
- 24.Ashby M N, Spear D H, Edwards P A. In: Proceedings of the 20th Steenbeck Symposium. Attie A D, editor. New York: Elsevier; 1990. pp. 27–35. [Google Scholar]
- 25.King H L, Rilling H C. Biochemistry. 1977;16:3815–3819. doi: 10.1021/bi00636a015. [DOI] [PubMed] [Google Scholar]
- 26.Blanchard L, Karst F. Gene. 1993;125:185–189. doi: 10.1016/0378-1119(93)90326-x. [DOI] [PubMed] [Google Scholar]
- 27.Cornforth J W, Cornforth R H, Popjak G, Yengoyan L. J Biol Chem. 1966;241:3970–3987. [PubMed] [Google Scholar]