

Abstract
NAG-1 (nonsteroidal anti-inflammatory drug-activated gene), a member of the transforming growth factor-β superfamily, is involved in many cellular processes, such as inflammation, apoptosis/survival, and tumorigenesis. Vitamin E succinate (VES) is the succinate derivative of α-tocopherol and has antitumorigenic activity in a variety of cell culture and animal models. In the current study, the regulation and role of NAG-1 expression in PC-3 human prostate carcinoma cells by VES was examined. VES treatment induced growth arrest and apoptosis as well as an increase in NAG-1 protein and mRNA levels in a time- and concentration-dependent manner. VES treatment induced nuclear translocation and activation of p38 kinase. Pretreatment with p38 kinase inhibitor blocked the VES-induced increase in NAG-1 protein and mRNA levels, whereas an inhibition of protein kinase C, Akt, c-Jun NH2-terminal kinase, or MEK activity had no effect on VES-induced NAG-1 levels. Forced expression of constitutively active MKK6, an upstream kinase for p38, induced an increase in NAG-1 promoter activity, whereas p38 kinase inhibitor blocked MKK6-induced increase in NAG-1 promoter activity. VES treatment resulted in >3-fold increase in the half-life of NAG-1 mRNA in a p38 kinase-dependent manner and transient transfection experiment showed that VES stabilizes NAG-1 mRNA through AU-rich elements in 3′-untranslated region of NAG-1 mRNA. The inhibition of NAG-1 expression by small interfering RNA significantly blocked VES-induced poly(ADP-ribose) polymerase cleavage, suggesting that NAG-1 may play an important role in VES-induced apoptosis. These results indicate that VES-induced expression of NAG-1 mRNA/protein is regulated by transcriptional/post-transcriptional mechanism in a p38 kinase-dependent manner and NAG-1 can be chemopreventive/therapeutic target in prostate cancer.
Introduction
Prostate cancer is the most common malignancy in men and the second leading cause of cancer-related deaths in men in the United States. Like most cancers, prostate cancer development/progression may result from interactions between genetic susceptibility and environmental exposures, such as diet or drugs. Vitamin E is a group of compounds, including different subtypes of tocopherols and tocotrienols, and α-tocopherol is known as the most abundant and active forms. Initially, α-Tocopherol β-Carotene Cancer Prevention study showed that the long-term intake of vitamin E substantially reduces prostate cancer incidence and mortality. Other epidemiologic and clinical studies also suggested that the intake of vitamin E may prevent prostate cancer (1, 2) and these findings formed the basis of the Selenium and Vitamin E Cancer Prevention Trial, which is in progress. Vitamin E succinate (VES) is the succinate derivative of α-tocopherol and has the most effective antitumorigenic activity in a variety of cell culture and animal models (3-8). The antitumorigenic activity of VES includes inhibition of proliferation (9, 10), invasion (11), and angiogenesis (12) and induction of apoptosis (13). VES induces apoptosis in many types of cancer cells, including breast, cervical, endometrial, colon, and prostate. However, VES does not induce apoptosis in normal human mammary epithelial cells (14) or normal human prostate epithelial cells (15). Although many studies have suggested the potential mechanism for antitumor activity of VES, the mechanism for antitumorigenic activity of VES is still not clearly understood. As VES treatment has multiple effects on cancer cells, it is possible that VES affects multiple pathways that regulate the growth/survival of cancer cells.
Nonsteroidal anti-inflammatory drug-activated gene-1 (NAG-1) is a distant member of transforming growth factor-β superfamily and encodes a secreted protein. NAG-1 protein is synthesized as a 308–amino acid propeptide with RXXR cleavage site and secreted as a 30-kDa dimeric mature protein after cleavage at RXXR site and formation of disulfide bond between cysteine residues. NAG-1 is highly expressed in human placenta and prostate. NAG-1 is also known as macrophage inhibitory cytokine-1, placental transforming growth factor-β, prostate-derived factor, novel placental bone morphogenic protein, or growth differentiation factor-15. NAG-1 was identified from a cyclooxygenase inhibitor (indomethacin)–treated HCT-116 colon cancer cell lines using subtractive hybridization. Many studies have shown that the expression of NAG-1 is induced by many chemopreventive/chemotherapeutic agent and NAG-1 negatively regulates cell proliferation or survival in a variety of cancer cells. For example, forced expression of NAG-1 inhibits proliferation of breast carcinoma cells, mink lung epithelial cells, and prostate carcinoma cells (16, 17). Treatment with NAG-1 induces a decrease in cell adhesion and an increase in apoptosis in the prostate cancer cell line (18) and forced expression of NAG-1 in HCT-116 colon cancer cells results in reduced soft agar growth and tumor growth in nude mice (19). We recently have shown that NAG-1 negatively affects cell survival during 12-O-tetradecanoylphorbol-13-acetate–induced apoptosis in prostate cancer cells (20). In addition, the study on NAG-1 transgenic mice showed that NAG-1 expression suppresses intestinal tumorigenesis (21).
The p38 kinase family is a group of serine/threonine kinase and there are four isoforms of p38 kinase (α, β, γ, and δ; refs. 22, 23). In response to cellular stress or external stimuli, p38 kinase is phosphorylated by its upstream kinase, MKK3/6 (24), resulting in the activation of p38 kinase. Many chemopreventive/chemotherapeutic chemicals, such as resveratrol (25), EGCG (26), Taxol (27), and cisplatin (28), have been shown to activate p38. The activated p38 kinase phosphorylates downstream target molecules, such as ATF-2, GADD153, MEF2, and p53, and induces cellular responses including growth arrest, apoptosis, cytokine synthesis, and differentiation in a cell type–specific manner. In addition to activating transcription factors, p38 kinase has been shown to increase expression of certain genes, such as tumor necrosis factor-α, interleukin-6, MIP-1 (29), and cyclooxygenase-2 (30), via stabilization of their mRNA.
In this study, we examined the regulation and the role of NAG-1 expression in VES-treated PC-3 human prostate carcinoma cells. We report that the VES induces NAG-1 expression via transcriptional and post-transcriptional mechanism in a p38-dependent manner and NAG-1 plays an important role in VES-induced apoptosis.
Materials and Methods
Materials
Akt antibody, phospho-Akt antibody (Ser473), extracellular signal-regulated protein (ERK), phospho-ERK antibody (Thr218/Tyr220), c-Jun NH2-terminal kinase (JNK), phospho-JNK antibody (Thr183/Tyr185), phospho-p38 antibody (Thr180/Tyr182), phospho-heat shock protein 27 (Hsp27) antibody (Ser82), and poly(ADP-ribose) polymerase (PARP) antibody were purchased from Cell Signaling Technology. p38 antibody was purchased Santa Cruz Biotechnology. Hsp27 antibody was purchased from StressGen. β-Actin antibody was purchased from Sigma. VES, Go6983, Ly294002, PD98059, SB203580, SB202190, and SP600125 were purchased from Calbiochem.
Cell Culture
PC-3 human prostate carcinoma cells were purchased from the American Type Culture Collection and cultured in RPMI 1640 supplemented with 10% fetal bovine serum (Hyclone) and 10 μg/mL gentamycin (Invitrogen). For the VES treatment, 2.5 × 105 cells were plated in P60 cell culture dish and 48 h later, cells were treated with indicated concentration of VES in RPMI 1640 supplemented with 2.5% fetal bovine serum.
Western Blot Analysis
Cells were lysed in radioimmunoprecipitation buffer and protein concentration was measured using BCA protein assay kit (Pierce). Equal amounts of protein were solubilized and heated at 65°C in LDS sample buffer (Invitrogen) with sample reducing agent (Invitrogen) for 10 min and then separated by SDS-PAGE. The separated proteins were transferred to an Immobilon-P membrane (Millipore). Following incubation in blocking buffer (TBS with 5% nonfat dry milk and 0.1% Tween 20) for 1 h at room temperature, the membranes were probed overnight at 4°C. The membranes were washed and then probed with a horseradish peroxidase–linked secondary antibody (Amersham Biosciences) for 1 h at room temperature. Detection was made with an enhanced chemiluminescence reagent (Amersham Biosciences) followed by exposure of membrane to film. Western blot analysis was done at least three times. For studies involving kinase inhibitors, the phosphorylation status of a substrate for each kinase was determined to confirm that inhibitors are effective (data not shown).
Northern Blot Analysis
Total RNA was isolated using SV total RNA isolation kit (Promega) or mRNA isolation kit (Qiagen). NAG-1 cDNA was labeled with [32P]dCTP by using Decaprime II kit (Ambion) according to the manufacturer's protocol. RNA was electrophoresed on agarose gel containing formaldehyde, transferred to Hybond XL membrane (Amersham Biosciences), and UV cross-linked. Blots were incubated at 65°C in Rapid Hybridization buffer (Amersham Biosciences) and sequentially washed with washing buffer 1 (0.5% SDS, 0.1× SSC) and washing buffer 2 (0.1% SDS, 0.1× SSC) at 65°C. Films were exposed to membranes at −80°C and developed. Northern blot analysis was done at least three times and data from a representative experiment are shown.
Plasmids, Transfection, and Luciferase Assay
pcDNA3-MKK6b(E) was the kind gift from Dr. Jiahuai Han (Department of Molecular Biology, The Scripps Research Institute). Construction of the NAG-1 reporter plasmid has been described previously (31). To generate pNAG-1 3′-untranslated region (UTR)-luciferase plasmid, NAG-1 3′-UTR was amplified with primers (forward primer: GGCCGATCTAGAGTTAACGCAGTCCTGGTCCTTCCACTGT and reverse primer: GGCCGATCTAGAAAACAGTTCAGACAGCTTTATT, XbaI site is italicized) and amplified PCR product was digested with XbaI and ligated to XbaI site between luciferase and SV polyadenylation site in pGL3 promoter vector (Promega). All plasmids were sequenced. For luciferase assay, PC-3 cells were plated at 0.65 × 105 per well in a 12-well culture plate. Forty-eight hours after plating, PC-3 cells were transfected in triplicate with the specified reporter plasmid as described in the text and phRL-null plasmid (Promega) using LipofectAMINE 2000 (Invitrogen) according to the manufacturer's protocol. Forty-eight hours later, cells were harvested and the firefly luciferase activity was determined and normalized to Renilla luciferase activity with a dual luciferase assay kit (Promega). Luciferase assay was done at least three times.
Immunofluorescence Staining
For immunofluorescence detection of p38 kinase, PC-3 cells were plated onto coverslips and treated with VES. Twenty-four hours after VES treatment, the cultures were fixed in 4% paraformaldehyde at 4°C for 10 min and incubated with p38 polyclonal antibody (Santa Cruz Biotechnolgy) in 0.2% Triton X-100 and 0.5% normal goat serum in PBS at 4°C overnight. After washing, the samples were incubated with FITC-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology) at room temperature for 1 h. After rinsing, glass coverslips were mounted with mounting medium (Vector Lab) and cells were examined with a Nikon microscope equipped with filter cubes for the detection of FITC fluorescence.
Reverse Transcription and Real-time Reverse Transcription-PCR
RNA was treated with 1 unit amplification grade DNase I (Invitrogen) per microgram RNA at room temperature for 15 min to remove genomic DNA followed by inactivation of the DNase I with 2.5 mmol/L EDTA (pH 8.0) and incubation at 65°C for 5 min. Reverse transcription was done with 5 μg total RNA using SuperScript II reverse transcription system (Invitrogen) according to the manufacturer's instructions. Real-time reverse transcription-PCR was done in triplicate with individual time-matched vehicle-treated controls using with Power SYBR Green PCR Master Mix (Applied Biosystems). Real-time PCR analysis was done at least three times and data from a representative experiment are shown.
Generation of NAG-1 Small Interfering RNA Expressing PC-3 Cells
The NAG-1 small interfering RNA (siRNA) vector (pSuper-retro-puro-si NAG-1) was constructed using a pSuper-retro-puro and a synthetic oligonucleotide targeting 5′-ACATGCACGCGCAGATCAA-3′ corresponding to positions 780 to 798 on NAG-1 mRNA. ϕNX cells were cultured in DMEM plus 10% fetal bovine serum and transfected with pSuper-retro-puro-si NAG-1 by LipofectAMINE 2000 (Invitrogen) according to the manufacturer's protocol. Virus-containing medium was collected from ϕNX cell cultures three times, 6 h apart, filtered through a 0.45-μm filter, and overlaid onto PC-3 cells with 10 μg/mL Polybrene (Sigma). Cultures were maintained with viral-containing medium overnight, and 24 h later, cells were shifted to a selection medium containing 2 μg/mL puromycin (Sigma). Puromycin-resistant PC-3 cells were pooled and used for Western blot analysis.
Results
VESInduces Growth Arrest and Cell Deathin PC-3 Cells
VES induces growth arrest and apoptosis in a variety of cancer cells including prostate cancer cells (32-35). We first determined if VES treatment induces growth arrest or cell death in PC-3 cells. PC-3 cells were plated and treated with varying concentrations of VES for 48 h. As shown in Fig. 1A and B, VES treatment induced a concentration-dependent increase in cell death and growth arrest in PC-3 cells. VES-induced growth arrest/cell death was also observed in androgen-sensitive LNCaP human prostate carcinoma cells (data not shown).
Figure 1.

VES induces cell death and growth arrest in PC-3 cells. A, PC-3 cells were plated on a six-well plate and each group (three wells) was treated with various concentrations of VES for 48 h. After VES treatment, cells were trypsinized and combined with floating cells. The number of trypan blue–stained cells was counted. B, PC-3 cells were plated on a six-well plate and each group (three wells) was treated with various concentrations of VES for 48 h. After treatment, attached cells were harvested by trypsinization and counted under the microsccope.
VES Induces NAG-1 Protein and mRNA Expression in PC-3 Cells in a Concentration- and Time-Dependent Manner
NAG-1 has been known to induce growth arrest or apoptosis in a variety of cancer cells and we have shown previously that forced expression of NAG-1 resulted in growth arrest/apoptosis in PC-3 cells (36). To study if NAG-1 is involved in the antiproliferative/proapoptotic effect of VES, we determined the effect of VES on NAG-1 levels in PC-3 cells. PC-3 cells were treated with various concentrations of VES for 48 h and NAG-1 protein levels were determined by Western blot analysis. As shown in Fig. 2A and B, VES induced an increase in NAG-1 protein levels in a concentration- and time-dependent manner. To determine whether this increase in NAG-1 protein levels results from increased NAG-1 mRNA levels, we performed Northern blot analysis for NAG-1. Figure 2C and D shows that VES treatment increased NAG-1 mRNA levels in a concentration- and time-dependent manner, indicating that an increase in NAG-1 protein levels by VES is due to an increase in NAG-1 mRNA levels. To determine if VES treatment activates the NAG-1 promoter, PC-3 cells were transfected with a luciferase reporter gene fused to NAG-1 promoter and treated with VES. As shown in Fig. 2E, VES treatment resulted in an increase in NAG-1 promoter activity, suggesting that VES activates NAG-1 promoter. However, VES treatment increased NAG-1 promoter luciferase activity 3-fold over control, whereas VES treatment induced an increase in NAG-1 mRNA >3-fold (Fig. 2C and D), suggesting that other mechanism(s) are involved in VES-induced NAG-1 protein/ mRNA expression.
Figure 2.

VES induces NAG-1 protein/mRNA expression in a concentration- and time-dependent manner. A, PC-3 cells were treated with the indicated concentrations of VES for 48 h and total lysates were subjected to Western blot analysis for NAG-1. For Western analysis, 39-kDa form of NAG-1 protein (proform) was shown. The membrane was stripped and reprobed for β-actin to confirm equal loading. B, PC-3 cells were treated with 20 μmol/L VES for the indicated amount of time and total cell lysates were subjected to Western blot analysis for NAG-1. For Western analysis, 39-kDa form of NAG-1 protein (proform) was shown. The membrane was stripped and reprobed for β-actin to confirm equal loading. C, PC-3 cells were treated with the indicated concentrations of VES for 48 h and mRNA was subjected to Northern blot analysis for NAG-1. The membrane was stripped and reprobed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to confirm equal loading of RNA. D, PC-3 cells were treated with 20 μmol/L VES for indicated amount of time and mRNA was subjected to Northern blot analysis for NAG-1. The membrane was stripped and reprobed for glyceraldehyde-3-phosphate dehydrogenase to confirm equal loading of RNA. E, PC-3 cells were transfected with NAG-1 promoter reporter plasmid (−966/70) and phRL-null plasmid. Twenty-four hours after transfection, cells were treated with 20 μmol/L VES for 48 h. NAG-1 promoter reporter activity was normalized to Renilla luciferase activity.
VES Induces NAG-1 Expression in p38 Kinase-Dependent Manner
NAG-1 expression is regulated by several kinases, such as Akt/GSK3 (37), ERK (38), and protein kinase C (20), in a cell type–specific manner. VES treatment induces changes in activity of some kinases, including JNK (39) and ERK (40). Therefore, we examined if VES treatment induces changes in activation status of kinases, which have been known to be involved in NAG-1 expression or which have been shown to be regulated by VES. PC-3 cells were treated with VES for various time points and total cell lysates were subjected for Western blot analysis for phospho-ERK, phospho-Akt, phospho-JNK, and phospho-p38 to determine if VES treatment activates or inhibits these kinases. As shown in Fig. 3A, VES treatment resulted in increased level of phospho-Akt and phospho-ERK until 6 h after VES treatment, whereas phospho-Akt and phospho-ERK levels are significantly reduced at 24 h after VES treatment. Phosphorylation of JNK was induced 6 h after VES treatment and maintained until 48 h, whereas phosphorylation of p38 was significantly induced at 48 h after VES treatment. These results suggest that either the inhibition of Akt or ERK or the activation of JNK or p38 may be involved in VES-induced NAG-1 expression. To test this possibility, we determined the effect of kinase inhibitors on VES-induced NAG-1 expression. If an inhibition of Akt or ERK is responsible for VES-induced NAG-1 expression, treatment with Akt or ERK inhibitor should induce NAG-1 expression. If an activation of JNK or p38 kinase is responsible for VES-induced NAG-1 expression, pretreatment with JNK or p38 kinase inhibitor should block VES-induced NAG-1 expression. PC-3 cells were pretreated with specific inhibitor for MEK inhibitor (PD98059), phosphatidylinositol-3-kinase inhibitor (Ly294002), JNK inhibitor (SP600125), or p38 kinase inhibitor (SB203580). Because NAG-1 expression is also regulated by protein kinase C, we included an inhibitor for protein kinase C (Go6983). PC-3 cells were then treated with VES and total lysates were subjected to Western blot analysis. Whereas SB203580 pretreatment significantly blocked VES-induced NAG-1 expression, the inhibitors for other kinases failed to block VES-induced NAG-1 expression (Fig. 3B), suggesting that VES induces NAG-1 expression in a p38 kinase-dependent mechanism. To confirm that VES induces NAG-1 expression in a p38 kinase-dependent mechanism, PC-3 cells were pretreated with SB202190, another p38 kinase inhibitor, and treated with VES. Pretreatment with SB202190 significantly blocked VES-induced NAG-1 protein expression (Fig. 3C), showing that VES induces NAG-1 protein expression in a p38 kinase-dependent mechanism. Because VES induced an increase in NAG-1 mRNA levels (Fig. 2C and D), we examined if an inhibition of p38 kinase blocks VES-induced increase in NAG-1 mRNA levels. PC-3 cells were pretreated with SB203580 and treated with VES. Forty-eight hours after VES treatment, RNA was isolated and subjected to real-time PCR analysis. As shown in Fig. 3D, VES induced NAG-1 mRNA levels by >9-fold, whereas the inhibition of p38 kinase blocked VES-induced increase in NAG-1 mRNA levels, suggesting that VES induces an increase in NAG-1 mRNA levels in a p38 kinase-dependent mechanism.
Figure 3.

VES induces NAG-1 protein/mRNA expression in a p38 kinase-dependent mechanism. A, PC-3 cells were treated with 20 μmol/L VES for the indicated amount of time, and total cell lysates were subjected to Western blot analysis for NAG-1. For Western analysis, 39-kDa form of NAG-1 protein (proform) was shown. Membranes was stripped and reprobed with indicated antibody. B, PC-3 cells were pretreated with 2.5 μmol/L Go6983, 20 μmol/L PD980509, 20 μmol/L Ly294002, 20 μmol/L SP600125, or 20 μmol/L SB203580 for 50 min and treated with 20 μmol/L VES for 48 h. Total cell lysates were subjected to Western blot analysis for NAG-1. For Western analysis, 39-kDa form of NAG-1 protein (proform) was shown. C, PC-3 cells were pretreated with 20 μmol/L SB203580 or SB202190 for 50 min and treated with 20 μmol/L VES for 48 h. Total cell lysates were subjected to Western blot analysis for NAG-1. For Western analysis, 39-kDa form of NAG-1 protein (proform) was shown. CTL, vehicle treated. D, PC-3 cells were pretreated with 20 μmol/L SB203580 for 50 min and treated with 20 μmol/L VES for 48 h. Total RNA was purified and 5 μg total RNA was reverse transcribed. The level of NAG-1 cDNA was analyzed with real-time PCR analysis. CTL, vehicle treated; SB203580, treated with SB203580; SB203580/VES, pretreated with SB203580 before VES treatment; VES, VES treated.
VES Increases p38 KinaseActivity and Induces Nuclear Translocation of p38
Because VES treatment induced the expression of NAG-1 protein/mRNA in a p38 kinase-dependent manner, we examined if VES treatment activates p38 kinase pathway. Hsp27 is known to be phosphorylated at Ser15, Ser78, and Ser82 by MAPKAP kinase 2 as a result of the activation of the p38 mitogen-activated protein kinase pathway (41, 42). To determine if p38 kinase is activated by VES treatment, PC-3 cells were treated with VES in the presence or absence of SB203580 and cell lysates were subjected to Western blot analysis for phospho-Hsp27. As shown in Fig. 4A, VES treatment significantly increased the phosphorylation of Hsp27, and VES-induced increase in Hsp27 phosphorylation was blocked by p38 kinase inhibitor treatment, suggesting that VES does activate p38 pathway in PC-3 cells.
Figure 4.

VES induces p38 kinase activation and nuclear translocation. A, PC-3 cells were pretreated with 20 μmol/L SB203580 for 50 min and treated with 20 μmol/L VES for 48 h. Total cell lysates were subjected to Western blot analysis for phospho-Hsp27. Membrane was stripped and reprobed for total Hsp27. CTL, vehicle treated; SB203580, treated with SB203580; SB203580/VES, pretreated with SB203580 before VES treatment; VES, VES treated. B, PC-3 cells were plated on coverslip and treated with indicated concentration of VES for 24 h. Cells are stained by p38 antibody followed by FITC-conjugated anti-rabbit antibody.
Members of the mitogen-activated protein kinase family are translocated to the nucleus where they are thought to phosphorylate their target transcription factors and p38 kinase has been shown to be concentrated in nuclei (43). To determine if VES treatment induces nuclear localization of p38, we did immunofluorescence staining for p38 (Fig. 4B). PC-3 cells were plated on coverslip and treated with VES for 24 h before immunofluorescence staining as 48-h treatment with VES resulted in almost complete loss of attached cells. p38 localizes diffusively in cytoplasm and nuclei in vehicle-treated cells. However, p38 kinase started to be concentrated in nuclei in some of 10 μmol/L VES-treated cells and nuclear localization of p38 was significantly increased in 20 μmol/L VES-treated cells, indicating VES treatment induces nuclear localization of p38.
Constitutively Active MKK6, Upstream Kinase p38, Activates NAG-1 Promoter in a p38 Kinase-Dependent Manner
Mitogen-activated protein kinase activation is achieved by kinase cascade and it is believed that MKK3 and MKK6 are the two main MKKs that activate p38α (44, 45). To determine the direct role of p38 on NAG-1 expression, PC-3 cells were cotransfected with constitutively active MKK6 and NAG-1 promoter reporter plasmid. As shown in Fig. 5A, constitutively active MKK6 induced NAG-1 promoter activity by >4-fold, suggesting that NAG-1 promoter may be regulated by p38 kinase-activated transcription factor. MKK6-induced increase in NAG-1 promoter activity was significantly repressed by pretreatment with p38 kinase inhibitor, SB203580, indicating that the activation of p38 kinase is important for NAG-1 promoter activation. Because VES treatment induced NAG-1 mRNA by >4.5-fold (Fig. 2C and D), there may be additional transcription factor binding site(s), which is activated by p38 kinase. To determine if there are additional transcription factor binding sites that are activated by p38, we cotransfected PC-3 cells with constitutively active MKK6 and various length of NAG-1 promoter reporters. However, we were not able to find additional increase in NAG-1 promoter reporter activity by cotransfection with constitutively active MKK6, whereas we found that NAG-1 promoter region from −133 to −474 is responsible for an increase in NAG-1 promoter activity by constitutively active MKK6 (Fig. 5B), suggesting the existence of far-upstream or downstream regulatory site(s) or other mechanism for an increase in mRNA levels.
Figure 5.

MKK6 activates NAG-1 promoter in a p38 kinase-dependent mechanism. A, PC-3 cells were cotransfected with NAG-1 promoter reporter plasmid (−966/70) and pcDNA3.1-lacZ or pcDNA3-MKK6b(E). As an internal control, the phRL-null vector was used to correct for transfection efficiency. Twenty-four hours after transfection, medium was changed and cells were incubated for another 24 h in the presence or absence of 20 μmol/L SB203580. Luciferase activity was measured 48 h after transfection and firefly luciferase activity was normalized to Renilla luciferase. B, PC-3 cells were cotransfected with indicated length of NAG-1 promoter reporter plasmid and pcDNA3.1-lacZ or pcDNA3-MKK6b(E). As an internal control, the phRL-null vector was used to correct for transfection efficiency. Luciferase activity was measured 48 h after transfection and firefly luciferase activity was normalized to Renilla luciferase.
VES Stabilizes NAG-1 mRNA in a p3 8 Kinase-Dependent Manner
p38 kinase increases the expression of certain genes, such as tumor necrosis factor-α, interleukin-6, MIP-1 (29), and cyclooxygenase-2 (30) via stabilization of mRNA. Because NAG-1 has four copies of an AU-rich element (ARE) in the 3′-UTR of its mRNA (46), it is possible that VES-induced p38 kinase activity may contribute to increased NAG-1 mRNA transcription as well as mRNA stabilization. To determine if VES stabilizes NAG-1 mRNA, we amplified 3′-UTR of NAG-1 mRNA using PCR and inserted it between luciferase and poly(A) signal in pGL3 promoter construct (pNAG-1 3′-UTR-luciferase). When PC-3 cells were transfected with pNAG-1 3′-UTR-luciferase and treated with VES, luciferase activity was increased 3-fold (Fig. 6A) over pGL3 promoter which does not have 3′-UTR of NAG-1, suggesting that VES stabilizes 3′-UTR of NAG-1 mRNA. In addition, forced expression of constitutively active MKK6 activated pNAG-1 3′-UTR-luciferase, suggesting that p38 stabilizes 3′-UTR of NAG-1 mRNA (Fig. 6B). To obtain direct evidence for the role of VES on the stability of NAG-1 mRNA, PC-3 cells were treated with VES in the presence or absence of SB203580 for 48 h followed by actinomycin D treatment to block new mRNA synthesis. RNA was isolated at various time points after actinomycin D treatment and subjected to real-time PCR analysis to determine the level of NAG-1 mRNA. As shown in Fig. 6C, the half-life of NAG-1 mRNA was ∼7 h whereas VES treatment significantly increased the half-life of NAG-1 (∼21 h). VES-induced increase in the half-life of NAG-1 mRNA was blocked by SB203580 pretreatment, suggesting that VES-induced activation of p38 is important in stabilization of NAG-1 mRNA.
Figure 6.

NAG-1 mRNA is stabilized by VES treatment in a p38 kinase-dependent mechanism. A, PC-3 cells were transfected with pGL3 promoter or pGL3 promoter plasmid with 3′-UTR of NAG-1. As an internal control, the phRL-null vector was used to correct for transfection efficiency. Twenty-four hours after transfection, cells were treated with or without VES for 48 h. Luciferase activity was measured and firefly luciferase activity was normalized to Renilla luciferase. B, PC-3 cells were cotransfected with pGL3 promoter or pGL3 promoter plasmid with 3′-UTR of NAG-1 and pcDNA3.1-lacZ or pcDNA3-MKK6b(E). As an internal control, the phRL-null vector was used to correct for transfection efficiency. Luciferase activity was measured 48 h after transfection and firefly luciferase activity was normalized to Renilla luciferase. C, PC-3 cells were treated with or without VES in the presence or absence of SB203580. Forty-eight hours after VES treatment, cells were treated with 5 μg/mL actinomycin D for the times indicated and NAG-1 mRNA levels adjusted to actin were measured by real-time reverse transcription-PCR analysis. The level of NAG-1 mRNA was expressed as the percentage of the NAG-1 mRNA at the 0 h of actinomycin D treatment in each group.
Inhibition of NAG-1 Expression Suppresses VES-Induced Apoptosis in PC-3 Cells
To determine direct role of NAG-1 in VES-induced apoptosis in PC-3 cells, we generated PC-3 cells that stably express NAG-1 siRNA using retroviral system. Empty vector or NAG-1 siRNA-infected PC-3 cells were selected in the presence of puromycin and were pooled. Each cell line was treated with VES for 48 h and total lysates were subjected to Western blot analysis for PARP and cleaved PARP. PARP cleavage is used as a marker for apoptosis and as shown in Fig. 7, the stable expression of NAG-1 siRNA significantly blocked VES-induced PARP cleavage, indicating that NAG-1 expression plays an important role in VES-induced apoptosis. Additionally, SB203580 pretreatment significantly blocked VES-induced cell death in PC-3 cells (data not shown).
Figure 7.
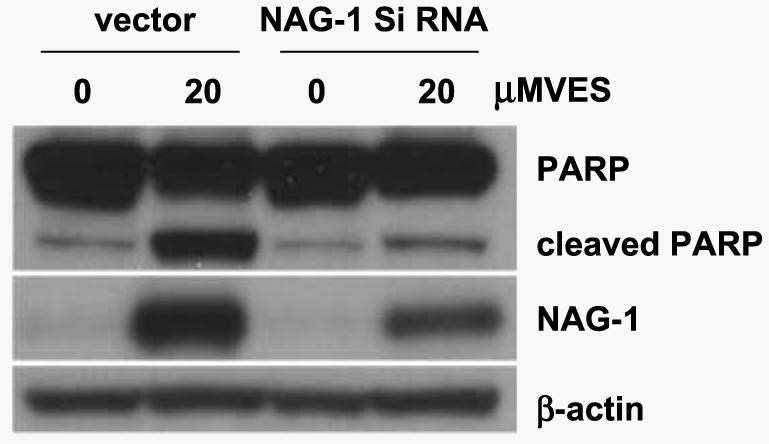
NAG-1 siRNA-expressing PC-3 cells are resistant to VES-induced apoptosis. Empty vector or NAG-1 siRNA-expressing PC-3 cells were treated with 20 μmol/L VES for 48 h. Total lysates were subjected to Western blot analysis for PARP and NAG-1. For Western analysis, 39-kDa form of NAG-1 protein (proform) was shown. The membrane was stripped and reprobed for β-actin to confirm equal loading.
Discussion
In this report, we found that p38 kinase regulates VES-induced NAG-1 mRNA/protein expression via transcriptional/post-transcriptional mechanism and NAG-1 expression is important in VES-induced apoptosis.
Yu et al. (47) reported that the activation of ERK and JNK but not p38 is required for VES-induced apoptosis of human breast cancer cells. We observed the increased level of phospho-JNK after VES treatment (Fig. 3A); however, pretreatment with a JNK inhibitor did not block VES-induced NAG-1 (Fig. 3B) and cell death (data not shown). This discrepancy may be due to different time points (12 versus 48 h), different VES concentration (20 μg/mL = 37 versus 20 μmol/L), or different cell type (MDA-MB-435 human breast cancer cells versus PC-3 human prostate carcinoma cells). In addition, we have determined if an inhibition of protein kinase A affect VES-induced NAG-1 expression using protein kinase A inhibitor (H-89). However, an inhibition of protein kinase A did not affect VES-induced NAG-1 expression, although H-89 treatment blocked phosphorylation of CREB (data not shown).
Because VES treatment induced NAG-1 promoter reporter activity, we tried to determine the transcription factor that activates NAG-1 promoter in response to VES treatment. NAG-1 has been shown to be a target gene for several transcription factors, such as p53, Egr-1, ATF-3, and Sp1. In addition, p38 kinase has been shown to phosphorylate and activate several transcription factors, such as MEF2, ATF-2, CHOP, Ets-1, Elk-1, and p53. We have examined if VES treatment activates or induces expression of above-mentioned transcription factors in a p38 kinase-dependent manner. Based on gel shift, Western blot analysis, or NAG-1 promoter reporter assays, none of the known p38 target transcription factors were responsible for VES-induced NAG-1 expression (data not shown). Furthermore, the transcriptional activity or the expression level of Egr-1 or Sp1 was not changed by VES treatment. Interestingly, the level of ATF-3 expression was significantly increased by VES treatment. However, the increase in ATF-3 expression by VES was p38 kinase independent but protein kinase A dependent (data not shown), indicating that ATF-3 is not responsible for VES-induced NAG-1 expression. These results suggest that VES may activate or induce expression of novel p38 target transcription factor.
Although NAG-1 expression is induced by many stimuli via transcriptional activation, some chemopreventive/chemotherapeutic chemicals, such as MCC-555 (46) and 5F-203 (48), have been shown to increase NAG-1 mRNA levels by stabilizing NAG-1 mRNA levels. The regulation of mRNA stability is the important mechanism to control certain gene expression where the stability of mRNA is determined by interactions between specific sequences within mRNA (cis-acting elements) and cellular RNA-binding proteins (trans-acting factors). One of the best characterized cis-acting elements is the ARE which exists in the 3′-UTR of mRNAs of cytokines or growth factors and NAG-1 has four well-conserved copies of ARE (46). Because VES treatment activates p38 kinase and p38 kinase regulates the stability of certain mRNAs, such as tumor necrosis factor-α and Cox-2, we hypothesized that VES may stabilize NAG-1 mRNA and found that VES stabilize NAG-1 mRNA. ARE regulates the stability of mRNAs via the interaction with RNA-binding proteins. There are several known ARE-binding proteins, such as HuR, AUF1, TIA-1, and TTP, which bind to ARE and stabilize/destabilize mRNA. We examined the level of these mRNA-binding proteins after VES treatment. HuR is a member of RNA-binding proteins (49) and known to bind/stabilize AREs in a p38 kinase-dependent manner. However, based on Western blot analysis, the level of HuR protein was not changed significantly and nuclear/cytoplasmic distribution of HuR protein was not changed by VES treatment (data not shown). The level of TTP mRNA, ARE-binding protein that destabilizes mRNA, was increased by VES treatment (data not shown), which is consistent with the finding that p38 pathway up-regulate TTP expression by stabilizing ARE in TTP mRNA (50). However, the forced expression of TTP did not affect endogenous level of NAG-1 (data not shown). In addition, the level of AUF1 mRNA was decreased by VES treatment (data not shown). Because VES treatment affected the level of these ARE-binding proteins, it is possible that VES may regulate the expression of growth/apoptotic genes by changing their mRNA stability. Further studies will focus which ARE-binding proteins are involved in VES-induced stabilization of NAG-1 mRNA.
Our results indicate that VES-induced NAG-1 expression could contribute the antitumorigenic activity of VES. The level of NAG-1 expression was only significant at 10 and 20 μmol/L of VES treatment (Fig. 2A and C), at which significant growth arrest and cell death occurs (Fig. 1A and B), indicating the possible role of NAG-1 in VES-induced growth arrest or cell death. NAG-1-expressing PC-3 cells that stably express NAG-1 resulted in ∼50% reduction in final tumor weight1 in nude mice xenograft experiment, which is consistent with previous findings (36). Furthermore, the expression of NAG-1 siRNA significantly blocked VES-induced apoptosis (Fig. 7). However, VES seems to affect many other signaling molecules which are important for cell growth/survival because NAG-1 siRNA expression was not able to completely block VES-induced apoptosis. In summary, we have shown that VES, which is a chemopreventive compound in prostate cancer, induces NAG-1 expression in a p38 kinase-dependent mechanism and these findings suggest NAG-1 as a possible therapeutic target against the prostate cancer.
Acknowledgments
Grant support: NIH, NIEHS Intramural Program.
Footnotes
Unpublished data.
References
- 1.Chan JM, Stampfer MJ, Ma J, Rimm EB, Willett WC, Giovannucci EL. Supplemental vitamin E intake and prostate cancer risk in a large cohort of men in the United States. Cancer Epidemiol Biomarkers Prev. 1999;8:893–9. [PubMed] [Google Scholar]
- 2.Weinstein SJ, Wright ME, Pietinen P, et al. Serum α-tocopherol and γ-tocopherol in relation to prostate cancer risk in a prospective study. J Natl Cancer Inst. 2005;97:396–9. doi: 10.1093/jnci/dji045. [DOI] [PubMed] [Google Scholar]
- 3.Malafa MP, Neitzel LT. Vitamin E succinate promotes breast cancer tumor dormancy. J Surg Res. 2000;93:163–70. doi: 10.1006/jsre.2000.5948. [DOI] [PubMed] [Google Scholar]
- 4.Neuzil J, Weber T, Schroder A, et al. Induction of cancer cell apoptosis by α-tocopheryl succinate: molecular pathways and structural requirements. FASEB J. 2001;15:403–15. doi: 10.1096/fj.00-0251com. [DOI] [PubMed] [Google Scholar]
- 5.Wu K, Shan YJ, Zhao Y, Yu JW, Liu BH. Inhibitory effects of RRR-α-tocopheryl succinate on benzo(a)pyrene (B(a)P)-induced forestomach carcinogenesis in female mice. World J Gastroenterol. 2001;7:60–5. doi: 10.3748/wjg.v7.i1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malafa MP, Fokum FD, Mowlavi A, Abusief M, King M. Vitamin E inhibits melanoma growth in mice. Surgery. 2002;131:85–91. doi: 10.1067/msy.2002.119191. [DOI] [PubMed] [Google Scholar]
- 7.Kogure K, Manabe S, Hama S, Tokumura A, Fukuzawa K. Potentiation of anti-cancer effect by intravenous administration of vesiculated α-tocopheryl hemisuccinate on mouse melanoma in vivo. Cancer Lett. 2003;;192:19–24. doi: 10.1016/s0304-3835(02)00683-3. [DOI] [PubMed] [Google Scholar]
- 8.Barnett KT, Fokum FD, Malafa MP. Vitamin E succinate inhibits colon cancer liver metastases. J Surg Res. 2002;106:292–8. doi: 10.1006/jsre.2002.6466. [DOI] [PubMed] [Google Scholar]
- 9.Israel K, Sanders BG, Kline K. RRR-α-tocopheryl succinate inhibits the proliferation of human prostatic tumor cells with defective cell cycle/differentiation pathways. Nutr Cancer. 1995;24:161–9. doi: 10.1080/01635589509514404. [DOI] [PubMed] [Google Scholar]
- 10.Turley JM, Funakoshi S, Ruscetti FW, et al. Growth inhibition and apoptosis of RL human B lymphoma cells by vitamin E succinate and retinoic acid: role for transforming growth factor β. Cell Growth Differ. 1995;6:655–63. [PubMed] [Google Scholar]
- 11.Zhang M, Altuwaijri S, Yeh S. RRR-α-tocopheryl succinate inhibits human prostate cancer cell invasiveness. Oncogene. 2004;23:3080–8. doi: 10.1038/sj.onc.1207435. [DOI] [PubMed] [Google Scholar]
- 12.Malafa MP, Fokum FD, Smith L, Louis A. Inhibition of angiogenesis and promotion of melanoma dormancy by vitamin E succinate. Ann Surg Oncol. 2002;9:1023–32. doi: 10.1007/BF02574523. [DOI] [PubMed] [Google Scholar]
- 13.Yu W, Sanders BG, Kline K. RRR-α-tocopheryl succinate inhibits EL4 thymic lymphoma cell growth by inducing apoptosis and DNA synthesis arrest. Nutr Cancer. 1997;27:92–101. doi: 10.1080/01635589709514508. [DOI] [PubMed] [Google Scholar]
- 14.Yu W, Israel K, Liao QY, Aldaz CM, Sanders BG, Kline K. Vitamin E succinate (VES) induces Fas sensitivity in human breast cancer cells: role for Mr 43,000 Fas in VES-triggered apoptosis. Cancer Res. 1999;59:953–61. [PubMed] [Google Scholar]
- 15.Israel K, Yu W, Sanders BG, Kline K. Vitamin E succinate induces apoptosis in human prostate cancer cells: role for Fas in vitamin E succinate-triggered apoptosis. Nutr Cancer. 2000;36:90–100. doi: 10.1207/S15327914NC3601_13. [DOI] [PubMed] [Google Scholar]
- 16.Tan M, Wang Y, Guan K, Sun Y. PTGF-β, a type β transforming growth factor (TGF-β) superfamily member, is a p53 target gene that inhibits tumor cell growth via TGF-β signaling pathway. Proc Natl Acad Sci U S A. 2000;97:109–14. doi: 10.1073/pnas.97.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li PX, Wong J, Ayed A, et al. Placental transforming growth factor-β is a downstream mediator of the growth arrest and apoptotic response of tumor cells to DNA damage and p53 overexpression. J Biol Chem. 2000;275:20127–35. doi: 10.1074/jbc.M909580199. [DOI] [PubMed] [Google Scholar]
- 18.Liu T, Bauskin AR, Zaunders J, et al. Macrophage inhibitory cytokine 1 reduces cell adhesion and induces apoptosis in prostate cancer cells. Cancer Res. 2003;63:5034–40. [PubMed] [Google Scholar]
- 19.Baek SJ, Kim KS, Nixon JB, Wilson LC, Eling TE. Cyclooxygenase inhibitors regulate the expression of a TGF-β superfamily member that has proapoptotic and antitumorigenic activities. Mol Pharmacol. 2001;59:901–8. [PubMed] [Google Scholar]
- 20.Shim M, Eling TE. Protein kinase C-dependent regulation of NAG-1/placental bone morphogenic protein/MIC-1 expression in LNCaP prostate carcinoma cells. J Biol Chem. 2005;280:18636–42. doi: 10.1074/jbc.M414613200. [DOI] [PubMed] [Google Scholar]
- 21.Baek SJ, Okazaki R, Lee SH, et al. Nonsteroidal anti-inflammatory drug-activated gene-1 over expression in transgenic mice suppresses intestinal neoplasia. Gastroenterology. 2006;131:1553–60. doi: 10.1053/j.gastro.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 22.Olson JM, Hallahan AR. p38 MAP kinase: a convergence point in cancer therapy. Trends Mol Med. 2004;10:125–9. doi: 10.1016/j.molmed.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 23.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 24.Enslen H, Raingeaud J, Davis RJ. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J Biol Chem. 1998;273:1741–8. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- 25.She QB, Bode AM, Ma WY, Chen NY, Dong Z. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res. 2001;61:1604–10. [PubMed] [Google Scholar]
- 26.Saeki K, Kobayashi N, Inazawa Y, et al. Oxidation-triggered c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein (MAP) kinase pathways for apoptosis in human leukaemic cells stimulated by epigallocatechin-3-gallate (EGCG): a distinct pathway from those of chemically induced and receptor-mediated apoptosis. Biochem J. 2002;368:705–20. doi: 10.1042/BJ20020101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deacon K, Mistry P, Chernoff J, Blank JL, Patel R. p38 Mitogen-activated protein kinase mediates cell death and p21-activated kinase mediates cell survival during chemotherapeutic drug-induced mitotic arrest. Mol Biol Cell. 2003;14:2071–87. doi: 10.1091/mbc.E02-10-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Losa JH, Parada Cobo C, Viniegra JG, Sanchez-Arevalo Lobo VJ, Ramon y, ajal S, Sanchez-Prieto R. Role of the p38 MAPK pathway in cisplatin-based therapy. Oncogene. 2003;22:3998–4006. doi: 10.1038/sj.onc.1206608. [DOI] [PubMed] [Google Scholar]
- 29.Wang SW, Pawlowski J, Wathen ST, Kinney SD, Lichenstein HS, Manthey CL. Cytokine mRNA decay is accelerated by an inhibitor of p38-mitogen-activated protein kinase. Inflamm Res. 1999;48:533–8. doi: 10.1007/s000110050499. [DOI] [PubMed] [Google Scholar]
- 30.Dean JL, Brook M, Clark AR, Saklatvala J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem. 1999;274:264–9. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 31.Baek SJ, Wilson LC, Eling TE. Resveratrol enhances the expression of non-steroidal anti-inflammatory drug-activated gene (NAG-1) by increasing the expression of p53. Carcinogenesis. 2002;23:425–34. doi: 10.1093/carcin/23.3.425. [DOI] [PubMed] [Google Scholar]
- 32.Shiau CW, Huang JW, Wang DS, et al. α-Tocopheryl succinate induces apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 function. J Biol Chem. 2006;281:11819–25. doi: 10.1074/jbc.M511015200. [DOI] [PubMed] [Google Scholar]
- 33.Alleva R, Benassi MS, Tomasetti M, et al. α-Tocopheryl succinate induces cytostasis and apoptosis in osteosarcoma cells: the role of E2F1. Biochem Biophys Res Commun. 2005;331:1515–21. doi: 10.1016/j.bbrc.2005.04.080. [DOI] [PubMed] [Google Scholar]
- 34.Malafa MP, Fokum FD, Andoh J, et al. Vitamin E succinate suppresses prostate tumor growth by inducing apoptosis. Int J Cancer. 2006;118:2441–7. doi: 10.1002/ijc.21689. [DOI] [PubMed] [Google Scholar]
- 35.Zu K, Hawthorn L, Ip C. Up-regulation of c-Jun-NH2-kinase pathway contributes to the induction of mitochondria-mediated apoptosis by α-tocopheryl succinate in human prostate cancer cells. Mol Cancer Ther. 2005;4:43–50. [PubMed] [Google Scholar]
- 36.Lambert JR, Kelly JA, Shim M, et al. Prostate derived factor in human prostate cancer cells: gene induction by vitamin D via a p53-dependent mechanism and inhibition of prostate cancer cell growth. J Cell Physiol. 2006;208:566–74. doi: 10.1002/jcp.20692. [DOI] [PubMed] [Google Scholar]
- 37.Yamaguchi K, Lee SH, Eling TE, Baek SJ. Identification of nonsteroidal anti-inflammatory drug-activated gene (NAG-1) as a novel downstream target of phosphatidylinositol 3-kinase/AKT/GSK-3β pathway. J Biol Chem. 2004;279:49617–23. doi: 10.1074/jbc.M408796200. [DOI] [PubMed] [Google Scholar]
- 38.Subramaniam S, Strelau J, Unsicker K. Growth differentiation factor-15 prevents low potassium-induced cell death of cerebellar granule neurons by differential regulation of Akt and ERK pathways. J Biol Chem. 2003;278:8904–12. doi: 10.1074/jbc.M210037200. [DOI] [PubMed] [Google Scholar]
- 39.Wu K, Zhao Y, Li GC, Yu WP. c-Jun N-terminal kinase is required for vitamin E succinate-induced apoptosis in human gastric cancer cells. World J Gastroenterol. 2004;10:1110–4. doi: 10.3748/wjg.v10.i8.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Y, Wu K, Yu Y, Li G. Roles of ERK1/2 MAPK in vitamin E succinate-induced apoptosis in human gastric cancer SGC-7901 cells. Wei Sheng Yan Jiu. 2003;32:573–5. [PubMed] [Google Scholar]
- 41.Landry J, Lambert H, Zhou M, et al. Human HSP27 is phosphorylated at serines 78 and 82 by heat shock and mitogen-activated kinases that recognize the same amino acid motif as S6 kinase II. J Biol Chem. 1992;267:794–803. [PubMed] [Google Scholar]
- 42.Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–77. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 43.Bulavin DV, Saito S, Hollander MC, et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999;18:6845–54. doi: 10.1093/emboj/18.23.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. Characterization of the structure and function of a novel MAP kinase kinase (MKK6) J Biol Chem. 1996;271:2886–91. doi: 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- 45.Derijard B, Raingeaud J, Barrett T, et al. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–5. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 46.Yamaguchi K, Lee SH, Eling TE, Baek SJ. A novel peroxisome proliferator-activated receptor γ ligand, MCC-555, induces apoptosis via posttranscriptional regulation of NAG-1 in colorectal cancer cells. Mol Cancer Ther. 2006;5:1352–61. doi: 10.1158/1535-7163.MCT-05-0528. [DOI] [PubMed] [Google Scholar]
- 47.Yu W, Liao QY, Hantash FM, Sanders BG, Kline K. Activation of extracellular signal-regulated kinase and c-Jun-NH(2)-terminal kinase but not p38 mitogen-activated protein kinases is required for RRR-α-tocopheryl succinate-induced apoptosis of human breast cancer cells. Cancer Res. 2001;61:6569–76. [PubMed] [Google Scholar]
- 48.Martinez JM, Sali T, Okazaki R, et al. Drug-induced expression of nonsteroidal anti-inflammatory drug-activated gene/macrophage inhibitory cytokine-1/prostate-derived factor, a putative tumor suppressor, inhibits tumor growth. J Pharmacol Exp Ther. 2006;318:899–906. doi: 10.1124/jpet.105.100081. [DOI] [PubMed] [Google Scholar]
- 49.Robinow S, Campos AR, Yao KM, White K. The elav gene product of Drosophila, required in neurons, has three RNP consensus motifs. Science. 1988;242:1570–2. doi: 10.1126/science.3144044. [DOI] [PubMed] [Google Scholar]
- 50.Tchen CR, Brook M, Saklatvala J, Clark AR. The stability of tristetraprolin mRNA is regulated by mitogen-activated protein kinase p38 and by tristetraprolin itself. J Biol Chem. 2004;279:32393–400. doi: 10.1074/jbc.M402059200. [DOI] [PubMed] [Google Scholar]