

Summary
Cytokines play pivotal roles in immunity and inflammation, and targeting cytokines and their receptors is an effective means of treating such disorders. Type I and II cytokine receptors associate with Janus family kinases (JAKs) to effect intracellular signaling. These structurally unique protein kinases play essential and specific roles in immune cell development and function. One JAK, JAK3, has particularly selective functions. Mutations of this kinase underlie severe combined immunodeficiency, indicative of its critical role in the development and function of lymphocytes. Because JAK3 appears not to have functions outside of hematopoietic cells, this kinase has been viewed as an excellent therapeutic target to for the development a new class of immunosuppressive drugs. In fact, several companies are developing JAK3 inhibitors and Phase II studies are underway. Mutations of Tyk2 cause autosomal recessive hyperIgE syndrome and in principle, Tyk2 inhibitors might also be useful as immunosuppressive drugs. JAK2 gain-of-function mutations (V617F) underlie a subset of disorders collectively referred to as myeloproliferative diseases and phase 2 trials using JAK inhibitors are underway in this setting. Thus, we are learning a great deal about the feasibility and effectiveness of targeting Janus kinases and it appears likely that this will be a fruitful strategy in a variety of settings.
Introduction
Cytokines have critical functions in regulating many aspects of immunity and inflammation, ranging from the development and differentiation of immune cells to the suppression of immune responses. Type I and II cytokine receptors represent a structurally distinct class of receptors that lack intrinsic enzymatic activity capable of mediating signal transduction. Reversible protein phosphorylation is a fundamental aspect of signaling by many receptors and is a major means by which information is transmitted from outside the cell and between components within the cell. Protein kinases or phosphotransferases catalyze the transfer of γ phosphate of a purine nucleotide triphosphate, (ATP or GTP) to the hydroxyl groups of their protein substrates and are classified by the amino acid substrate preference. Thus, protein kinases are designated serine/threonine kinases, tyrosine kinases and dual kinases (meaning that both serine/threonine and tyrosine residues are phosphorylated).
Cytokine stimulation of cells was found to induce tyrosine phosphorylation of various cellular substrates, but initially the protein tyrosine kinases (PTKs) responsible were unknown. We now know that the human genome comprises 518 protein kinases and the PTK subfamily has 90 members. The mystery of how cytokine receptors signaled was solved with the discovery of a new class of PTKs known as the Janus family of kinases (JAKs). The JAKs, which include Tyk2, JAK1, JAK2, and JAK3, were identified using PCR-based strategies and low-stringency hybridization [1-7]. The completion of the human genome project has indeed verified that, in fact, only four JAKs are present in the human kinome [8].
Evidence for a critical role of JAKs in cytokine signaling initially came from studies using mutagenized cell lines defective in interferon (IFN) signaling, which revealed that Tyk2 is essential for IFN signaling. This observation was quickly followed by studies showing that the other JAKs were activated by various cytokines and associated with different cytokine receptors. The essential function of this family of kinases has been now been established by generating knockout mice for each of the various JAKs [9-17].
Parallel to the discovery of the role of JAKs in cytokine signaling, the Stat family of transcription factors was also discovered; together these advances provided a new paradigm in cell signaling, and is now known as the JAK/Stat pathway.
Structure of JAKs
The structure of a complete JAK molecule has yet to be solved; however, we know that the carboxy-terminus of Janus kinases contains the catalytic or domain. Structurally, the catalytic domains of all typical protein kinases consist of two lobes (N-lobe and C-lobe) that surround the nucleotide binding site. The structure of the isolated JAK3 kinase domain has been solved and shows that the overall structure is like other PTKs, but also has unique features [18]. Mutations in this domain inhibit kinase activity and/or protein expression [19].
Like other protein tyrosine kinases, JAK have a so-called “activation loop” that regulates kinase activity and is a major site of autophosphorylation. Of note, phosphorylation in the JAK activation also loop allows one member of a family of negative regulators termed suppressors of cytokine signaling (SOCS), SOCS1, to bind and inhibit JAK activity. Deficiency of SOCS1 can enhance signaling by γc cytokines [20-22]. In addition, SOCS1 forms an E3 ubiquitin ligase complex with elongins B and C, Cullin-5 (Cul-5), and Rbx1 to mediate the ubiquitination of JAKs, which may promote proteasome-dependent degradation [23,24].
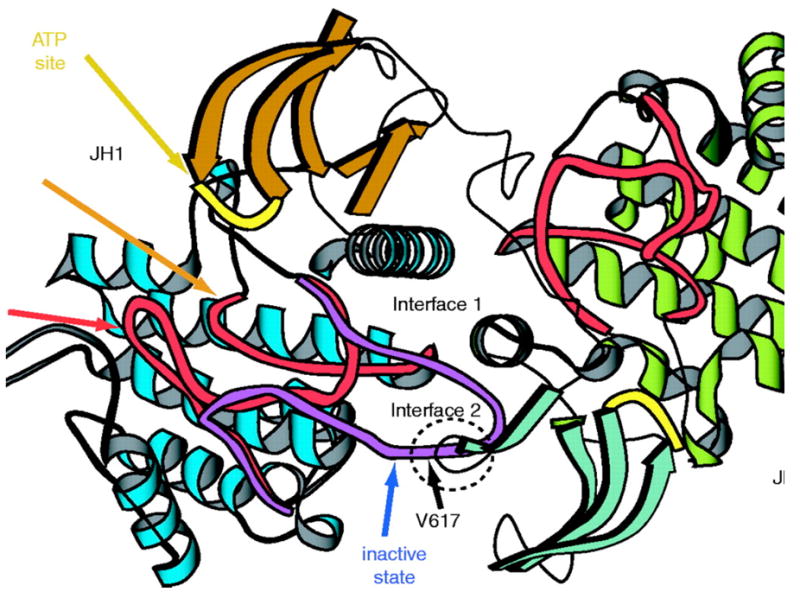
What makes the JAKs structurally unique is the existence of an enzymatically inactive pseudokinase domain immediately N-terminal to the kinase domain (Fig. 1). The pseudokinase domain has a high degree of sequence similarity to the kinase domain, but several residues required for phosphotransferase activity are altered from the canonical motifs. This unique domain gives JAKs their name. Like the Roman god Janus, JAKs are “two-faced” – that is, they have a kinase and kinase-like domains. Though the pseudokinase domain itself lacks catalytic activity, this domain has critical functions in regulating catalytic activity [25,26]. As discussed below, mutations of JAK2 V617 underlie the spectrum of diseases known as the chronic myeloproliferative disorders (MPD), which include polycythemia vera (PV), essential thrombocytosis (ET) and myelofibrosis (MF) [27-31]. This mutation is thought to result in a constitutively active kinase and thus, the pseudokinase domain appears to have essential positive and negative regulatory roles.
Figure 1.
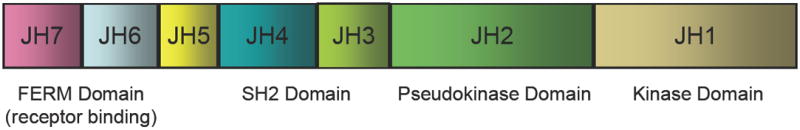
Structure of Janus kinases. A. Janus kinases have a carboxy-terminal kinase domain that is homologous to other PTKs, although some features are unique. What makes JAKs distinctive is the existence of a pseudokinase or kinase-like domain. This domain itself does not have catalytic activity, but it is essential in regulating enzymatic activity. JAKs have a src homology 2 (SH2) but its function is not known. The amino terminus of the JAKs comprises a band 4.1, ezrin, radixin, moesin domain, which binds to cytokine receptors and also regulates kinase activity. B. Though the crystal structure of the JAKs is lacking, a putative model can be generated.
JAKs are also phosphorylated on other sites that appear to be important for regulation of catalytic activity. For instance, Y785 and Y813 in JAK3 and JAK2 respectively are other prominent sites of autophosphorylation [32]. These sites serve to recruit SH-2 domain containing adapter molecules such as SH-2Bβ. For JAK2, this is a mechanism to enhance catalytic activity. In contrast, another related adapter, APS, also binds JAK2, but it negatively regulates activity [33]. The role of SH2Bβ and APS in regulating JAK3 activity is less clear. JAK2 and JAK3 have also been reported to complex with STAM- AMSH, hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) proteins [34-36]. The STAM/EAST/Hbp family of proteins comprises eight members well conserved from yeast to mammals. These proteins become tyrosine-phosphorylated by a variety of cytokines and growth factors and are thought to play a role in intracellular protein trafficking and the regulation of actin cytoskeleton. However, the precise role of these adapters in cytokine signaling has not been elucidated [37,38].
The amino terminus of JAKs comprises a FERM (band four point one, ezrin, radixin, moesin) domain and mutations identified in this region have established that this domain mediates association with the cognate cytokine receptor and regulates kinase activity[39-41]. JAKs also have an SH2 domain, but its function remains unclear.
Immunologic functions of JAK3
Because JAKs selectively associate with different cytokine receptors, they have functions that can be attributed to these various cytokines. JAK3 probably has the most discrete function, as it associate with only one cytokine receptor - the common gamma chain or γc. This is a shared receptor subunit that pairs with other ligand-specific subunits to form the receptors for interleukin (IL)-2, IL-4, IL-7, IL-9, IL-15 and IL-21. Germline deletion of γc in mice or mutations of γc in humans (denoted IL2RG) results in a disorder known as severe combined immunodeficiency (SCID). In humans, this characteristic phenotype is designated as T-B+NK-SCID meaning that T and NK cells are absent. B cells are present but their function is not normal. The association of γc and JAK3 led to the speculation that mutations of JAK3 might also be a cause of SCID and, in fact, analysis of humans with autosomal recessive T-B+NK-SCID revealed such mutations. In aggregate, mutations of γc/JAK3 account for the majority of all cases of SCID, and as best we can tell, mutations of γc (IL2RG) or JAK3 result in the identical clinical phenotype. While it is clear that mutations of JAK3 have profound effects on the immune system, it is also equally clear that these patients do not have other deficits. Moreover, hematopoietic stem cell transplants are curative, arguing for very discrete functions of JAK3.
The immune abnormalities associated with JAK3 deficiency were confirmed with the generation of JAK3 knockout mice; similar to humans, these mice have SCID that resembles γc deficiency with small thymuses, absence of lymph nodes and reduced numbers of α/β and γ/δ T cells[12-14]. JAK3-/- mice also have a profound reduction in thymic progenitor cells and reduced ability to reconstitute T cell development [42]. In the periphery, CD8+ T and NK cells are severely reduced. JAK3- (and γc) deficient mice also have severely defective B cell development [12-14,43,44], a phenotype that is not evident in humans. The reason for the difference between mice and humans with respect to the roles of γc and JAK3 in B cell development is unclear at this time but presumably relates to species-specific cytokine usage.
No effect on the development of myeloid or erythroid cells was noted in JAK3-deficient mice consistent with observations in humans and indicative of a specific effect on lymphoid precursors. However, murine JAK3 deficiency is associated with a T cell-dependent autoimmune disease characterized by infiltration of tissues by mononuclear cells, splenomegaly, and expansion of neutrophils and monocytic cells. This autoimmune disease is T cell dependent [45].
In terms of the SCID phenotype, inhibition of IL-7 receptor explains many of the abnormalities associated with deficiency of γc and JAK3. In mice, IL-7R deficiency disrupts thymocyte development at the double negative (CD4-CD8-) stage prior to productive T cell receptor rearrangement, which results in marked reduction in thymocytes and peripheral T cells, including γδ T cells. Furthermore, mutations of IL7R underlie about 10% of autosomal recessive SCID patients. In the mouse, the IL-7R is also critical for B cell development, in part due to its role in regulating the transcription factor Pax-5 [46]. However, B cells are present in patients with JAK3, γc, and IL-7R mutations, indicating that IL-7/IL-7R is dispensable for human B cell development [47]. IL-7/IL-7R signaling is not essential for NK cell development and therefore, the phenotype associated with Il7R mutations is distinct from JAK3-SCID and X-SCID. Specifically, patients with IL7R mutations lack T cells but do have NK and B cells (T-B+ SCID). IL-7’s importance is not just limited to lymphoid development, it is also important for the homeostasis of mature peripheral lymphocytes and T cell memory. In concert with IL-15, IL-7 promotes the survival of CD8+ memory cells. IL-7 is less critical for the generation and survival of CD4+.
IL-2 is the prototypic T cell growth factor, and although it was expected this cytokine would be essential for development and function of T cells, this is not the case. In fact, mice lacking either IL-2 or IL-2R (IL-2Rα and β) are normal with respect to thymus development and peripheral T cell subset composition [48-50], but surprisingly, exhibit severe lymphoproliferative disorder. One reason for this is that IL-2 is an important growth factor for T regulatory cells (see below) [51,52]. In addition, IL-2 inhibits Th17 cell differentiation (see below). Patients with mutations of the IL-2Rα subunit also have extensive lymphocytic infiltration and inflammation [53,54]. In principle therefore, lack of IL-2 signaling in JAK3-SCID patients could result in autoimmunity. In fact, not all patients with γc and JAK3 mutations have profound lymphopenia [55-58]. If T cells are generated, it is possible that autoimmune manifestations could occur and a JAK3- deficient patient with a mixed picture of immunodeficiency and autoimmunity has been identified. It is possible that the lack of IL-2 signaling could be one contributor to this clinical presentation [56].
Another γc cytokine, IL-4 is not essential for T cell development but is important for the differentiation of CD4+ T cells to the T helper 2 (Th2) phenotype. Th2 cells themselves produce IL-4 along with IL-5 and IL-13. These cytokines are important for host defense against helminthes but in addition contribute to the pathogenesis of allergies and asthma. IL-4 also activates B cells and promotes class switching. IL-9 is also a γc-using cytokine, but its absence does not result in major developmental effects on immune cells [59,60].
IL-21 has recently come to the fore as a critical immunoregulatory cytokine. Selectively produced by Th17 cells, IL-21 further promotes Th17 differentiation in a manner analogous to IL-4 and Th2 differentiation[61-64]. In contrast, IL-2 inhibits Th17 differentiation [65]. IL-4 and IL-21 also work in concert to regulate B cell function [66].
Another critical subset of CD4+ T cells is the regulatory T cell or Treg cell. Absence of Treg cells is associated with fatal autoimmunity in mice and man[51,67-69]. In vitro, IL-2 and transforming growth factor β (TGFβ)-1 induce Treg differentiation; however, deficiency of IL-2 alone does not completely abrogate Treg differentiation. However, deficiency of γc or JAK3 profoundly impair Treg differentiation implying a need for some γc cytokine in the development of these critical cells[70,71].
The clear genetic evidence in mouse and man implies that JAK3 is only essential and non-redundant with respect to its role in the immune response. This apparent specificity of JAK3 function in the immune system has important implications for the development of a new class of immunosuppressive drugs. Despite the unequivocal genetic evidence, it is possible that JAK3 is expressed in cells other immune cells – indeed databases that catalogue gene expression suggest that JAK3 can be expressed in nonhematopoietic cells. Nonetheless, JAK3-SCID patients and Jak3-/- mice indicate that this kinase does not have essential roles outside hematopoietic cells.
Function of other JAKs
As mentioned, TYK2 was the prototype that linked this family of kinases with cytokine signaling and abundant evidence from cell lines pointed to a critical role of Tyk2 in signaling by Type IFNs. Accordingly, generation of Tyk2 gene targeted mice revealed that absence of this gene resulted in increased viral susceptibility[16]. However the effect was less dramatic than expected in that IFNα/β signaling was reduced but not abolished. Tyk2 is also involved in signaling by IL-12 and IL-23. A naturally occurring mutant of Tyk2 was identified in B10.Q/J mice, which were found have increased resistance to collagen-induced arthritis and susceptibility to infection with Toxoplasma gondii[72-74]. Tyk2 had been reported to be activated by a large number of other cytokines, but their signaling was noted to be intact Tyk2-/- mice. A single patient with autosomal recessive hyperimmunoglobulin E (AR-HIES) or Job’s syndrome has been described with mutations of TYK2[75,76]. This patient suffered from multiple opportunistic infections. In contrast to what was reported in the mouse, signaling by a wide variety of cytokines including Type I IFNs, IL-6, IL-10, IL-12, and IL-23 was found to be impaired. Thus the requirement for Tyk2 for various cytokines remains somewhat unclear and probably reflects cell- and possibly species-specific effects.
In contrast, to TYK2 and JAK3, deficiency of JAK1 and JAK2 is lethal. Jak2-/- mice die as embryos due to the absence of definitive erythropoiesis. The cells fail to respond to erythropoietin, thrombopoietin, IFN-γ, IL-3, or granulocyte/macrophage colony-stimulating factor (GM-CSF). In contrast, the response to granulocyte-CSF was reportedly unaffected. Jak1-/- mice are runted at birth, fail to nurse, and die perinatally. Jak1-/- cells are unresponsive to three distinct families of cytokine receptors – all class II cytokine receptors (IFNs and IL-10 related cytokines), γc-cytokines, as well as IL-6 and other gp130-using cytokines.
Jak mutations and leukemia
Fusion proteins comprising the oligomerisation domain of the transcription repressor, translocated ets leukemia (TEL) and JAK2 have been reported in both myeloid and lymphoid leukemias [77,78]. Fusion proteins of all the JAK family kinases with TEL have been reported to produce active kinases that constitutively phosphorylate STAT5 and induce growth factor independence in Ba/F3 cells [79]. Finally, JAK2 inhibitors have shown activity against primary human acute myeloid [80] and lymphobastic [77,78] leukemias suggesting an important role for this kinase in the patho-physiology of these diseases.
Individuals with Downs syndrome (DS) are predisposed to develop acute megakaryoblastic leukemia (AMKL). The subsequent development of leukemia in DS is often preceded earlier on in life by a transient myeloproliferative disorder (TMD). Acquired mutations JAK3 have been reported in both DS patients with TMD or AMPK, and non-DS patients with AMKL[81,82]. Walters et al have gone on to show that the point mutations in JAK3 affect the pseudokinse domain and render the kinase constitutively active. It is of note that murine bone marrow transfected with different mutations of JAK3 induced widely differing forms of leukemia with the A572V mutation leading to a T cell lymphoproliferative disorder, where as the V617F mutation lead to myeloproliferative disease.
The BCR-Abl negative classical myeloproliferative diseases, PV, ET and MF, have associated with a high incidence of mutations in JAK2 [29-31,83]. The incidence of the V617F JAK2 mutation in PV is a near universal 95% and is analogous to the presence of BCR-Abl in CML. Most of the remaining 5% of PV patients that are V617F negative are positive for other activating mutations in JAK2. While less significant, the V617F JAK2 mutation is seen in 50% of patients with MF and ET. [29-31,83]. In contrast to the TEL-JAK2 fusion protein, the V617F mutation is not seen in any human lymphoid malignancy.
Development of protein kinase inhibitors
Before discussing the generation of a selective JAK3 inhibitor, it is worth considering the general aspects related to the development of PTK inhibitors. In terms of its catalytic role, the kinase domain has three functions: the binding of the ATP (or GTP) phosphate donor as a complex with divalent cation (usually Mg2+ or Mn2+), the binding of the protein substrate, and the transfer of the γ-phosphate from ATP or GTP to the protein substrate. However, protein kinases are not the only kinases – there are lipid kinases, nucleotide kinases, etc, as well as many other ATP-binding proteins in cells. Moreover, despite the many potential ways of designing a small molecule kinase inhibitor, in practice, most successful kinase inhibitors interact with ATP binding pocket. Structurally the kinase domain of all typical protein kinases is highly conserved and consists of two lobes (N-lobe and C-lobe) that surround the nucleotide binding site. Of the more than 500 human kinases, many serve critical cellular functions. Thus, it was rather unclear that it would be possible to generate selective PTK inhibitors that would have not have substantial toxicity. For all these reasons, the idea that targeting protein kinase would be a useful strategy was not universally embraced.
However, this skepticism appears to be unfounded as we now have a number of FDA-approved kinase inhibitors. The first FDA-approved protein kinase inhibitor is imatinib (Table I). The mutated form of the Abl tyrosine kinase, BCR-Abl represents a fusion protein consisting of the oligomerization domain of a transcription factor (BCR) and the kinase domain of Abl. This is the result of a pathognomonic chromosomal translocation (Philadelphia chromosome) seen chronic myeloid leukemia (CML) [84] and has been one of the most intensively studied protein tyrosine kinases. Because this kinase was uniquely present in malignant transformation it was thought to be an ideal target for the generation of a selective kinase inhibitor and indeed, imatinib has revolutionized the treatment of CML. Not only is it remarkably effective but is also well tolerated with minimal side effects [85]. The epidermal growth factor receptor is another intensively studied PTK, which is highly expressed and/or mutated in cancers. It too has been considered a good therapeutic target and gefitonib was the first selective EGFR inhibitor. Gefitonib was not efficacious in a nonsmall cell lung cancer trial; however, erolotinib, another EGFR inhibitor has been approved by the FDA for non-small cell lung cancer and pancreatic cancer. Given its importance primary and metastatic tumors, targeting angiogenesis therapeutically is an intense area of research. Accordingly, the vascular endothelial growth factor receptor another family of PTK that has received great attention as therapeutic targets. At present, there are seven FDA-approved PTK inhibitors (Table 1).
Table 1.
FDA-approved kinase inhibitors.
| Compound | Kinase target | Clinical Use |
|---|---|---|
| Desatinib | multikinase | CML, ALL |
| Erlotinib | EGFR | |
| Gefitinib | EGFR | |
| Imatinib | BCR-Abl | CML
Gastrointestinal Stromal Tumors Hypereosinophilic syndrome |
| Lapatinib | EGFR, Erb2 | Breast cancer, other tumors |
| Lestaurinib | FLT3 | AML |
| Sorafenib | multikinase | Renal cell cancer |
Abbreviations: ALL – acute lymphocytic leukemia, CML – chronic myelogenous leukemia, EGFR – epidermal growth factor receptor, Erb2 – FLT3 – Fms-like tyrosine kinase 3,
The generation of therapeutically successful PTK inhibitors begs reconsideration of a number of issues that initially appeared to be serious impediments. Though the conserved structure of the ATP binding pocket was initially envisioned as a major barrier to designing kinase inhibitors, in practice this has not happened for a number of reasons. Although different kinases are structurally similar in their active, ATP-bound, confirmation, the inactive confirmations are distinct [86]. The ATP binding region is made up of six polar amino acid residues, which are invariant among kinases. Similarly, a number of lipophillic residues are also highly conserved but this critical region also contains an amino acid termed ‘the gate keeper residue’ [87]. In the active state, the amide carbonyl of this residue binds to N-6 of adenine. However, in the inactive state, side chain of the gatekeeper residue occludes the reaction pocket. Within the kinase superfamily almost any amino acid can serve as the gate-keeper although in practice it is typically a bulky non-polar residue (methionine, tyrosine, phenylalanine, lysine) [87]. As the side chain is not involved in direct ATP binding it varies among kinases, and variation of this gatekeeper residue is exploited by a number of inhibitors that bind the inactive confirmation of specific kinases. For instance, the Abl kinase the gatekeeper residue is threonine, which binds directly to a methyl group of the phenyl ring of the Abl kinase inhibitor imatinib [86]. In principle, detailed structural information should allow investigators to capitalize upon subtle differences in structure to improve potency and specificity.
A second major surprise in the emerging field of kinase inhibition is that absolute specificity is not necessary for an effective and safe drub. Even imatinib, which was designed to be highly specific inhibitor for Abl kinase, has activity against other PTKs; in fact, this has turned out to be a benefit [88,89]. Specifically, it has been found to be useful in the treatment of that has led to its successful use in the treatment of gastrointestinal stromal tumor, and hypereosinophilic syndrome through its effects on c-kit receptor tyrosine kinase and FIP1L1-PDGFRA [90]. Perhaps even more surprising is that partial inhibition of multiple kinases probably contributes efficacy and is less toxic than originally feared. For instance, dasatinib and sunitinib are FDA approved multi-targeted PTK inhibitor approved for cancer treatment. Sorafenib inhibits receptor tyrosine kinases including VEGFR and the platelet-derived growth factor receptor, but also inhibits the serine/threonine kinase RAF. In the recent screen, sunitinib and dasatinib were found bind >15% of kinases tested with Kd < 100 nM. Nonetheless, these drugs evidently have acceptable toxicity in the setting of treatment of malignancy [91].
The development of a selective JAK3 antagonist
The selective but essential functions of JAK3 made it a very attractive candidate for the development of inhibitors with the idea that interfering with JAK3 function could be the basis of a novel class of immunosuppressants or anti-cancer drugs. In principle, a highly selective JAK3 inhibitor could have very limited and specific effects, as JAK3-SCID patients do not exhibit pathology outside the immune system. This contrasts with many of the widely used immunosuppressive drugs (calcineurin inhibitors, mTOR inhibitors, anti-metabolites and corticosteroids), which target ubiquitously expressed molecules, and thereby have attendant toxicities. The adverse effects of these drugs remain a major cause of morbidity and mortality in the treatment of transplant rejection and autoimmune disease, since treatment is usually lifelong. Consequently, a potent and selective JAK3 inhibitor would be expected to have significant advantages over current regimens.
A number of compounds have been reported to selectively inhibit JAK3 [92] and several pharmaceutical companies have ongoing programs to target JAK3. The compound that has progressed furthest in clinical studies is CP-690,550 [93]. CP-690,550 has nanomolar potency in in vitro JAK3 kinase assays, with similar potency in various cell-based assays that are dependent upon JAK3 activity. The in vivo efficacy of the drug as monotherapy was established by its prevention of transplant rejection in a murine heterotopic heart transplant model, and in nonhuman primate renal transplant model. Prolongation of graft survival was found to correlate well with the inhibition of cytokine-inducible genes in vivo. The total number of T lymphocytes did not diminish in animals treated with CP-690,550, but there was a trend toward a reduction of CD8+ T cells, consistent with the documented effects of γc cytokines on CD4+ and CD8+ lymphocytes [93]. A modest decline in NK cells was also observed in treated animals, presumably due to inhibition of IL-15 signaling. Mild anemia was observed with highest doses. Similarly, in a rodent model of aorta transplantation CP-690,550 was also found to prevent allograft vasculopathy [94]. Likewise, in a rat model of trachea transplantation, the development of obliterative bronchiolitis was prevented by CP-690,550 treatment [95]. When administered in conjunction mycophenolate mofetil (MMF) in a nonhuman primates model of kidney transplantation, graft survival was significantly enhanced by the addition of CP-690,550 [96]. CP-690,550 was also studied in rodent arthritis models including collagen-induced arthritis (CIA) and adjuvant-induced arthritis. CP-690,550 significantly reduced inflammation and joint damage in these models [97].
Clinical trials with Jak3 antagonists
The therapeutic effects of CP 690,550 have been evaluated clinical trials in psoriasis, rheumatoid arthritis and kidney transplantation. In a randomized, double-blind, placebo-controlled, dose-escalation Phase 1 study, 6 dose levels of CP-690,550 were assessed in 59 otherwise healthy adults with psoriasis. Except for the lowest dose level, significant improvement in psoriasis was noted. The improvement was dose-dependent and was confirmed histologically [98].
A Phase 2A trial involving 264 patients with rheumatoid arthritis (RA) has also been completed. This was a six week, randomized, double blind, placebo-controlled study in which patients who had previously failed other disease-modifying anti-rheumatic drugs were treated with varying doses of CP-690,550. Patient response was assessed by using the American College of Rheumatology (ACR) criteria and at week 6, an ACR 20 response was achieved in roughly 80% of patients receiving CP-690,550. ACR 50 and ACR 70 responses occurred in 33% to 54% and 13% to 28% respectively of patients receiving the drug. Dose-dependent neutropenia and anemia were observed, and more patients withdrew from the study due to adverse events in the highest dose cohort (manuscript in preparation; data presented at the 70th American College of Rheumatology meeting in 2006). Ongoing studies with CP-690,550 in RA are assessing the safety and efficacy of CP-690,550 compared to placebo in combination with methotrexate and in comparison to adalimumab.
CP-690,550 has been evaluated in stable as well as de novo renal allograft recipients. In a randomized placebo-controlled, Phase 1 study, escalating doses of CP-690,550 were evaluated in 28 stable kidney transplant recipients, whose background immunosuppression included mycophenolate mofetil. The most prevalent adverse events were infections and gastrointestinal disorders. CP-690,550 at 15 mg BID and 30 mg BID were associated with a mean decrease in hemoglobin of 11% and a mean decrease in absolute natural killer cell counts of 50%. Higher doses of CP-690,550 were associated with an increase in absolute CD19+ B-lymphocytes (130%) a decrease of CD4+CD25bright+ T cells (38%) and a decrease in IFN-γ production by mononuclear cells (41%). There were no changes in the number of neutrophils, total lymphocytes, platelets, or total numbers of CD4+ or CD8+ T-cells. (manuscript submitted, [98].
In a Phase 2A multicenter, randomized trial, 2 dose levels of CP-690,550 were compared to tacrolimus in 61 de novo kidney transplants, who also received anti-CD25 antibody induction, mycophenolate mofetil and corticosteroids. Efficacy was observed in all treatment arms, with 12-month incidence of biopsy-proven acute rejection in the CP-690,550 15 mg BID, 30 mg BID and tacrolimus arms of 5.3%, 21.1% and 9.8%, respectively. By 6 months posttransplant, significantly more subjects who received the higher dose of CP-690,550 developed clinically significant infections and cytomegalovirus disease than in the control arm. Additionally, in 4/20 subjects receiving CP-690,550 30 mg BID developed polyomavirus-associated nephropathy but this did occur in the other arms. White blood counts and hemoglobin levels were not significantly different among the treatment groups at six months; additional clinical trials in renal allograft recipients are ongoing.
R348, a JAK3 inhibitor produced by Rigel Pharmaceuticals is also being tested in a Phase 1 trial in RA. Other companies with JAK3 programs include Wyeth/Pharmacopeia and Cytopia/Novartis, but clinical trials have not yet commenced (Table II).
Table 2.
| Company | Compound | Target | Status | Indication |
|---|---|---|---|---|
| Pfizer | CP-690,550 | JAK3 | Phase 2b | Psoriasis
Rheumatoid Arthritis Transplantation |
| Rigel | R-348 | JAK3 | Phase 1 | Rheumatoid arthritis
Autoimmune disease Transplant rejection |
| Cytopia Ltd/Novartis | JAK3 | Discovery | Autoimmune disease,
Transplantation Chronic Obstructive Pulmonary Disease |
|
| Pharmacopeia/Wyeth | PS-608504
PS-020613 |
JAK3 | Discovery | Transplant
Rejection |
| Cephalon | Lestaurtinib | multikinase | Phase 1 | PV, ET, MPD |
| Incyte | INCB18424 | JAK2 | Phase 1 | PV, ET, MPD, RA |
| Exelixis Inc | XL-019 | JAK2 | Phase 1 | PV, ET, MPD |
| AstraZeneca | AZ-01
AZ-60 |
JAK2 | Discovery | Hematological malignancy |
Jak2 inhibitors and the treatment of myeloproliferative disorders
The finding of gain-of-function JAK2 mutations in PV and other MPDs spurred the testing of JAK2 inhibitors in these disorders. In fact, at the time of writing two clinical trials are now underway and more are commencing. Lestaurtinib (Cephalon) is a FMS-like tyrosine kinase/Trk receptor tyrosine kinase inhibitor, which is FDA-approved approved for the treatment of acute myeloid leukemia. However, lestaurinib has also been reported to have activity against JAKand is currently being tested in a Phase 2 study in PV and ET[99].
Another JAK inhibitor, INCB18424 (Incyte) has been studied in Phase 1 and 2 trials in patients with PV, ET and MF. Patients receiving the drug were reported to have benefit irrespective of the presence or absence of the JAK2 mutation, as evidence by reduction in splenomegaly, normalization of leukocytosis or thrombocytosis, as well as symptomatic improvement (data presented in abstract at the 49th American Society of Hematology). Higher doses of the drug resulted thrombocytopenia. Patients on the drug also exhibited reduction in inflammatory cytokines and chemokines including IL-6, IL-1 and TNF, associated with increases in hematopoeitic growth factors such as erythropoietin, IL-3 and GM-CSF. Exposure to the drug was associated with reduced phosphorylated STAT3. Interestingly, INCB18424 is also being tested in RA (RA) and psoriasis.
XL019 (Exelixis) is another JAK inhibitory being used in the setting of myeloproliferative disorders and a Phase 1 dose-escalation trial is underway.
Emerging issues in the development of Jak antagonists
Initially, the “holy grail” of kinase inhibition was specificity; however, we now know that many inhibitors – even the wildly successful imatinib – are not as specific as we thought. However, in a recent survey of kinase inhibitors against a large panel of kinases, CP-690,550 faired surprisingly well – it appears to be a rather selective JAK inhibitor with little activity towards most other kinases [91]. Nonetheless, absolute specificity is apparently not required clinical utility as evidenced by the use of multi-kinase inhibitors. These drugs have acceptable toxicity profiles at least in the setting of cancer therapy. Still some putative Jak inhibitors are nonspecific and pre-clinical and in vitro data need to be interpreted with caution [100]. ---These compounds have been used in numerous publications in the transplant and autoimmune disease literature, their specificity should be considered when interpreting these results. Other JAK antagonists in clinical trials were not assessed in the recent screen; however, rigorous screens of inhibitors against comprehensive panels of kinases is clearly welcome in assessing the promise of potential antagonists.
In the recent survey, the potency of CP-690,550 for JAK2 and JAK3 binding were similar; although the drug was much less active against TYK2. The binding to JAK1 was not assessed. . Role of Selectivity in contribution to efficacy and side effects will need to be followed closely with all of the JAK antagonists. Generation of a selective TYK2 inhibitor is also of potential interest, given that we know the consequences of mutations of this kinase in humans. The relative effectiveness and safety of such a compound compared to other JAK inhibitors would need to be determined.
Another important issue is to understand the mode of action of JAK3 in RA and other inflammatory diseases. As delineated, γc cytokines have an array of actions on developing and mature lymphocytes. It will be important to determine whether JAK3 antagonists are acting principally on T or B cells and if the former, which T cell subset? Is the effect predominantly on memory or naïve cells? Since JAK3 is important for IL-21 signaling, the question arises whether generation of Th17 cells are blocked by JAK3 antagonists. If drugs like CP-690,552 and INCB18424 inhibit JAK2 in vivo, it is conceivable that IL-23 and IL-12 signaling is also blocked. In principle, this could be efficacious in autoimmune disease.
Clearly immense progress has been made in bringing JAK inhibitors to the clinic. It will be very exciting to assess their efficacies and to ascertain the diseases in which they are effective. Equally, it will be critical to understand how they are working and to carefully examine their toxicities.
Figure 2.
References
- 1.Firmbach-Kraft I, et al. tyk2, prototype of a novel class of non-receptor tyrosine kinase genes. Oncogene. 1990;5(9):1329–1336. [PubMed] [Google Scholar]
- 2.Harpur AG, et al. JAK2, a third member of the JAK family of protein tyrosine kinases. Oncogene. 1992;7(7):1347–1353. [PubMed] [Google Scholar]
- 3.Krolewski JJ, et al. Identification and chromosomal mapping of new human tyrosine kinase genes. Oncogene. 1990;5(3):277–282. [PubMed] [Google Scholar]
- 4.Wilks AF. Two putative protein-tyrosine kinases identified by application of the polymerase chain reaction. Proc Natl Acad Sci U S A. 1989;86(5):1603–1607. doi: 10.1073/pnas.86.5.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilks AF. Cloning members of protein-tyrosine kinase family using polymerase chain reaction. Methods Enzymol. 1991;200:533–546. doi: 10.1016/0076-6879(91)00169-w. [DOI] [PubMed] [Google Scholar]
- 6.Wilks AF, et al. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11(4):2057–2065. doi: 10.1128/mcb.11.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawamura M, et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc Natl Acad Sci U S A. 1994;91(14):6374–6378. doi: 10.1073/pnas.91.14.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manning G, et al. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 9.Rodig SJ, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93(3):373–383. doi: 10.1016/s0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 10.Neubauer H, et al. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93(3):397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 11.Parganas E, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93(3):385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 12.Nosaka T, et al. Defective lymphoid development in mice lacking Jak3. Science. 1995;270(5237):800–802. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- 13.Park SY, et al. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity. 1995;3(6):771–782. doi: 10.1016/1074-7613(95)90066-7. [DOI] [PubMed] [Google Scholar]
- 14.Thomis DC, et al. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995;270(5237):794–797. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- 15.Shimoda K, et al. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity. 2000;13(4):561–571. doi: 10.1016/s1074-7613(00)00055-8. [DOI] [PubMed] [Google Scholar]
- 16.Karaghiosoff M, et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity. 2000;13(4):549–560. doi: 10.1016/s1074-7613(00)00054-6. [DOI] [PubMed] [Google Scholar]
- 17.Boulay JL, et al. Molecular phylogeny within type I cytokines and their cognate receptors. Immunity. 2003;19(2):159–163. doi: 10.1016/s1074-7613(03)00211-5. [DOI] [PubMed] [Google Scholar]
- 18.Boggon TJ, et al. Crystal structure of the Jak3 kinase domain in complex with a staurosporine analog. Blood. 2005;106(3):996–1002. doi: 10.1182/blood-2005-02-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mella P, et al. Eleven novel JAK3 mutations in patients with severe combined immunodeficiency-including the first patients with mutations in the kinase domain. Hum Mutat. 2001;18(4):355–356. doi: 10.1002/humu.1199. [DOI] [PubMed] [Google Scholar]
- 20.Chong MM, et al. Suppressor of cytokine signaling-1 is a critical regulator of interleukin-7-dependent CD8+ T cell differentiation. Immunity. 2003;18(4):475–487. doi: 10.1016/s1074-7613(03)00078-5. [DOI] [PubMed] [Google Scholar]
- 21.Sporri B, et al. JAB/SOCS1/SSI-1 is an interleukin-2-induced inhibitor of IL-2 signaling. Blood. 2001;97(1):221–226. doi: 10.1182/blood.v97.1.221. [DOI] [PubMed] [Google Scholar]
- 22.Cornish AL, et al. Suppressor of cytokine signaling-1 regulates signaling in response to interleukin-2 and other gamma c-dependent cytokines in peripheral T cells. J Biol Chem. 2003;278(25):22755–22761. doi: 10.1074/jbc.M303021200. [DOI] [PubMed] [Google Scholar]
- 23.Ungureanu D, et al. Regulation of Jak2 through the ubiquitin-proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1. Mol Cell Biol. 2002;22(10):3316–3326. doi: 10.1128/MCB.22.10.3316-3326.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamizono S, et al. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J Biol Chem. 2001;276(16):12530–12538. doi: 10.1074/jbc.M010074200. [DOI] [PubMed] [Google Scholar]
- 25.Candotti F, et al. Structural and functional basis for JAK3-deficient severe combined immunodeficiency. Blood. 1997;90(10):3996–4003. [PubMed] [Google Scholar]
- 26.Chen M, et al. Complex effects of naturally occurring mutations in the JAK3 pseudokinase domain: evidence for interactions between the kinase and pseudokinase domains. Mol Cell Biol. 2000;20(3):947–956. doi: 10.1128/mcb.20.3.947-956.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao R, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280(24):22788–22792. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steensma DP, et al. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and the myelodysplastic syndrome. Blood. 2005 doi: 10.1182/blood-2005-03-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levine RL, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 30.Kralovics R, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 31.James C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 32.Kurzer JH, et al. Tyrosine 813 is a site of JAK2 autophosphorylation critical for activation of JAK2 by SH2-B beta. Mol Cell Biol. 2004;24(10):4557–4570. doi: 10.1128/MCB.24.10.4557-4570.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Brien KB, et al. SH2-B family members differentially regulate JAK family tyrosine kinases. J Biol Chem. 2002;277(10):8673–8681. doi: 10.1074/jbc.M109165200. [DOI] [PubMed] [Google Scholar]
- 34.Asao H, et al. Hrs is associated with STAM, a signal-transducing adaptor molecule. Its suppressive effect on cytokine-induced cell growth. J Biol Chem. 1997;272(52):32785–32791. doi: 10.1074/jbc.272.52.32785. [DOI] [PubMed] [Google Scholar]
- 35.Takeshita T, et al. STAM, signal transducing adaptor molecule, is associated with Janus kinases and involved in signaling for cell growth and c-myc induction. Immunity. 1997;6(4):449–457. doi: 10.1016/s1074-7613(00)80288-5. [DOI] [PubMed] [Google Scholar]
- 36.Endo K, et al. STAM2, a new member of the STAM family, binding to the Janus kinases. FEBS Lett. 2000;477(12):55–61. doi: 10.1016/s0014-5793(00)01760-9. [DOI] [PubMed] [Google Scholar]
- 37.Lohi O, Lehto VP. STAM/EAST/Hbp adapter proteins--integrators of signalling pathways. FEBS Lett. 2001;508(3):287–290. doi: 10.1016/s0014-5793(01)03079-4. [DOI] [PubMed] [Google Scholar]
- 38.Lohi O, et al. VHS domain -- a longshoreman of vesicle lines. FEBS Lett. 2002;513(1):19–23. doi: 10.1016/s0014-5793(01)03287-2. [DOI] [PubMed] [Google Scholar]
- 39.Cacalano NA, et al. Autosomal SCID caused by a point mutation in the N-terminus of Jak3: mapping of the Jak3-receptor interaction domain. Embo J. 1999;18(6):1549–1558. doi: 10.1093/emboj/18.6.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen M, et al. The amino terminus of JAK3 is necessary and sufficient for binding to the common gamma chain and confers the ability to transmit interleukin 2-mediated signals. Proc Natl Acad Sci U S A. 1997;94(13):6910–6915. doi: 10.1073/pnas.94.13.6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou YJ, et al. Unexpected effects of FERM domain mutations on catalytic activity of Jak3: structural implication for Janus kinases. Mol Cell. 2001;8(5):959–969. doi: 10.1016/s1097-2765(01)00398-7. [DOI] [PubMed] [Google Scholar]
- 42.Baird AM, et al. A profound deficiency in thymic progenitor cells in mice lacking Jak3. J Immunol. 2000;165(7):3680–3688. doi: 10.4049/jimmunol.165.7.3680. [DOI] [PubMed] [Google Scholar]
- 43.Cao X, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity. 1995;2(3):223–238. doi: 10.1016/1074-7613(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 44.DiSanto JP, et al. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc Natl Acad Sci U S A. 1995;92(2):377–381. doi: 10.1073/pnas.92.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grossman WJ, et al. Dysregulated myelopoiesis in mice lacking Jak3. Blood. 1999;94(3):932–939. [PubMed] [Google Scholar]
- 46.Nutt SL, et al. Essential functions of Pax5 (BSAP) in pro-B cell development: difference between fetal and adult B lymphopoiesis and reduced V-to-DJ recombination at the IgH locus. Genes Dev. 1997;11(4):476–491. doi: 10.1101/gad.11.4.476. [DOI] [PubMed] [Google Scholar]
- 47.Puel A, et al. Defective IL7R expression in T(-)B(+)NK(+) severe combined immunodeficiency. Nat Genet. 1998;20(4):394–397. doi: 10.1038/3877. [DOI] [PubMed] [Google Scholar]
- 48.Schorle H, et al. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 1991;352(6336):621–624. doi: 10.1038/352621a0. [DOI] [PubMed] [Google Scholar]
- 49.Kundig TM, et al. Immune responses in interleukin-2-deficient mice. Science. 1993;262(5136):1059–1061. doi: 10.1126/science.8235625. [DOI] [PubMed] [Google Scholar]
- 50.Nelson BH. Interleukin-2 signaling and the maintenance of self-tolerance. Curr Dir Autoimmun. 2002;5:92–112. doi: 10.1159/000060549. [DOI] [PubMed] [Google Scholar]
- 51.Shevach EM. Regulatory T cells in autoimmmunity*. Annu Rev Immunol. 2000;18:423–449. doi: 10.1146/annurev.immunol.18.1.423. [DOI] [PubMed] [Google Scholar]
- 52.Malek TR. The main function of IL-2 is to promote the development of T regulatory cells. J Leukoc Biol. 2003;74(6):961–965. doi: 10.1189/jlb.0603272. [DOI] [PubMed] [Google Scholar]
- 53.Sharfe N, et al. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc Natl Acad Sci U S A. 1997;94(7):3168–3171. doi: 10.1073/pnas.94.7.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roifman CM. Human IL-2 receptor alpha chain deficiency. Pediatr Res. 2000;48(1):6–11. doi: 10.1203/00006450-200007000-00004. [DOI] [PubMed] [Google Scholar]
- 55.Sharfe N, et al. An interleukin-2 receptor gamma chain mutation with normal thymus morphology. J Clin Invest. 1997;100(12):3036–3043. doi: 10.1172/JCI119858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frucht DM, et al. Unexpected and variable phenotypes in a family with JAK3 deficiency. Genes Immun. 2001;2(8):422–432. doi: 10.1038/sj.gene.6363802. [DOI] [PubMed] [Google Scholar]
- 57.Mella P, et al. Development of autologous T lymphocytes in two males with X-linked severe combined immune deficiency: molecular and cellular characterization. Clin Immunol. 2000;95(1 Pt 1):39–50. doi: 10.1006/clim.2000.4842. [DOI] [PubMed] [Google Scholar]
- 58.Brugnoni D, et al. Development of autologous, oligoclonal, poorly functioning T lymphocytes in a patient with autosomal recessive severe combined immunodeficiency caused by defects of the Jak3 tyrosine kinase. Blood. 1998;91(3):949–955. [PubMed] [Google Scholar]
- 59.Townsend JM, et al. IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity. 2000;13(4):573–583. doi: 10.1016/s1074-7613(00)00056-x. [DOI] [PubMed] [Google Scholar]
- 60.McMillan SJ, et al. The absence of interleukin 9 does not affect the development of allergen-induced pulmonary inflammation nor airway hyperreactivity. J Exp Med. 2002;195(1):51–57. doi: 10.1084/jem.20011732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Korn T, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448(7152):484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nurieva R, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448(7152):480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 63.Wei L, et al. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282(48):34605–34610. doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou L, et al. IL-6 programs T(H)-17 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8(9):967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 65.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26(3):371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 66.Ozaki K, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298(5598):1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 67.Lyon MF, et al. The scurfy mouse mutant has previously unrecognized hematological abnormalities and resembles Wiskott-Aldrich syndrome. Proc Natl Acad Sci U S A. 1990;87(7):2433–2437. doi: 10.1073/pnas.87.7.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bennett CL, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 69.Wildin RS, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 70.Fontenot JD, et al. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6(11):1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 71.Mayack SR, Berg LJ. Cutting edge: an alternative pathway of CD4+ T cell differentiation is induced following activation in the absence of gamma-chain-dependent cytokine signals. J Immunol. 2006;176(4):2059–2063. doi: 10.4049/jimmunol.176.4.2059. [DOI] [PubMed] [Google Scholar]
- 72.Ortmann R, et al. A heritable defect in IL-12 signaling in B10.Q/J mice. I. In vitro analysis. J Immunol. 2001;166(9):5712–5719. doi: 10.4049/jimmunol.166.9.5712. [DOI] [PubMed] [Google Scholar]
- 73.Yap GS, et al. A heritable defect in IL-12 signaling in B10.Q/J mice. II. Effect on acute resistance to Toxoplasma gondii and rescue by IL-18 treatment. J Immunol. 2001;166(9):5720–5725. doi: 10.4049/jimmunol.166.9.5720. [DOI] [PubMed] [Google Scholar]
- 74.Shaw MH, et al. A natural mutation in the Tyk2 pseudokinase domain underlies altered susceptibility of B10.Q/J mice to infection and autoimmunity. Proc Natl Acad Sci U S A. 2003;100(20):11594–11599. doi: 10.1073/pnas.1930781100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Minegishi Y, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25(5):745–755. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 76.Watford WT, O’Shea JJ. Human tyk2 kinase deficiency: another primary immunodeficiency syndrome. Immunity. 2006;25(5):695–697. doi: 10.1016/j.immuni.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 77.Peeters P, et al. Fusion of ETV6 to MDS1/EVI1 as a result of t(3;12)(q26;p13) in myeloproliferative disorders. Cancer Res. 1997;57(4):564–569. [PubMed] [Google Scholar]
- 78.Lacronique V, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278(5341):1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 79.Lacronique V, et al. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood. 2000;95(6):2076–2083. [PubMed] [Google Scholar]
- 80.Grandage VL, et al. Go6976 is a potent inhibitor of the JAK 2 and FLT3 tyrosine kinases with significant activity in primary acute myeloid leukaemia cells. Br J Haematol. 2006;135(3):303–316. doi: 10.1111/j.1365-2141.2006.06291.x. [DOI] [PubMed] [Google Scholar]
- 81.Walters DK, et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. 2006;10(1):65–75. doi: 10.1016/j.ccr.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 82.De Vita S, et al. Loss-of-function JAK3 mutations in TMD and AMKL of Down syndrome. Br J Haematol. 2007;137(4):337–341. doi: 10.1111/j.1365-2141.2007.06574.x. [DOI] [PubMed] [Google Scholar]
- 83.Baxter EJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 84.Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999;340(17):1330–1340. doi: 10.1056/NEJM199904293401706. [DOI] [PubMed] [Google Scholar]
- 85.Mughal TI, Goldman JM. Chronic myeloid leukemia: current status and controversies. Oncology (Williston Park) 2004;18(7):837–844. 847. discussion 847-850, 853-834. [PubMed] [Google Scholar]
- 86.Schindler T, et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289(5486):1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 87.Adams J, et al. A strategy for the design of multiplex inhibitors for kinase-mediated signalling in angiogenesis. Curr Opin Chem Biol. 2002;6(4):486–492. doi: 10.1016/s1367-5931(02)00357-5. [DOI] [PubMed] [Google Scholar]
- 88.Buchdunger E, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295(1):139–145. [PubMed] [Google Scholar]
- 89.Druker BJ, Lydon NB. Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Invest. 2000;105(1):3–7. doi: 10.1172/JCI9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cools J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348(13):1201–1214. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 91.Karaman MW, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26(1):127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 92.O’Shea JJ, et al. A new modality for immunosuppression: targeting the JAK/STAT pathway. Nat Rev Drug Discov. 2004;3(7):555–564. doi: 10.1038/nrd1441. [DOI] [PubMed] [Google Scholar]
- 93.Changelian PS, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302(5646):875–878. doi: 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]
- 94.Rousvoal G, et al. Janus kinase 3 inhibition with CP-690,550 prevents allograft vasculopathy. Transpl Int. 2006;19(12):1014–1021. doi: 10.1111/j.1432-2277.2006.00387.x. [DOI] [PubMed] [Google Scholar]
- 95.Lau M, et al. Prevention of obliterative bronchiolitis by JAK3 inhibition with CP-690,550 is accompanied by a distinct TGFβ3/TGFβ1 gene expression profile. 3rd International Congress on Immunosuppression. 2004 Vol. Abstract P-244. [Google Scholar]
- 96.Borie DC, et al. Immunosuppression by the JAK3 inhibitor CP-690,550 delays rejection and significantly prolongs kidney allograft survival in nonhuman primates. Transplantation. 2005;79(7):791–801. doi: 10.1097/01.tp.0000157117.30290.6f. [DOI] [PubMed] [Google Scholar]
- 97.Milici AJ, et al. Cartilage preservation by inhibition of Janus Kinase 3 in two rodent models of rheumatoid arthritis. Arthritis Res Ther. 2008;10(1):R14. doi: 10.1186/ar2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wilkinson B, et al. Improvement in psoriatic lesions during a 14-day trial of CP-690,550 (CP), an orally active inhibitor of Janus Kinase 3 (JAK3) Ann Rheum Dis. 2007;66(Suppl II):155. [Google Scholar]
- 99.Hexner EO, et al. Lestaurtinib (CEP701)is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2007 doi: 10.1182/blood-2007-04-083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Changelian PS, et al. The specificity of JAK3 kinase inhibitors. Blood. 2008;111(4):2155–2157. doi: 10.1182/blood-2007-09-115030. [DOI] [PubMed] [Google Scholar]