

Abstract
A new synthesis of 3,3′-bioxindoles is reported that is well suited for the preparation of unsymmetrical structures. In the key step, 3-hydroxy-3,3′-bioxindoles are constructed by Mukaiyama aldol reaction of 2-siloxyindoles with isatins These tertiary carb inols are formed in high diastereoselectivities, with substitution at various positions of the isatin and the 2-siloxyindole being tolerated.
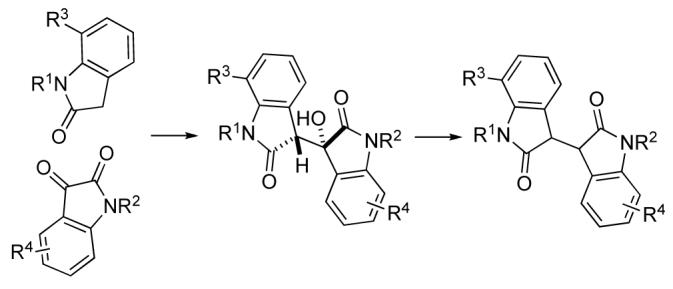
3,3′-Bioxindoles (1) are precursors to a variety of complex nitrogen heterocycles (Figure 1). In the alkaloid field, they have been used to synthesize a number of cyclotryptamine alkaloids,1,2 exemplified by (+)-chimonanthine (2),3 meso-calycanthine (3)4 and (-)-idiospermuline (4).5 3,3′-Bioxindoles have also been employed to access heterocycles containing other polycyclic motifs, such as hexahydrodiazachrysene 5.6 In our laboratories, the reaction of dienolates of 3,3′-bioxindoles (1) with enantiomerically pure tartrate-derived dielectrophiles has been employed for the enantioselective construction of contiguous quaternary carbon stereocenters.7,8
Figure 1.
3,3′-Bioxindole and representative heterocycles prepared from these intermediates
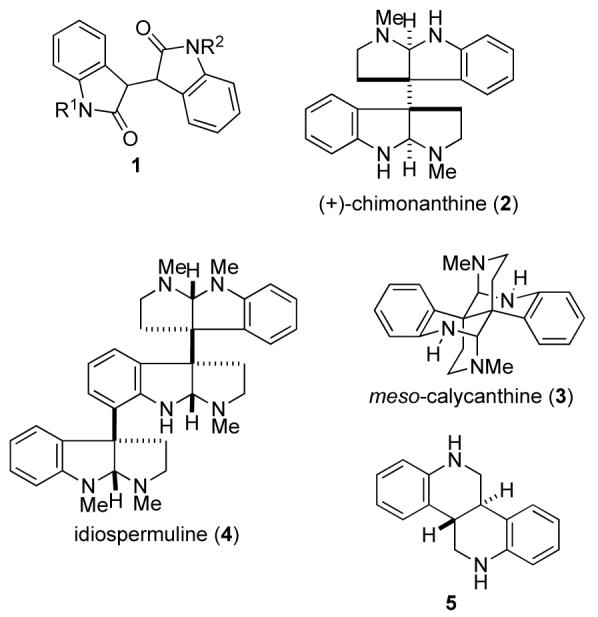
3,3′-Bioxindoles (1) are commonly prepared by acidpromoted condensation of an isatin and an oxindole to generate an isoindigo, 8, followed by saturation of the double bond (Scheme 1).5–9 In ongoing studies, we required access to a series of unsymmetrical 3,3′-bioxindoles (1) having variation in both the nitrogen protecting group and the substituents on the two aromatic rings.10 As some of the substituents are acid sensitive, the classical sequence illustrated in Scheme 1 was not viable. We report herein that the Mukaiyama aldol reaction11 between 2-siloxyindoles12 and isatins provides access to a variety of 3-hydroxy-3,3′-bioxindoles,13 intermediates that are readily converted to 3,3′-bioxindoles. We also disclose that the aldol addition occurs with high stereoselectivity to preferentially generate the syn aldol stereoisomer.
Scheme 1.
Typical Synthesis of 3,3′-Bioxindoles
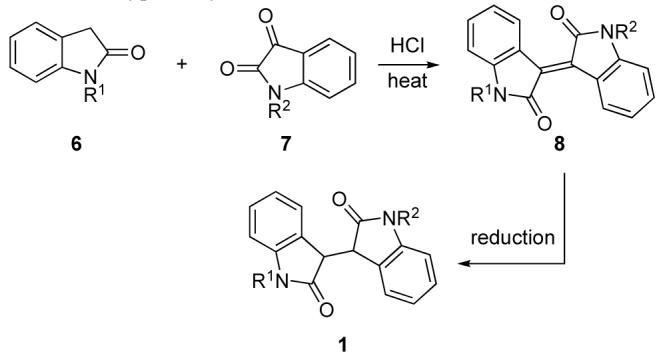
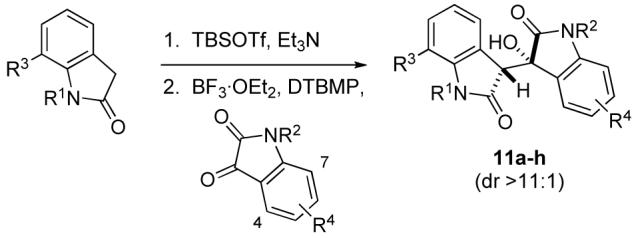
Our initial studies focused on the union of (2-(trimethylsilyl)ethoxy)methyl (SEM)-protected oxindole 9 and 4-vinylisatin 10 (Scheme 2). Addition of the lithium enolate of oxindole 9 to isatin 10 generated aldol adduct 11a and its epimer (3:1 dr, syn:anti);14 however, the yield was low. In contrast, Mukaiyama aldol reaction of the 2-siloxyindole derivative of 9 with 2 equiv of isatin 10 in the presence of excess BF3·OEt2 (8 equiv) and 2,6-di-tert-butyl-4-methylpyridine (DTBMP, 7 equiv) beginning at −78 °C, then warming to −50 °C, yielded aldol adduct 11a in 78% yield and high diastereoselectivity (>19:1 syn:anti). Further investigation revealed that the optimal conditions for this transformation involved the use of 1.0 equiv of the isatin, 2.0 equiv of BF3·OEt2, and 2.5 equiv of DTBMP at −78 to −50 °C, conditions that provided adduct 11a in 77% yield.
Scheme 2.
Aldol Reaction of 2-Siloxyoxindoles with Isatins
The scope of this aldol reaction is illustrated by the examples summarized in Table 1. Crude 2-siloxyindoles were employed, which were isolated by adding a small volume of methanol to the silylation reaction mixture, removing volatiles under reduced pressure, extracting the resultant oil with pentane, and concentration. N-Methyl, N-benzyl, and N-SEM substituents were tolerated on both the oxindole and the isatin. Substitution at each position on the aromatic ring of the isatin had no adverse effect on the aldol reaction (entries 1, 4–8). Substitution at the 7-position of the oxindole was also viable (entries 1, 2, 8). Additionally, the Mukaiyama aldol reaction was effective for a substrate containing a pyridine appended to the isatin (entry 7), although the yield in this case was slightly lower. In all reactions, diastereoselectivity was high (>11:1).15 The reactions summarized in Table 1 were carried out at various scales (0.50 mmol to 40 mmol), with the best result being observed in the largest scale reaction (entry 2). Although only oxindoles having substituents at C7 of the aromatic ring were included in this study, we anticipate that oxindoles having substituents at C4—C6 will also successfully undergo Mukaiyama aldol condensation with isatins.
Table 1.
Mukaiyama Aldol Reactions of 2-Siloxyindoles and Isatinsa
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | R3 | R2 | R4 | 11 | Yield (%)b |
| 1 | SEM | H | Me | 4-vinyl | 11a | 77 |
| 2 | Me | MeO | SEM | H | 11b | 87 |
| 3 | Me | MeO | Bn | H | 11c | 80 |
| 4 | SEM | H | Bn | H | 11d | 76 |
| 5 | Bn | H | SEM | 7-F | 11e | 83 |
| 6 | SEM | H | Bn | 6-phenyl | 11f | 84 |
| 7 | SEM | H | Bn | 5-(3-pyridyl) | 11g | 60 |
| 8 | Me | MeO | Me | 4-vinyl | 11h | 52c |
Conditions: Oxindole (1.0 equiv), isatin (1.0 equiv), BF3·Et2O (2.0 equiv), DTBMP (2.5 equiv), –78 °C to –50 °C.
Yield over two steps of the aldol adduct after purification by silica gel flash column chromatography.
The starting oxindole (15–20%) was recovered.
The syn relative configuration of aldol adduct 11c was established by single crystal X-ray analysis. 16 The pre-transition state arrangement 12 of the 2-siloxyindole and the isatin, depicted in Figure 2, would be consistent with the observed syn stereoselection. This orientation places the bulky silyl group away from the aromatic ring of the isatin, and also allows for some degree of π-overlap of the two aromatic moieties.17,18
Figure 2.
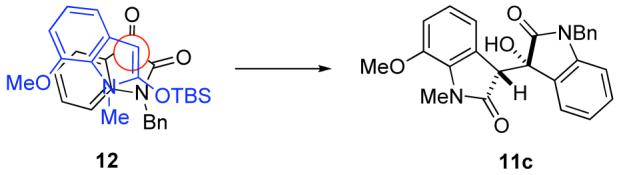
Pre-transition state model rationalizing syn selectivity
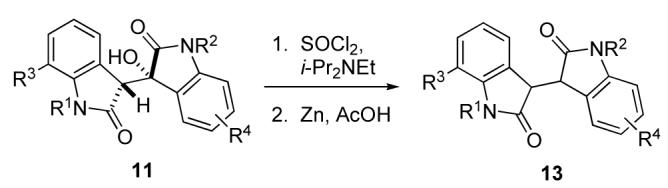
The conversion of the aldol adducts to the corresponding 3,3′-bioxindole was best accomplished by reaction with SOCl2 and diisopropylethylamine19 to give the corresponding isoindigo, which without purification was reduced with zinc and acetic acid.20–22 Representative examples are shown in Table 2.
Table 2.
3,3′-Bioxindole Formation from 3-Hydroxy-3,3′-Bioxindolesa
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | 11 | R1 | R3 | R2 | R4 | 13 | Yield (%)b |
| 1 | 11b | Me | MeO | SEM | H | 13a | 87c |
| 2 | 11c | Me | MeO | Bn | H | 13b | 89 |
| 3 | 11d | SEM | H | Bn | H | 13c | 85 |
| 4 | 11e | Bn | H | SEM | 7-F | 13d | 79 |
| 5 | 11f | SEM | H | Bn | 6-Ph | 13e | 90 |
Conditions: step 1: 11 (1.0 equiv), SOCl2 (1.2 equiv), i-Pr2NEt (3.0 equiv), 0 °C to rt; step 2: Zn (30 equiv), AcOH (18 equiv), 0 °C to rt.
Yield over two steps after purification of 13 by silica gel flash column chromatography.
Reduction performed with a modified procedure: Zn (6.0 equiv), AcOH (5.6 equiv), 0 °C to rt.
In conclusion, Mukaiyama aldol reaction of 2-siloxyindoles with isatins takes place in good yield and with high stereoselectivity to give syn diastereomers of 3-hydroxy-3,3′-bioxindoles. These aldol products can be reduced in good yield to give the corresponding 3,3′-bioxindoles. By use of this method, a variety of substituents, including those that are acid-sensitive, can be introduced at various positions of the 3,3′-bioxindole product.
Experimental Section
Representative Procedure for Generating 2-Siloxyindole Intermediates and Their Mukaiyama Aldol Reaction with Isatins. Preparation of 3-Hydroxy-7′-methoxy-N’-methyl-N-((2-(trimethylsilyl)ethoxy)methyl)-3,3′-biindoline-2,2′-dione (11b)
To a stirring solution of 7-methoxy-N-methyloxindole (S5) (6.60 g, 37.3 mmol, 1.0 equiv), triethylamine (15.6 mL, 112 mmol, 3.0 equiv) and dichloromethane (700 mL) at 0 °C was added dropwise tert-butyldimethylsilyl trifluoromethanesulfonate (12.8 mL, 55.9 mmol, 1.5 equiv). The resultant solution was maintained at 0 °C for 25 min and then allowed to warm to rt. Methanol (1.5 mL, 37 mmol, 1.0 equiv) was added, and the solution was concentrated under reduced pressure (∼30 mmHg). The resulting biphasic mixture was extracted with pentanes (3 × 120 mL), and the combined organic extracts were concentrated under reduced pressure to provide the corresponding 2-siloxyindole as a pale red/pink solid, which was used in the subsequent reaction without further purification.
A solution of the crude 2-siloxyindole, N-((2-(trimethylsilyl)ethoxy)methyl)isatin (10.4 g, 37.6 mmol, 1.01 equiv), 2,6-di-tert-butyl-4-methylpyridine (19.1 g, 93.2 mmol, 2.5 equiv), and dichloromethane (630 mL) was cooled to −78 °C, and boron trifluoride diethyl ether complex (9.20 mL, 74.6 mmol, 2.0 equiv) was added dropwise. After 2 h at −78 °C, the solution was allowed to warm to −50 °C where it was maintained overnight. This solution was then poured into water (250 mL) and the layers were separated. The aqueous layer was extracted with dichloromethane (3 × 250 mL). The combined organic extracts were washed with brine (40 mL), dried over Na2SO4, and concentrated under reduced pressure to provide a brown oil. Purification of this oil by silica gel flash column chromatography (dry loaded on Celite, gradient: 1:6 EtOAc-hexanes to 1:3 EtOAc-hexanes) provided alcohol 11b (14.8 g, 87%) as a pale brown solid: mp 129–132 °C; Rf 0.54 (1:4 EtOAc-hexanes); 1H NMR (500 MHz, CDCl3) δ 7.54 (d, J = 7.4, 1H), 7.42 (ddd, J = 8.8, 8.0, 7.7, 1H), 7.24 (t, J = 7.6, 1H), 7.02 (d, J = 7.7, 1H), 6.76 (d, J = 8.3, 1H), 6.67 (dd, J = 8.3, 7.7, 1H), 6.45 (br s, 1H), 5.60 (d, J = 7.6, 1H), 4.90 (d, J = 11.4, 1H), 4.82 (d, J = 11.4, 1H), 3.91(s, 1H), 3.80 (s, 3H), 3.55 (s, 3H), 3.10 (ddd, J = 10.4, 9.9, 6.4, 1H), 3.02 (ddd, J = 10.4, 9.9, 6.1, 1H), 0.80–0.66 (m, 2H), −0.07 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 176.0 (C), 175.5 (C), 145.5 (C), 143.0 (C), 132.8 (C), 130.8 (CH), 127.5 (C), 124.5 (CH), 124.1 (C), 123.9 (CH), 123.1 (CH), 116.6 (CH), 112.8 (CH), 110.2 (CH), 77.4 (C), 69.2 (CH2), 65.6 (CH2), 55.8 (CH3), 49.3 (CH), 29.8 (CH3), 17.5 (CH2), −1.3 (CH3); IR (film) 3365, 2952, 1733, 1713, 1615 cm−1; HRMS-ESI (m/z) [M + Na]+ calcd for C24H30N2NaO5Si: 477.1822; found: 477.1823.
Representative Procedure for Converting Aldol Adducts to 3,3′-Bioxindoles. Preparation of 7′-Methoxy-N’-methyl-N-(2-(trimethylsilyl)ethoxymethyl)-3,3′-dihydroisoindigo (13a)
To a solution of tertiary alcohol 11b (14.8 g, 32.5 mmol, 1.0 equiv) and dichloromethane (275 mL) at 0 °C was added over 5 min diisopropylethylamine (17.0 mL, 97.5 mmol, 3.0 equiv) and then freshly distilled thionyl chloride (2.85 mL, 39.0 mmol, 1.2 equiv). The resulting solution was maintained for 25 min at 0 °C, before being allowed to warm to rt where it was maintained for 45 min. The reaction mixture was then poured into a saturated aqueous solution of sodium hydrogen carbonate (150 mL). The aqueous layer was separated and extracted with dichloromethane (4 × 200 mL). The combined organic layers were dried over MgSO4 and concentrated under reduce pressure to yield a brown oil that was used without further purification in the subsequent reaction.
In a separate experiment, purification of this product by silica gel flash column chromatography (1:9 EtOAc-hexanes) gave the corresponding isoindigo S20 as a dark burgundy solid: mp 84–85 °C; 1H NMR (500 MHz, CDCl3) δ 9.17 (d, J = 8.0, 1H), 8.73 (m, 1H), 7.36 (td, J = 7.7, 0.9, 1H), 7.09 (d, J = 7.5, 1H), 7.01 (d, J = 7.8, 1H), 6.96 (m, 2H), 5.24 (s, 2H), 3.87 (s, 3H), 3.62 (t, J = 8.2, 2H), 3.57 (s, 3H), 0.94 (t, J = 8.2, 2H), −0.02 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 168.2, 168.0, 144.8, 143.8, 134.8, 134.3, 133.7, 133.3, 132.6, 129.6, 122.9, 122.7, 122.3, 121.6, 116.8, 109.1, 69.5, 66.2, 56.3, 29.7, 17.9, −1.4; IR (film) 2954, 1698, 1609 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C24H29N2O4Si, 437.1897; found, 437.1912.
The unpurified isoindigo intermediate was dissolved in THF (315 mL) and cooled to 0 °C. To this solution was added zinc dust (12.8 g, 195 mmol, 6.0 equiv) and glacial acetic acid (3.2 mL, 180 mmol, 5.6 equiv). The resulting suspension was vigorously stirred at 0 °C for 1.5 h, and then allowed to warm to rt over 45 min. The reaction mixture was then filtered through a pad of Celite eluting with EtOAc (250 mL). The organic solution was washed with saturated aqueous sodium hydrogen carbonate (200 mL), dried over MgSO4, and concentrated under reduced pressure to yield a black foamy oil. This residue was further purified by silica gel flash column chromatography (dry loaded on Celite, gradient: 1:4 EtOAchexanes to 1:1 EtOAc-hexanes) to afford bioxindole 13a as a 1.6:1 mixture of epimers (12.4 g, 87%) as an orange foam: Rf 0.20 (1:1 EtOAc-hexanes); 1H NMR (500 MHz, CDCl3) δ 7.28 (t, J = 7.8, 0.6H), 7.12 (t, J = 7.7, 0.4H), 7.06–7.02 (m, 0.6H), 6.99 (t, J = 7.3, 0.6H), 6.93–6.80 (m, 3.0H), 6.74–6.65 (m, 0.8H), 6.55 (d, J = 7.2, 0.3H), 6.33 (m, 0.5H), 5.22 (d, J = 10.8, 0.4H), 5.18 (d, J = 10.9, 0.4H), 5.04 (s, 1.2H), 4.34 (d, J = 3.5, 0.3H), 4.25 (m, 0.8H), 4.12 (d, J = 3.3, 0.6H), 3.87–3.81 (m, 1.9H), 3.80–3.74 (m, 1.1H), 3.59–3.52 (m, 1.1H), 3.47 (s, 0.5H), 3.55–3.44 (m, 2.7H), 0.97 (m, 0.8H), 0.84 (t, J = 7.8, 1.3H), 0.10 (s, 3.2H), −0.05 (s, 5.8H); 13C NMR (125 MHz, CDCl3) δ 176.4 (C), 175.9 (C), 175.0 (C), 174.8 (C), 145.2 (C), 145.1 (C), 143.4 (C), 142.7 (C), 132.9 (C), 131.9 (C), 128.7 (CH), 128.3 (CH), 127.1 (C), 126.3 (C), 125.5 (C), 124.4 (C), 123.4 (CH), 123.3 (CH), 122.9 (CH), 122.82 (CH), 122.78 (CH), 122.75 (CH), 116.1 (CH), 115.9 (CH), 112.6 (CH), 112.3 (CH), 109.8 (CH), 109.4 (CH), 69.8 (CH2), 69.3 (CH2), 66.6 (CH2), 65.8 (CH2), 55.74 (CH3), 55.71 (CH3), 46.5 (CH), 46.2 (CH), 46.0 (CH), 29.6 (CH3), 29.5 (CH3), 17.9 (CH2), 17.6 (CH2), −1.4 (CH3), −1.5 (CH3); IR (film) 2950, 1716, 1612 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C24H31N2O4Si, 439.2053; found, 439.2048.
Supplementary Material
Acknowledgment
This research was supported by the NIH National Institutes of General Medical Sciences (GM-30859) and by postdoctoral fellowships to J.M.E. (GM082113) from NIGMS and to H.R.T. from Merck. NMR and mass spectra were determined at UC Irvine using instruments acquired with the assistance of NSF and NIH shared instrumentation grants. We thank Dr. Joe Ziller for the X-ray analysis of 11c.
References
- (1)(a).Hino T. Chem. Pharm. Bull. 1961;9:979–988. [Google Scholar]; (b) Hino T, Yamada S. Tetrahedron Lett. 1963;4:1757–1760. [Google Scholar]
- (2).For a recent brief review, see: Steven A, Overman LE. Angew. Chem., Int. Ed. 2007;46:5488–5508. doi: 10.1002/anie.200700612.
- (3).Overman LE, Larrow JF, Stearns BA, Vance JM. Angew. Chem., Int. Ed. 2000;39:213–215. doi: 10.1002/(sici)1521-3773(20000103)39:1<213::aid-anie213>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- (4).Link JT, Overman LE. J. Am. Chem. Soc. 1996;118:8166–8167. [Google Scholar]
- (5).Overman LE, Peterson EA. Angew. Chem., Int. Ed. 2003;42:2525–2528. doi: 10.1002/anie.200351260. [DOI] [PubMed] [Google Scholar]
- (6).Papageorgiou C, Borer X. Helv. Chim. Acta. 1988;71:1079–1083. [Google Scholar]
- (7)(a).Hoyt SB, Overman LE. Org. Lett. 2000;2:3241–3244. doi: 10.1021/ol006494m. [DOI] [PubMed] [Google Scholar]; (b) Overman LE, Peterson EA. Angew. Chem., Int. Ed. 2003;42:2525–2528. doi: 10.1002/anie.200351260. [DOI] [PubMed] [Google Scholar]
- (8).3,3′-Bioxindoles have also been employed in double Steglich rearrangements for the diastereoselective synthesis of contiguous quaternary stereocenters, see: Menozzi C, Dalko PI, Cossy J. Heterocycles. 2007;72:199–204.
- (9).Hansen CW. Ann. Chim. (Paris) 1924:94–134. [Google Scholar]
- (10)(a).Only two unsymmetrical 3,3′-bioxindoles were found by a substructure search of SciFinder conducted on 8/20/08.For one example of the challenges involved in preparing unsymmetrical 3,3′-bioxindoles, see: Overman LE, Peterson EA.Tetrahedron 2003596905–6919.
- (11).Mukaiyama T, Banno K, Narasaka K. J. Am. Chem. Soc. 1974;96:7503–7509. [Google Scholar]
- (12)(a).Sawada T, Fuerst DE, Wood JL. Tetrahedron Lett. 2003;44:4919–4921. [Google Scholar]; (b) Nicolaou KC, Hao J, Reddy MV, Rao PB, Rassias G, Snyder SA, Huang X, Chen DY-K, Brenzovich WE, Giuseppone N, Giannakakou P, O’Brate A. J. Am. Chem. Soc. 2004;126:12897–12906. doi: 10.1021/ja040093a. [DOI] [PubMed] [Google Scholar]; (c) Alcaraz M-L, Atkinson S, Cornwall P, Foster AC, Gill DM, Humphries LA, Keegan PS, Kemp R, Merifield E, Nixon RA, Noble AJ, O’Beirne D, Patel ZM, Perkins J, Rowan P, Sadler P, Singleton JT, Tornos J, Watts AJ, Woodland IA. Org. Process Res. Dev. 2005;9:555–569. [Google Scholar]; (d) Trost BM, Brennan MK. Org. Lett. 2006;8:2027–2030. doi: 10.1021/ol060298j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Overman LE, Shin Y. Org. Lett. 2007;9:339–341. doi: 10.1021/ol062801y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Poulsen TB, Bernardi L, Aleman J, Overgaard J, Jorgensen KA. J. Am. Chem. Soc. 2007;129:441–449. doi: 10.1021/ja067289q. [DOI] [PubMed] [Google Scholar]; (g) Corkey BK, Toste FD. J. Am. Chem. Soc. 2007;129:2764–2765. doi: 10.1021/ja068723r. [DOI] [PubMed] [Google Scholar]
- (13).For an earlier report of the utility of 2-siloxyindoles in Mukaiyama aldol reactions, see: Adhikari S, Caille S, Hanbauer M, Ngo VX, Overman LE. Org. Lett. 2005;7:2795–2797. doi: 10.1021/ol051172+.
- (14).Relative configuration is assigned in analogy to 11c (vide infra).
- (15).1H NMR analysis of the crude reaction product prior to purification typically showed the presence of only one stereosiomer.
- (16).The relative configurations of other aldol adducts were assigned in analogy to 11c
- (17).Open transition states with the enoxysilane and carbonyl π-bonds antiperiplanar are typically proposed for Mukaiyama aldol reactions, see: Marhwald R. Chem. Rev. 1999;99:1095–1120. doi: 10.1021/cr980415r.
- (18).The yield of the aldol reaction was somewhat lower when the 7-position of the oxindole and the 4-position of the isatin were both substituted (entry 8). The 7-position of the oxindole and 4-position of the isatin would be in close proximity in pre-transition state ensemble 12
- (19).Heymann H, Fieser LF. J. Am. Chem. Soc. 1951;73:5252–5265. [Google Scholar]
- (20).Additional methods have been demonstrated for the conversion of 3-hydroxy-3,3′-bioxindoles to 3,3′-bioxindoles. These include: acid-mediated dehydration of the tertiary alcohol to afford an isoindigo that is subsequently reduced by catalytic hydrogenation,6–9 and direct reduction with trimethylsilyl iodide21 or HI.22
- (21)(a).Sakai T, Miyata K, Utaka M, Takeda A. Tetrahedron Lett. 1987;28:3817–3818.For an example of the reduction of a 3-hydroxy-3,3′-bioxindole, see: Peterson EA.Ph. D. Dissertation 2005University of California; Irvine
- (22).Metwally SAM, Younes MI, Abbas HH. Acta Chim. Hung. 1989;126:591–597. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.