

Abstract
The chaperonin GroEL is a large complex composed of 14 identical 57-kDa subunits that requires ATP and GroES for some of its activities. We find that a monomeric polypeptide corresponding to residues 191 to 345 has the activity of the tetradecamer both in facilitating the refolding of rhodanese and cyclophilin A in the absence of ATP and in catalyzing the unfolding of native barnase. Its crystal structure, solved at 2.5 Å resolution, shows a well-ordered domain with the same fold as in intact GroEL. We have thus isolated the active site of the complex allosteric molecular chaperone, which functions as a “minichaperone.” This has mechanistic implications: the presence of a central cavity in the GroEL complex is not essential for those representative activities in vitro, and neither are the allosteric properties. The function of the allosteric behavior on the binding of GroES and ATP must be to regulate the affinity of the protein for its various substrates in vivo, where the cavity may also be required for special functions.
Keywords: protein folding, hsp60, cpn60, barnase, minichaperone
The molecular chaperone GroEL facilitates the folding of some proteins (1). GroEL is composed of 14 identical 57-kDa subunits that form two rings with 7-fold symmetry, each with a central cavity. Each GroEL subunit in the 2.8 Å crystal structure has three domains (2, 3). Mutations in the apical domain lead to a decrease in polypeptide binding (4), suggesting that this domain is involved in the binding of polypeptides. Electron microscopy suggests that denatured protein binds to the inner side of the apical end of the GroEL-cylinder (5). The equatorial domain has been shown from the 2.4 Å crystal structure of ATP[γS]-ligated GroEL (6) and mutagenesis studies (4) to contain the nucleotide binding sites. Binding and hydrolysis of ATP is cooperative (7, 8) and lowers the affinity for polypeptides (9). Most of the intermolecular contacts between the subunits of GroEL are between the equatorial domains. The intermediate domain connects the other two domains, transmitting allosteric effects (2, 3).
Two features of the complex are usually assumed to be the keys to its activity: (i) the central cavity, which can act as a cage for folding, and (ii) the allosteric switching of GroEL from a state that has a high affinity for denatured peptides to one that has a low affinity on the binding of ATP and GroES (10, 11). It has been argued that the primary function of GroEL is to provide a cage in which a protein can fold in isolation (1), which has been supported by the finding that small proteins can fold fully while bound to GroEL (12). It has also been shown that GroEL can catalyze the unfolding of one of these proteins, barnase (10), which supports the idea of an annealing activity that causes misfolded proteins to unfold and then have further attempts at productive folding (13, 14) It has been suggested that the role of GroEL in vivo is to provide a combination of cage and annealing activities, with the allosteric switching controlling the balance of activities and the entry to the chaperoning cycle (15).
Does GroEL have to be a complex multimer to be functional? Is there a small part of GroEL that is responsible for its activity, while the rest of the protein is present for regulation and other biological functions? Yoshida and coworkers (16, 17) have reported that a 34-kDa proteolytic fragment of GroEL (GroEL150-456) and a 50-kDa fragment of cpn60 from Thermus thermophilus facilitate refolding of denatured rhodanese in the absence of GroES and ATP. In contrast, monomeric forms of GroEL that have been induced by site-directed mutagenesis do not affect the refolding of rhodanese (18). Further, monomers of GroEL can be induced by urea, treatment with ion-exchange resin, or pressure, but they are able to facilitate the refolding of rhodanese only after reassociation to form the central cavity (19–21). Although the proteolytic fragment GroEL150-456 elutes as a monomer during gel filtration (16), it contains parts of the intermediate and equatorial domains, which determine the intersubunit contacts of GroEL (2), and could thus be capable of transient formation of oligomers or even the central cavity.
To investigate these and other questions, we have functionally expressed the apical domain of GroEL and various of its fragments in Escherichia coli, allowing us to study the chaperone activity, folding, and crystal structure of the polypeptide binding domain in isolation.
MATERIALS AND METHODS
Production of Fragments.
The apical domain of GroEL (GroEL191-376) (2) and the various C-terminally truncated fragments of the apical domain were cloned by polymerase chain reaction (PCR) into the polylinker site of a pRSET A vector (Invitrogen), coding for an N-terminal histidine-tail, which contained an engineered thrombin cleavage site (M. Proctor and M. Bycroft, unpublished work) as in Fig. 1. The histidine-tail (ht) was composed of 36 amino acids (Invitrogen) or 17 amino acids (sht; M. Proctor and M. Bycroft, unpublished work). For the PCR reaction, we used the plasmid pOF39 (24) as a template and two primers flanking the respective GroEL fragment with BamHI and EcoRI restriction enzyme sites, which enabled us to clone the PCR fragment into the polylinker of pRSET A. PCR was performed with Pfu (Stratagene) to reduce the risk of undesired random mutations. The reaction was performed in 25-μl vol for 25 cycles with 400 nM primer and 200 μM each dNTP. The annealing temperature was 65°C. The following primers were used for the PCR: 5′-flanking, 5′-CGG ATC CGA AGG TAT GCA GTT CGA CCG; 3′-flanking (s)ht-GroEL191-376, 5′-CGA ATT CTT AAA CGC CGC CTG CCA GTT TCG; 3′-flanking (s)ht-GroEL191-345, 5′-CGA ATT CTT AAC GGC CCT GGA TTG CAG CTT C; 3′-flanking ht-GroEL191-337, 5′-CGA ATT CTT AAC CCA CGC CAT CGA TGA TAG TGG TG; 3′-flanking ht-GroEL191-328, 5′-CGA ATT CTT AGT CTT TGT TGA TCA CAA CAC GTT TAG CCT GAC; 3′-flanking ht-GroEL191-322, 5′-CGA ATT CTT AAC GTT TAG CCT GAC CCA GGT CTT CCA; 3′-flanking ht-GroEL191-298, 5′-CGA ATT CTT AAC CGC CAG TCA GGG TTG CGA TAT C. The following protocol was used for expression and purification of the fragments of GroEL in E. coli TG2 cells. Two liters of Luria broth medium plus ampicillin were inoculated 1:100 with an overnight culture of TG2 cells containing the respective plasmid. At OD600 = 0.3, expression was induced with isopropyl β-d-galactopyranoside (IPTG) to 0.2 mM final concentration and M13/T7-phage at a multiplicity of infection of 10 phage per cell. The cells were harvested 8 h after induction, centrifuged, and resuspended in 200 ml buffer A (50 mM Tris·HCl, pH 8.2/300 mM NaCl). After sonication and centrifugation, the soluble protein fraction was added to 20 ml nickel-NTA agarose resin and stirred for 10 min. The resin was washed with 200 ml buffer A, and the histidine-tail containing fusion protein was eluted with 50 ml buffer A containing 200 mM imidazole. The eluted protein was precipitated with 80% ammonium sulfate, redissolved in 4 ml of buffer B (50 mM Tris·HCl pH 8.2/150 mM NaCl), and was loaded on a HiLoad 26/60 Superdex 75 column (Pharmacia) that was equilibrated with buffer B. The fragment GroEL191-345 was produced by thrombin-cleavage of ht-GroEL191-376 before gel filtration. The cleaving reaction was carried out for several days in buffer B after addition of 250 μl of thrombin (Sigma, 1 unit/μl) to the protein solution eluted from the nickel-NTA column. The GroEL fragments were analyzed by quantitative amino acid analysis, N-terminal sequencing, and mass spectrometry. Our clone differs from the sequence in the data base by the mutations Leu-262 → Ala and Met-267 → Ile.
Figure 1.
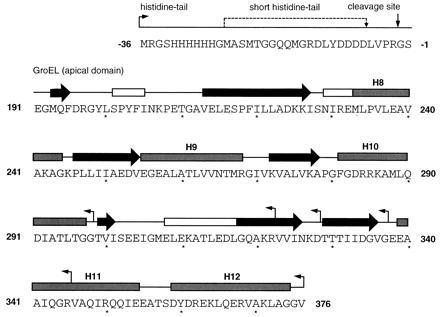
Cloning of the apical domain of GroEL and various of its fragments. The utilized expression vector coded for an N-terminal histidine-tail (ht) composed of 36 amino acids and containing a thrombin cleavage site (vertical arrow). Alternatively, a shorter version of this histidine-tail (sht) containing 17 amino acids was used. The N and C termini of the generated fusion proteins, namely ht-GroEL191-298, ht-GroEL191-322, ht-GroEL191-328, ht-GroEL191-337, ht-GroEL191-345, ht-GroEL191-376, sht-GroEL191-345, and sht-GroEL191-376 are indicated by horizontal arrows. Secondary structure is indicated by boxes and arrows for α-helix (shaded) or 310-helix (open) and β-sheet structure, respectively. Assignment of secondary structure of residues 191 to 336 was from the crystal structure of sht-GroEL191-345 using procheck (22) and the algorithm of Kabsch and Sander (23). Numbering of α-helices and secondary assignment of residues 337 to 376 according to Braig et al. (3).
Crystallization, Data Collection, and Solution of Structure.
Crystals were obtained from hanging drops initially containing sht-GroEL191-345 at 23 mg·ml−1, 11% PEG 4000, 50 mM Tris·HCl (pH 8.5), and 100 mM LiSO4, equilibrated against reservoirs consisting of 22% PEG 4000, 100 mM Tris·HCl (pH 8.5), and 200 mM LiSO4. X-ray data were collected from a capillary-mounted crystal at 4°C using a 30-cm MAR Research (Hamburg) image plate detector at station 9.6 of the Synchrotron Radiation Source at Daresbury, U.K. (λ = 0.87 Å). Unless stated otherwise, all data processing, data reduction, electron density syntheses, and structural analyses were carried out using ccp4 software (Daresbury Laboratory, Warrington, U.K.). Indexing and intensity measurements of diffraction data were carried out with the mosflm program suite (25). The asymmetric unit contains one protein molecule, corresponding to a solvent content of 51%. The structure was solved by conventional molecular replacement methods, using the program amore (26), and a search model consisting of residues 191–345 of the refined structure of GroEL (3). Model rebuilding was carried with the program o (27), and the structure was refined using x-plor (28) using Engh and Huber parameters (29). The current model contains eight water molecules and is complete with the following exceptions, which could not be modeled due to poor or nonexistent electron density: residues 302–307 and residues 337–345 from the C terminus. Electron density for the N-terminal His-tag is also not observed. No residues have disallowed backbone φ,ψ angles.
NMR.
Exchange experiments were carried out at 33°C in 20 mM imidazole buffer as described (10, 11).
Refolding Experiments.
Refolding assay of rhodanese was as described by Horowitz (30). To stop the refolding reaction in the kinetic experiments, rhodanese activity was assayed in the presence of 10 mM trans-1,2-cyclohexanediaminetetraacetate to inhibit GroEL activity or 0.5 mg/ml of casein to saturate the apical domain. Refolding of cyclophilin A was initiated by diluting 8 M urea denatured protein (100 μM) into 100 mM potassium phosphate buffer (pH 7.0) and 10 mM DTT to a final concentration of 1 μM. The final concentration of GroEL and apical domain in refolding buffer was 7 μM and 4 μM or 1 μM, respectively. Refolding temperature was 25°C. After incubation for the times indicated, cyclophilin activity was measured as described (36). Spontaneous refolding of cyclophilin A occurred to a yield of about 30%, and was finished in less than 1 min. Standard error was 5%.
Thermal Denaturation Experiments.
Thermal denaturation was measured with a Jasco (Easton, MD) model J720 spectropolarimeter interfaced with a Neslab Instruments (Portsmouth, NH) model RTE-110 water bath, using a thermostatted cuvette of 1-mm path length. The temperature was increased at a linear rate of 50°C per h. The protein concentration was 40 μM in 10 mM sodium phosphate buffer (pH 7.0). Data were fitted to a denaturation curve to determine Tm, the midpoint temperature of denaturation. Differential scanning calorimetric measurements were performed at a protein concentration of 88 ± 5 μM in 10 mM sodium phosphate buffer (pH 7.0), using a Microcal (Amherst, MA) model MC-2D instrument at a notional scan rate of 60°C per h. Sample preparation and data analysis were performed as described previously (31). Both proteins exhibit at least 50% reversibility in their thermal unfolding as judged from the area of endotherms obtained on rescanning samples. Higher levels of reversibility were obtained at lower concentrations or by stopping scans at temperatures closer to the main unfolding transition. The low temperature transition observed in sht-GroEL191-376 was completely reversible with scans limited to temperatures below 50°C.
RESULTS
Expression of Fragments.
The apical domain is expressed at >20 mg purified protein per liter of culture as are the smaller fragments lacking the C-terminal α-helices, H11, and H12. Further truncation before residue 329 leads to considerable destabilization of the apical domain. The fragment GroEL191-345, which is preceded by Gly-Ser and lacks the two C-terminal helices, was generated by thrombin cleavage of purified ht-GroEL191-376. The circular dichroism (CD) of sht-GroEL191-376, sht-GroEL191-345, and GroEL191-345 indicated native-like secondary and tertiary structure (data not shown). The apical domain and the fragment truncated at position 345 are monomeric at micromolar concentrations, as determined by ultracentrifugation experiments (R.Z., S. E. Harding, and A.R.F., unpublished work). However, the line widths in the NMR spectra of GroEL191-345 at higher concentration are larger than expected for a 17-kDa protein but smaller than for a stable dimer (data not shown), indicating a fast intermolecular interaction between monomers on the NMR time scale (32).
Chaperone Activity of Monomeric Fragments.
The GroEL fragments catalyze, under folding conditions, exchange of amide protons of native barnase (Fig. 2) that are known to exchange only from its fully unfolded state. Thus, like intact GroEL (10), the apical domain binds with high affinity to unfolded barnase. Helices H11 and H12 are not essential for polypeptide binding, and the presence of the N-terminal histidine-tail did not abolish binding activity. The mechanism of exchange at pD 6.6 is EX2, but changes to EX1 at higher pD (33). We can estimate the rate constant for dissociation of barnase from the apical domain as 2 s−1, which is less than 5-fold larger than that from intact GroEL (10, 11).
Figure 2.
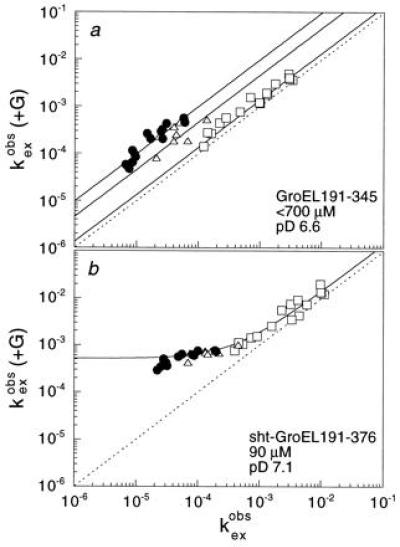
Catalysis of amide proton exchange of barnase
(2.4 mM) by the fragments GroEL191-345 (a) and
sht-GroEL191-376 (b). The results are very similar to
those described in refs. 10 and 11 for intact GroEL. The rate constants
(in units of min−1) for the exchange of individual NH
protons in barnase in the presence of fragment
[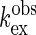 (+G)] are plotted against
those in the absence (
(+G)] are plotted against
those in the absence (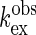 ). Amide
protons that exchange by global, mixed, and local unfolding
mechanisms are displayed by circles, triangles, and squares,
respectively. The plot for 90 μM sht-GroEL191-345 at pD 7.1 (not
shown) is virtually superimposable on that for sht-GroEL191-376
(b). (pD = pH measured in 2H2O.)
It is clearly seen that those protons that require global unfolding for
exchange have significantly increased rates, thus showing that the
fragments bind to the unfolded state of barnase and catalyze its
unfolding. We could only estimate the final concentration of GroEL
fragment in a since GroEL191-345 tended to crystallize
during the exchange experiment at the high initial protein
concentration.
). Amide
protons that exchange by global, mixed, and local unfolding
mechanisms are displayed by circles, triangles, and squares,
respectively. The plot for 90 μM sht-GroEL191-345 at pD 7.1 (not
shown) is virtually superimposable on that for sht-GroEL191-376
(b). (pD = pH measured in 2H2O.)
It is clearly seen that those protons that require global unfolding for
exchange have significantly increased rates, thus showing that the
fragments bind to the unfolded state of barnase and catalyze its
unfolding. We could only estimate the final concentration of GroEL
fragment in a since GroEL191-345 tended to crystallize
during the exchange experiment at the high initial protein
concentration.
Chaperone activity was confirmed directly from studies on rhodanese, which refolds in high yield in vitro only in the presence of GroEL, ATP, and the co-chaperonin GroES (Fig. 3a and refs. 30, 34, and 35). In the absence of GroES, rhodanese dissociates from nucleotide-ligated GroEL in a non-native state and aggregates (35). Sht-GroEL191-376 and sht-GroEL191-345 facilitate refolding of rhodanese in the absence of additional cofactors (Fig. 3a), indicating a transient binding of the GroEL fragments to the aggregation-prone intermediates during refolding of rhodanese, which prevents aggregation. The effect of the GroEL-fragments on refolding yield of rhodanese is lowered in the presence of GroES (Fig. 3a), indicating a competition between rhodanese and the co-chaperonin for binding to the apical domain or its truncated form. Interestingly, the refolding of rhodanese in the presence of sht-GroEL191-345 (Fig. 3c) or sht-GroEL191-376 (Fig. 3d) has a similar rate constant to that in the presence of GroEL, GroES, and ATP (Fig. 3b), the half time of refolding being about 15 min. Thus, the affinity of rhodanese for the apical domain seems to be similar to that of GroEL in the presence of GroES and ATP. The rate constant of refolding does not depend on the concentration of apical domain (Fig. 3 c and d), indicating that the rate-limiting step for refolding of rhodanese occurs in the complex. The refolding yield of rhodanese is generally lower in the presence of apical domain than in the presence of GroEL, GroES, and ATP (3 b and c), and the truncated domain is more active than the full length (3 b–d). The yield on refolding is maximal at molar ratios of apical domain to rhodanese of >1 because the binding is not sufficiently strong, so that there is not complete association, and we estimate a dissociation equilibrium constant of >10−7 M. We also tested the chaperone activity of sht-GroEL191-345 and sht-GroEL191-376 for a second protein. Cyclophilin A refolds only at low yield in the absence of chaperone (Fig. 3e), but refolding is facilitated in the presence of GroEL owing to transient complex formation (Fig. 3e) (36). There are similar rate constants for refolding of cyclophilin A in the presence of intact GroEL and in the presence of GroEL fragments, which within a factor of four do not depend on chaperone concentration (Fig. 3e). Maximal refolding yield was obtained at stoichiometric concentrations of cyclophilin A and apical domain, indicating the formation of a 1:1 complex between chaperone fragment and substrate protein during refolding.
Figure 3.
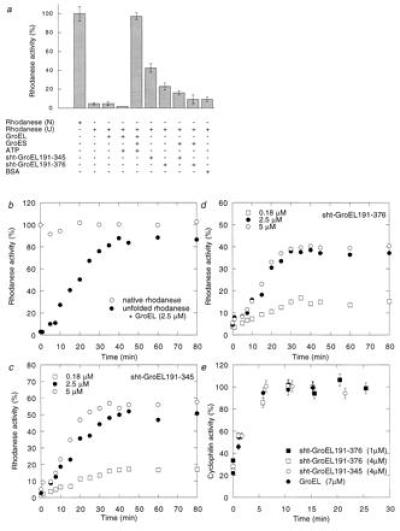
Refolding of rhodanese and cyclophilin A in the presence of sht-GroEL191-345 and sht-GroEL191-376. (a) Relative enzymatic activity of rhodanese (0.1 μM) after refolding in the presence (+) or absence (−) of GroEL (2.5 μM monomer), GroES (2.5 μM monomer), ATP (2 mM), sht-GroEL191-345 (2.5 μM), sht-GroEL191-376 (2.5 μM), or bovine serum albumin (45 μg/ml), from 8 M urea (U). One-hundred percent activity was obtained with native rhodanese (N). (b) Refolding kinetics of rhodanese in presence of GroEL, GroES, and ATP. The final concentrations are the same as in a. One-hundred percent activity was obtained with native rhodanese. (c and d) Refolding kinetics of rhodanese in the presence of 0.18 μM, 2.5 μM, or 5 μM sht-GroEL191-345 and sht-GroEL191-376, respectively. (e) Refolding of 1 μM cyclophilin A in the presence of 7 μM GroEL (monomer), 4 μM sht-GroEL191-345, 4 μM sht-GroEL191-376, or 1 μM sht-GroEL191-376. One-hundred percent activity was obtained with native cyclophilin A. Standard error bars are shown. The 30% spontaneous refolding of cyclophilin was complete in the dead time of the experiment.
Flexibility of C-Terminal Helices.
The apical domain and the fragment truncated at position 345 are reversibly denatured by temperature or urea, and the denaturation is not influenced by the N-terminal histidine-tail. There are two cooperative folding transitions, at 34°C and 67°C, during the thermal denaturation of sht-GroEL191-376, measured by far UV-CD (Fig. 4a), indicating the existence of two independent folding units within the apical domain. There is only one transition for unfolding of sht-GroEL191-345, at 67°C. At 45°C, the CD spectrum of the apical domain is identical to that of the truncated domain. The C-terminal α-helices must therefore melt at the lower temperature and separately from the “domain core.” The second cooperative transition associated with the extra 31 amino acids in sht-GroEL191-376 was confirmed by differential scanning calorimetry (Fig. 4b). The calorimetric data are also essentially consistent with the unfolding of the apical domain as a monomer. At physiological temperature, about 50% of helices H11 and H12 are in an unfolded conformation, and thus flexible.
Figure 4.
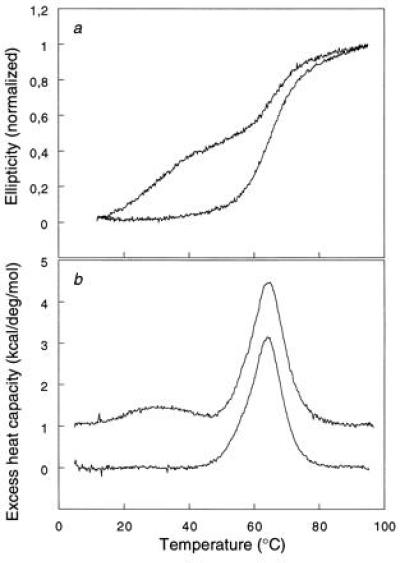
Thermal denaturation of sht-GroEL191-376 (upper trace) and of sht-GroEL191-345 (lower trace). (a) Monitored by far UV-CD at 222 nm. (b) Monitored by differential scanning calorimetry.
Three-Dimensional Structure of the Domain Core.
The crystallographic data are in Table 1 and the coordinates are deposited in the Brookhaven data base with the reference 1JON. Overall, sht-GroEL191-345 has the same fold as the corresponding region of the intact GroEL protein (Fig. 5 Upper Left): two orthogonal β-sheets forming a β-sandwich, flanked by three α-helices. The structure is more ordered and better resolved than is the apical domain of the intact protein (Fig. 5 Upper Right): the average B factor is 42 Å2, compared with 97 Å2 for residues 191–336 of the GroEL structure. Unfortunately, such unusually high disorder in the GroEL structure complicates the interpretation of a structural comparison, in the same way as it can be misleading to use an average of NMR structures for comparison with a crystal structure, and so we have not done so here. In essence, the structure can be described as a well-ordered β-sandwich scaffold, flanked by relatively flexible helical and loop regions. In particular, the B factors of most of the β-strand, α-helix, and loop structure are about 20, 40, and 60 Å2, respectively (Fig. 5 Upper Right). The quality of the electron density map is shown in Fig. 5 Lower. There are two regions of considerable disorder. First, electron density is not found for the C-terminal residues 337–345, which correspond to the first half of α-helix H11. This is in agreement with the results from folding experiments, described above. Second, electron density for residues 302–307 is very poor and fragmented. This region is part of the most disordered segment in the structure of intact GroEL (3).
Table 1.
Summary of crystallographic data
| Data collection statistics | |
| Unit cell dimensions, Å | a = b = 91.67, c = 38.33 |
| Space group | P3121 |
| Resolution, Å | 22.0–2.5 |
| Measured reflections | 21,762 |
| Unique reflections | 6,564 |
| Completeness of data (%)* | 99.4 (96.7) |
| Rmerge (%)*† | 9.9 (45.1) |
| 〈F/σF〉* | 19.8 (4.6) |
| Multiplicity* | 3.3 (3.0) |
| Refinement statistics | |
| Resolution, Å | 8.0–2.5 |
| R factor/free R factor (%), F > 0‡ | 21.4/29.1 |
| rmsd bond length, Å | 0.006 |
| rmsd bond angle, deg | 1.42 |
rmsd, Root-mean-square deviation.
Values given in parenthesis are for the highest resolution shell.
Agreement between intensities of repeated measurements of the same reflections and can be defined as: Σ (Ih,i − 〈Ih〉)/Σ Ih,i, where Ih,i are individual values and 〈Ih〉 is the mean value of the intensity of reflection h.
The free R factor was calculated with the 10% data omitted from the refinement [test set, prepared using dataman (40), using RFree SPhere option].
Figure 5.
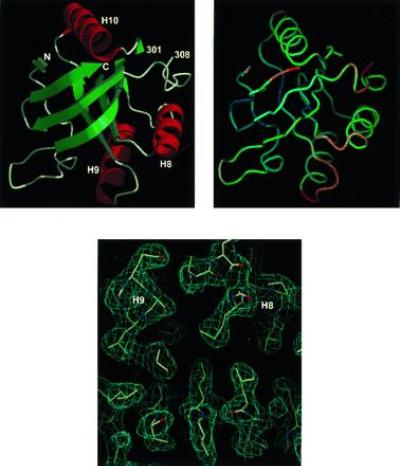
The three-dimensional structure of sht-GroEL191-345. Secondary structure representation is drawn with molscript (37) and raster3d (38). Helices are labeled as in Braig et al. (3). N and C refer to the N terminus (residue 191) and C terminus (residue 336) of the model, respectively. The backbone representation (Upper Right) is in the same orientation as Upper Left, color-coded according to B factor of main-chain atoms: blue (20 Å2) to red (60 Å2), drawn with program o (27). (Lower) Representative region of electron density, calculated using refined coordinates, viewed along the helices H8 and H9.
DISCUSSION
The apical domain of GroEL can be expressed as a stable protein and with high yield in E. coli, allowing its activity, folding, and structure to be studied separately from those of the equatorial and intermediate domains. The isolated, apical domain is functional in polypeptide binding (Fig. 2), and it facilitates protein folding, especially when truncated to remove its C-terminal α-helices, H11 and H12 (Fig. 3). Point mutagenesis studies support the passive role of the C-terminal part of the apical domain in polypeptide binding (4). The isolated apical domain and its active fragments thus function as “minichaperones.” The apical domain slows down refolding of cyclophilin by more than 10-fold (Fig. 3), and the folding of rhodanese is significantly more retarded by the minichaperones than by the longer proteolytic fragments of Yoshida et al. (16, 17). The increase in the yield of refolding of rhodanese and cyclophilin A in the presence of apical domain (Fig. 3), which is too small to reassociate and to form a cavity, clearly demonstrates the presence of an intrinsic chaperone activity in GroEL, which is independent of a central cavity. This is in agreement with the NMR experiments on barnase in the presence of apical domain (Fig. 2). The presence of μM concentrations of GroEL considerably broadens the resonance lines of barnase owing to fast association and dissociation of native barnase from the slowly tumbling GroEL (10, 11). If the apical domain and barnase formed a complex of large molecular size, e.g., containing 7 or 14 molecules of apical domain, one would expect a considerable degree of line broadening in the NMR spectra of barnase in the presence of apical domain. This is not observed at 8-fold higher concentration of apical domain than of GroEL (expressed in monomers). The complex between apical domain and barnase seems, therefore, to be of low molecular weight and probably has a 1:1 stoichiometry, which would be consistent with the binding stoichiometry of the apical domain and cyclophilin A (Fig. 3e).
The intrinsic chaperone activity of monomeric apical domain raises two interrelated questions: how does the fragment facilitate refolding of rhodanese when, at physiological conditions, GroES and ATP are required in the presence of intact GroEL; and what is the role of the central cavity? The role of GroES appears to be to weaken the affinity of GroEL for substrates, and to prevent premature dissociation of aggregation prone states: there has to be a balance between tight binding for the annealing activity and weaker binding to allow folding. The weaker binding of rhodanese to the fragment must be adequate for chaperoning activity and weak enough to allow folding (15). This is in agreement with the finding of similar rate constants for refolding of rhodanese in the presence of apical domain and in the presence of GroEL, GroES, and ATP (Fig. 3 b–d). The complex structure of GroEL and the modulation of its substrate affinity by GroES and nucleotides must be to allow the efficient folding of a wide range of proteins, with a wide variation of affinities for GroEL. Although the central cavity is clearly not essential for the refolding of cyclophilin A and rhodanese, it could still be important for refolding in vivo.
Helices H11 and H12 are much less stable than the domain core of the apical domain, and form a separate folding unit (Fig. 4). At physiological temperature, about 50% of the secondary structure of the low-melting helices is unfolded and thus becomes flexible. But, in vivo, the C terminus is not degraded, implying that the primary structure is not accessible to degradation by E. coli proteases. The low stability of the C-terminal helices may allow movement of the domain cores within the GroEL-ring. This flexible arrangement of the apical domain may be involved in the cooperative binding of unfolded polypeptide to the seven subunits of GroEL in each ring. GroEL is known to bind to at least half of E. coli proteins in their denatured states (39). It is thus likely that flexibility is required to bind a large variety of proteins with different primary, secondary, and tertiary structures. The low stability of the C terminus may also contribute to the large conformational change in GroEL on binding of GroES, which has been suggested to occur by rigid body movement through the β-sheet hinge region of the intermediate domain (2).
The production and crystallization of monomeric fragments of the apical domain has allowed us to determine the three-dimensional structure of the polypeptide binding part of GroEL with B factors much lower than those for the equivalent regions in GroEL (Fig. 5) (2, 3, 6), perhaps because of favorable packing interactions within the crystal, or the absence of static disorder that is present in crystals of intact GroEL, or a combination of both. The crystals of the minichaperone may provide a suitable paradigm for detailed studies of peptide–GroEL interactions.
Acknowledgments
A postdoctoral Liebig Fellowship to R.Z. is gratefully acknowledged.
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, Chemistry Department, Brookhaven National Laboratory, Upton, NY 11973 (reference 1JON).
References
- 1.Ellis R J, Hartl F U. FASEB J. 1996;10:20–26. doi: 10.1096/fasebj.10.1.8566542. [DOI] [PubMed] [Google Scholar]
- 2.Braig K, Otwinowski Z, Hegde R, Boisvert D C, Joachimiak A, Horwich A L, Sigler P B. Nature (London) 1994;371:578–586. doi: 10.1038/371578a0. [DOI] [PubMed] [Google Scholar]
- 3.Braig K, Adams P D, Brünger A T. Nat Struct Biol. 1995;2:1083–1094. doi: 10.1038/nsb1295-1083. [DOI] [PubMed] [Google Scholar]
- 4.Fenton W A, Kashi Y, Furtak K, Horwich A L. Nature (London) 1994;371:614–619. doi: 10.1038/371614a0. [DOI] [PubMed] [Google Scholar]
- 5.Chen S, Roseman A M, Hunter A S, Wood S P, Burston S G, Ranson N A, Clarke A R, Saibil H R. Nature (London) 1994;371:261–264. doi: 10.1038/371261a0. [DOI] [PubMed] [Google Scholar]
- 6.Boisvert D C, Wang J, Otwinowski Z, Horwich A L, Sigler P B. Nat Struct Biol. 1996;3:170–177. doi: 10.1038/nsb0296-170. [DOI] [PubMed] [Google Scholar]
- 7.Bochkareva E S, Lissin N M, Flynn G C, Rothman J E, Girshovich A S. J Biol Chem. 1992;267:6796–6800. [PubMed] [Google Scholar]
- 8.Gray T E, Fersht A R. FEBS Lett. 1991;292:254–258. doi: 10.1016/0014-5793(91)80878-7. [DOI] [PubMed] [Google Scholar]
- 9.Jackson G S, Staniforth R A, Halsall D J, Atkinson T, Holbrook J J, Clarke A R, Burston S G. Biochemistry. 1993;32:2554–2563. doi: 10.1021/bi00061a013. [DOI] [PubMed] [Google Scholar]
- 10.Zahn R, Perrett S, Stenberg G, Fersht A R. Science. 1996;271:642–645. doi: 10.1126/science.271.5249.642. [DOI] [PubMed] [Google Scholar]
- 11.Zahn R, Perrett S, Fersht A R. J Mol Biol. 1996;261:43–61. doi: 10.1006/jmbi.1996.0440. [DOI] [PubMed] [Google Scholar]
- 12.Gray T E, Fersht A R. J Mol Biol. 1993;232:1197–1207. doi: 10.1006/jmbi.1993.1471. [DOI] [PubMed] [Google Scholar]
- 13.Weissman J S, Kashi Y, Fenton W A, Horwich A L. Cell. 1994;78:693–702. doi: 10.1016/0092-8674(94)90533-9. [DOI] [PubMed] [Google Scholar]
- 14.Todd M J, Viitanen P V, Lorimer G H. Science. 1994;265:659–666. doi: 10.1126/science.7913555. [DOI] [PubMed] [Google Scholar]
- 15.Corrales F J, Fersht A R. Proc Natl Acad Sci USA. 1996;93:4509–4512. doi: 10.1073/pnas.93.9.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makino Y, Taguchi H, Yoshida M. FEBS Lett. 1993;336:363–367. doi: 10.1016/0014-5793(93)80838-l. [DOI] [PubMed] [Google Scholar]
- 17.Taguchi H, Makino Y, Yoshida M. J Biol Chem. 1994;269:8529–8534. [PubMed] [Google Scholar]
- 18.White Z W, Fisher K E, Eisenstein E. J Biol Chem. 1995;270:20404–20409. doi: 10.1074/jbc.270.35.20404. [DOI] [PubMed] [Google Scholar]
- 19.Mendoza J A, Demeler B, Horowitz P M. J Biol Chem. 1994;269:2447–2451. [PubMed] [Google Scholar]
- 20.Ybarra J, Horowitz P M. J Biol Chem. 1995;270:22962–22967. doi: 10.1074/jbc.270.39.22962. [DOI] [PubMed] [Google Scholar]
- 21.Gorovits B, Raman C S, Horowitz P M. J Biol Chem. 1995;270:2061–2066. doi: 10.1074/jbc.270.5.2061. [DOI] [PubMed] [Google Scholar]
- 22.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 23.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 24.Fayet O, Ziegelhoffer T, Georgopoulos C. J Bacteriol. 1989;171:1379–1385. doi: 10.1128/jb.171.3.1379-1385.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leslie A G W. Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography, No. 26. Warrington, U.K.: Daresbury Lab.; 1992. [Google Scholar]
- 26.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 27.Jones T A, Zou J-Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 28.Brünger, A. T. (1992) x-plor, A System for Crystallography and NMR (Yale Univ. Press, New Haven, CT), Version 3.1.
- 29.Engh R A, Huber R. Acta Crystallogr A. 1991;47:392–400. [Google Scholar]
- 30.Horowitz P M. In: Protein Stability and Folding. Shirley B A, editor. Clifton, NJ: Humana; 1995. pp. 361–368. [Google Scholar]
- 31.Johnson C M, Fersht A R. Biochemistry. 1995;34:6795–6804. doi: 10.1021/bi00020a026. [DOI] [PubMed] [Google Scholar]
- 32.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 33.Hvidt A, Nielsen S O. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 34.Mendoza J A, Rogers E, Lorimer G H, Horowitz P M. J Biol Chem. 1991;266:13044–13049. [PubMed] [Google Scholar]
- 35.Martin J, Langer T, Boteva R, Schramel A, Horwich A L, Hartl F-U. Nature (London) 1991;352:36–42. doi: 10.1038/352036a0. [DOI] [PubMed] [Google Scholar]
- 36.Zahn R, Spitzfaden C, Ottiger M, Plückthun A, Wüthrich K. Nature (London) 1996;368:261–265. doi: 10.1038/368261a0. [DOI] [PubMed] [Google Scholar]
- 37.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 38.Merrit E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 39.Viitanen P V, Gatenby A A, Lorimer G H. Protein Sci. 1992;1:363–369. doi: 10.1002/pro.5560010308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kleywegt G J, Jones T A. Acta Crystallogr D. 1994;50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]