

Post-translational modifications (PTM) of proteins occur in the secretory pathway of all eukaryotic organisms. For example, phosphorylation, acetylation, glycosylation, methylation and ubiquitinylation represent the major types of protein modifications.[1,2] Of these modifications, glycosylation is chemically the most complex, occurring either at an Asn residue (N-glycosylation)[3] in a conserved motif Asn–Xaa–Ser/Thr (in which Xaa can be any amino acid except Pro) or at a Ser/Thr residue (O-glycosylation) and playing a key role in the biosynthesis, folding, secretion and regulation of proteins.[4,5] Introduction of a β-d-glucosyl-N-acetyl group (β-GlcNAc) on a serine/threonine residue by O-N-acetyl-β-d-glucosaminyltransferases (OGT) in nuclear and cytoplasmic proteins compete with phosphorylation of the same site[6] and is associated with protein functional regulation or diseases such as cancer, Alzheimer's disease or diabetes.[7,8] The O-mannose-linked serine or threonine is common in yeast; fucose is essential in ABO blood-type antigen determination[9] and in Notch receptor signalling.[10] O-β-xylosyltransferases initiate proteoglycan biosynthesis.[11] Understanding the pattern and biological implications of post-translational glycosylation has been a major challenge, and in recent years the development of a platform for the general, fast and reliable identification of enzyme specificity has attracted considerable attention from researchers around the world.[12] Nagahori and Nishimura have developed a way of monitoring glycosyltransferase activity in solution on gold nanoparticles by using mass spectrometry.[13] Freire et al. have used surface-enhanced laser desorption/ionisation time-of-flight mass spectrometry (MALDI-ToF MS) for detecting enzymatic conjugation reactions in solution.[14] Whilst these studies have demonstrated the use of mass spectrometry as an excellent analytical tool, such enzyme assays in solution require additional purification/liquid handling steps and have a limited scope for further miniaturisation. The demands of high-throughput screening are better met by microarrays, in which the enzyme substrate remains immobilised on the array surface throughout enzymatic reaction and analysis. Here, we report such a (peptide) microarray platform for monitoring glycosyltransferases involved in protein O-glycosylation using the label-free MALDI-ToF MS analysis as a readout.[15,16]
For initial proof-of-principle studies we have chosen a member of the human UDP-N-acetyl-α-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase family (ppGalNAcT). ppGalNAcTs are type II membrane glycosyltransferases located on the lumenal side of the Golgi apparatus and initiate the biosynthesis of mucin-type glycoproteins by introducing an αGalNAc moiety on a serine or threonine residue of proteins.[17] To date more than 20 isoforms of the ppGalNAcT family have been identified, displaying both tissue-specific and spatial and temporal expression during development and in adults.[18] However, their substrate specificities remain largely unknown. The isoform T2[19] (541 amino acids, MW = 61.1 kDa) was chosen for this study. The gene was obtained from bacterial cDNA-clones. The N-terminal transmembrane domain was replaced by a hexahistidine tag and cloned into pPICzα, a Pichia pastoris vector used for expression and secretion.[20] Culture supernatants were assayed against different peptide substrates, and amongst the peptides tested, the previously reported GAGAPGPTPGPAGAGK sequence (1)[21] was found to be the optimal substrate giving a high rate of glycosylation in 2 h in a solution-phase assay (data not shown). The lysine residue on the C terminus of 1 allowed for easy immobilisation on gold surfaces using well established chemistry (Scheme 1).[22]
Scheme 1.

Schematic representation of the immobilisation of peptide 1 and subsequent enzymatic glycosylation with ppGalNAcT2. PBS=phosphate-buffered saline.
To immobilise the peptide substrate, the gold surfaces were first coated with a mixture of alkane thiolates presenting a carboxylic acid and tri(ethylene glycol) groups in a molar ratio of 1:20. After activation as N-hydroxysuccinimidyl (NHS) ester, the carboxylic acid was used to immobilise the peptide through coupling with the lysine residue. The tri(ethylene glycol) spacers are well known to prevent nonspecific protein adsorption.[23] MALDI-ToF MS analysis of the resulting monolayer (Figure 1 A) showed a main peak at m/z 2295, corresponding to the sodium adduct of the mixed disulfide formed by the peptide-terminated alkane thiol and the tri(ethylene glycol) alkane thiol. To a lesser extend, the sodium adduct of the peptide-terminated alkane thiol (m/z 1876) and of the peptide-terminated/carboxylic acid mixed disulfide (m/z 2485) were also detected. Such dimeric species are commonly observed on gold surfaces.[13] A mixture containing a crude extract of ppGalNAcT2, UDP-GalNAc and MnCl2 was then applied to the surface and left overnight at 37°C in a wet chamber to prevent evaporation. The resulting monolayer was then analysed by MALDI-ToF MS (Figure 1 B). The mass spectrum indicated a complete shift of the main peak by 203 mass units, corresponding to the addition of a GalNAc moiety (m/z 2498, sodium adduct of the GalNAc-peptide/tri(ethylene glycol) mixed disulfide). A secondary signal at m/z 2080 (GalNAc-peptide alkane thiol) was also seen. No signal was detected at m/z 2295, thus suggesting complete glycosylation of the peptide substrate. To demonstrate the applicability of this platform in multiple screening, an array was generated by spotting solutions of different peptides onto the same self-assembled monolayer (SAM)-coated slide. The use of PBS buffer in combination with 6 m guanidinium hydrochloride allowed discrete spotting of as little as 0.02 μL of each solution by preventing evaporation during the overnight coupling step and keeping the drops well separated on the surface.
Figure 1.

A) MALDI-ToF MS spectrum of immobilised peptide 1 on a mixed monolayer. B) MALDI-ToF MS spectrum of immobilised peptide 1 after enzymatic glycosylation with ppGalNAcT2. C) Structure and expected masses of the alkanethiols and mixed disulfides detected.
For this study, ten peptides (1–10) were synthesised and tested for ppGalNAcT2 activity (Table 1). Peptide 2 has the same sequence as the model peptide 1 except that the threonine was replaced by a serine and was chosen because serine glycosylation is reported to be slower than that of threonine residues. Peptides 3 and 4 are truncated sequences derived from 1. Peptides 5–7 were chosen to contain potential natural glycosylation sites, since they are fragments of a mucin (Muc1) tandem-repeat sequence PGSTAPPAHGVTSAPDTRPA, to which the flanking amino acids GA- and -AGK at the N and C termini, respectively, were added. 8 is a known substrate for OGT (O-GlcNAc transferase) derived from the RNA polymerase II CTD[24] and 9 (so-called EA2 peptide) was chosen as presenting several Ser and Thr potential glycosylation sites. 10, a fragment from Drosophila syndecan, has previously been described as a substrate for O-xylosyltransferase.[25]
Table 1.
Peptides synthesised and tested for ppGalNAcT2 activity.
| Peptide | Sequence | m/z [a] | +ppGalNAcT2 UDPGalNAc[a] |
|---|---|---|---|
| 1 | GAGAPGPTPGPAGAGK | 2295 | 2498 |
| 2 | GAGAPGPSPGPAGAGK | 2281 | 2484 |
| 3 | GAPGPTPGPAGK | 2039 | 2241 |
| 4 | AcPTPGPAGK | 1799 | 2018** |
| 5 | GAPGSTAPPAGK | 2043 | 2246 |
| 6 | GAHGVTSAPAGK | 2085 | 2085, 2288 |
| 7 | GAAPDTRPAAGK | 2144 | 2144 |
| 8 | YSPTSPSKR | 2033* | 2236* |
| 9 | PTTDSTTPAPTTK | 2353 | 2756, 2959, 3162 |
| 10 | DDDSIEGSGGR | 2246* | 2246* |
Masses indicated (m/z) correspond to the sodium adduct of the mixed disulfide formed by the (glyco)peptide-terminated and the tri(ethylene glycol)-terminated alkanethiols except for
proton adduct
potassium adduct of the species was the main signal detected.
All peptides were printed in duplicate in array format and could be reproducibly identified by MALDI-ToF MS analysis (Table 1). After treatment with the enzyme ppGalNAcT2, MALDI-ToF MS analysis showed a full conversion of 1–4 to their corresponding glycopeptides. Analysis of the glycosylation of the Muc1 fragments 5–7 (Figure 2) revealed an order of substrate specificity: peptide 5 appeared to be the best substrate among the three, since it was fully glycosylated after overnight reaction (m/z 2246). Next, 6 appeared to be only partially glycosylated, with both the starting peptide and the newly-formed glycopeptide detected (m/z 2085 and 2288, respectively) on the array. Finally, peptide 7 was not glycosylated at all, and only the starting peptide was observed after overnight incubation (m/z 2144). These results are consistent with other study of Muc1 glycosylation showing that ppGalNAcT2 glycosylates Thr in GSTAP more efficiently than Thr at GVTSA.[26]
Figure 2.

MALDI-ToF MS spectra of immobilised Muc1 fragments before (left) and after (right) enzymatic reaction. A) GAPGSTAPPAGK (5), B) GAHGVTSAPAGK (6), C) GAAPDTRPAAGK (7). D) Size of the 4 × 5 array compared to a five-pence coin.
MALDI-ToF MS analysis also revealed that peptide 8, previously described as a substrate for OGT, was recognised by ppGalNAcT2 and can thus incorporate either a βGlcNAc or an αGalNAc residue depending on the enzyme. The EA2 peptide 9 showed a more complex glycosylation pattern, with a mixture of two, three or four αGalNAc residues added under the tested conditions (Figure 3). Finally, peptide 10 was not a substrate for the enzyme.
Figure 3.
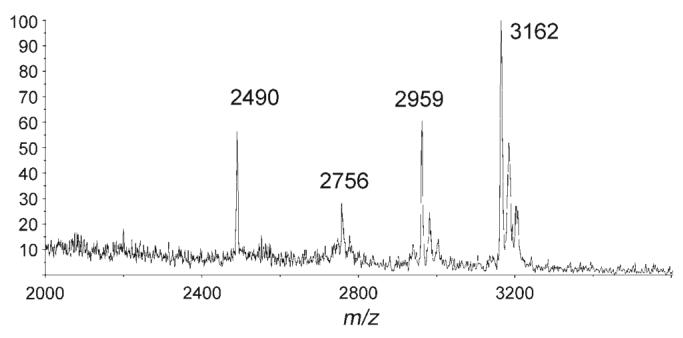
MALDI-ToF MS spectrum of the EA2 peptide 9 after enzymatic reaction. m/z 2756, 2959 and 3162 correspond to the di-, tri- and tetraglycosylated peptides, respectively. The peak at m/z 2490 is unknown.
In conclusion, we have demonstrated that peptide arrays on SAM-coated gold surfaces can be interrogated reliably by MALDI-ToF MS analysis and can be efficiently glycosylated with glycosyltransferases. It is interesting to note that glycosylation of immobilised fragments derived from natural mucin reflected a preferential order of glycosylation in good agreement with other reported assays. These data suggest that the arrays give similar results to other in vitro assay system. Obtaining quantitative data with MALDI-Tof MS will be more challenging because of differences in ionisation efficiency, although Nagahori et al. have described a quantitative inhibition essay using MS.[13] By using a peptide with several potential glycosylation sites like the EA2 peptide 9, direct evaluation of the number of GalNAc residues added was possible.
The methodology described here provides a valuable platform for easy detection of glycosyltransferase activity and specificity, which is label-free and can be performed with small quantities of material in a high-throughput format. Systematic studies of donor and acceptor specificity of several ppGalNAcTs isoforms are part of the work currently in progress in our group. In addition, the methodology can also be used for the efficient chemo-enzymatic synthesis of novel glycopeptide arrays by a combination of chemical synthesis and immobilisation and subsequent glycosylation. This is particularly useful for such linkages as αGalNAc-serine and -threonine, which are chemically difficult to synthesise. Such glycopeptide arrays will be useful for further screening of protein–carbohydrate interactions, in particular by using surface plasmon resonance spectroscopy.
Experimental Section
Synthesis of the peptides 1–10
Synthesis was performed under standard Fmoc-solid-phase peptide synthesis conditions with a CEM Liberty peptide synthesizer. Amino acid (5 equiv), benzotriazol-1-yl-oxytripyrro-lidinophosphonium hexafluorophosphate (PyBOP; 5 equiv) and N,N-diisopropylethylamine (DIPEA; 10 equiv) were used for the coupling steps, 20% piperidine/DMF for the Fmoc-deprotection steps. After completion, the resin-bound peptide was thoroughly washed with DMF, DMF/MeOH (1:1) and dichloromethane, then cleaved from the resin with trifluoroacetic acid/triisopropylsilane/water (95:2.5:2.5, v/v/v). Precipitation in cold diethyl ether, centrifugation and removal of the solvent afforded the desired peptide as a white solid.
Preparation of gold-coated slides
Microscope glass cover slips (13 mm diameter, no. 2 thickness) were obtained from Agar Scientific. The cover slips were cleaned in “Piranha” solution (H2SO4/H2O2 5:1, CAUTION! very reactive oxidising agent) for 20 min, rinsed with distilled water and dried under a stream of nitrogen. An adhesion layer of 5 nm of chromium and subsequent 100 nm of gold were sputtered onto the glass cover slips with a Denton Vacuum Desk III sputter coater.
Preparation of the monolayers on gold surface
Before SAM formation the gold-coated glass slides were cleaned with piranha solution and thoroughly rinsed with deionised water and ethanol and dried under a stream of nitrogen. The substrates were then immersed overnight in a DMSO solution of thiols presenting a carboxylic acid and a tri(ethylene glycol) group (final concentration 0.1 mg mL−1, molar ratio 1:20). The monolayers were rinsed with DMSO, ethanol and dried under a stream of nitrogen. N-hydroxysuccinimide (NHS)-activation of the carboxylic acid was performed by dipping the surfaces into a DMF solution of EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide [EtN = C = N(CH2)3NMe2·HCl] (0.2 m) and NHS (0.05 m) for 2 h followed by washing and drying as above.
Peptide immobilisation
Peptides were dissolved in a 100 mm phosphate buffer solution containing 6 m guanidinium hydrochloride, and the pH was adjusted with 2 m NaOH (pH 8, final concentration 50 mm). The NHS-activated monolayers were treated overnight with 50 μL of peptide solution and were then rinsed and dried as above. In array format, 0.02 μL of solution were manually spotted with an Eppendorf pipette.
Cloning and expression of ppGalNAcT2
The human ppGalNAcT2-gene was obtained from Geneservice Ltd., UK (IMAGE-clone #5553465). Cloning and production of soluble human ppGalNAcT2 was performed in the yeast Pichia pastoris according to Bourgeaux et al.[20] with slight modifications. The coding sequence corresponding to amino acids 51 to 571 was inserted in 3′ of the α-factor signal sequence of a pPICZαB expression vector (Invitrogen, UK, Paisley) modified to introduce an N-terminal hexahistidine tag. After transformation into E. coli TOP10F′ cells by heat-shock and plating the cells on LB/half-salt agar containing zeocin (25 μg mL−1; Invitrogen, UK, Paisley), positive clones were selected after PCR screening and by sequencing to confirm the reading frame. 10 μg of the expression construct plasmid DNA was linearised according to the supplier's instructions and used to electroporate Pichia pastoris-competent cells (GS115), which were then plated on YPDS agar containing zeocin (100 μg mL−1). Selected colonies of recombinant Pichia pastoris were inoculated into 50 mL of BMGY medium containing 100 μg mL−1 Zeocin. After overnight incubation at 30 °C with continuous shaking (200 rpm) in baffled flasks, cells were collected by centrifugation at 1500 g. The cells were washed once in BMMY and resuspended in 400 mL of methanol-containing BMMY medium to a final OD600 of 1.0 and incubated at 16 °C. Every 24 h samples of the cultures were removed and extra methanol was added to maintain a concentration of 1 % (v/v). The culture medium was concentrated 50-fold by using Vivaspin concentrators (Mr 30 000 cut-off).
Enzymatic glycosylation
SAMs presenting the peptide substrate were incubated overnight at 37 °C with a mixture consisting of ppGalNAcT2 (5 μL of a 50-fold concentrated solution obtained as described above), UDP-GalNAc (5 mm stock solution, 10 μL), 2-amino-2-methylpropane-1,3-diol (AMPD) buffer (pH 7.4, 5 μL), MnCl2 (1 m stock solution, 0.5 μL) and sterilised water (29.5 μL) and then rinsed with deionised water and ethanol and dried under a stream of nitrogen.
MALDI-ToF MS analysis
The targets were attached to a modified MALDI-ToF sample plate using adhesive tape, coated with a solution of 2,4,6-trihydroxyacetophenone (THAP; 10 mg. mL−1 in acetone) and loaded into a Voyager-DE STR Biospectrometry MALDI-ToF mass spectrometer (PerSeptive Biosystems) operating with a 337 nm nitrogen laser. Mass spectra were acquired using reflector ToF, positive-ion mode with an accelerating voltage of 20 kV and an extraction delay of 200 ns.
Acknowledgements
This work was funded by grants from the BBSRC, RCUK, The Wellcome Trust and The Royal Society (to S.L.F.). We thank Prof. Rob Field (John Innes Centre, Norwich, UK), Prof. Jeremy Turnbull and Dr. Zheng-Lianglane Zhi (University of Liverpool, UK) for helpful discussions.
References
- 1.Walsh CT, Garneau-Tsodikova S, Gatto GJ., Jr Angew. Chem. 2005;117:7508–7539. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 2.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 3.Helenius A, Aebi M. Annu. Rev. Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 4.Dwek RA. Chem. Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 5.Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamemura K, Hart GW. Prog. Nucleic Acid Res. Mol. Biol. 2003;73:107–136. doi: 10.1016/s0079-6603(03)01004-3. [DOI] [PubMed] [Google Scholar]
- 7.Slawson C, Hart GW. Curr. Opin. Struct. Biol. 2003;13:631–636. doi: 10.1016/j.sbi.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Whelan SA, Hart GW. Circ. Res. 2003;93:1047–1058. doi: 10.1161/01.RES.0000103190.20260.37. [DOI] [PubMed] [Google Scholar]
- 9.Becker DJ, Lowe JB. Glycobiology. 2003;13:41R–53R. doi: 10.1093/glycob/cwg054. [DOI] [PubMed] [Google Scholar]
- 10.Haines N, Irvine KD. Nat. Rev. Mol. Cell Biol. 2003;4:786–797. doi: 10.1038/nrm1228. [DOI] [PubMed] [Google Scholar]
- 11.Rodén LAS, Campbell P, Curenton T, Ekborg G, Manzella S, Pillon D, Meezan E. Adv. Exp. Med. Biol. 1992;313:1–20. doi: 10.1007/978-1-4899-2444-5_1. [DOI] [PubMed] [Google Scholar]
- 12.a) Leavy TM, Bertozzi CR. Bioorg. Med. Chem. Lett. 2007;17:3851–3854. doi: 10.1016/j.bmcl.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pratt MR, Hang HC, Ten Hagen GG, Rarick J, Gerken TA, Tabak LA, Bertozzi CR. Chem. Biol. 2004;11:1009–1016. doi: 10.1016/j.chembiol.2004.05.009. [DOI] [PubMed] [Google Scholar]; c) Shin I, Lee M-R. Chem. Eur. J. 2005;11:2894–2901. doi: 10.1002/chem.200401030. [DOI] [PubMed] [Google Scholar]; d) Houseman BT, Mrksich M. Chem. Biol. 2002;9:443–454. doi: 10.1016/s1074-5521(02)00124-2. [DOI] [PubMed] [Google Scholar]; e) Seibel J, Hellmuth H, Hofer B, Kicinska A-M, Schmalbruch B. ChemBioChem. 2006;7:310–320. doi: 10.1002/cbic.200500350. [DOI] [PubMed] [Google Scholar]; f) Love KR, Seeberger PH. Angew. Chem. 2002;114:3733–3736. [Google Scholar]; Angew. Chem. Int. Ed. 2002;41:3583–3586. doi: 10.1002/1521-3773(20021004)41:19<3583::AID-ANIE3583>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; g) Park S, Shin I. Org. Lett. 2007;9:1675–1678. doi: 10.1021/ol070250l. [DOI] [PubMed] [Google Scholar]
- 13.Nagahori N, Nishimura S-I. Chem. Eur. J. 2006;12:6478–6485. doi: 10.1002/chem.200501267. [DOI] [PubMed] [Google Scholar]
- 14.Freire T, D'Alayer J, Bay S. Bioconjugate Chem. 2006;17:559–564. doi: 10.1021/bc0502661. [DOI] [PubMed] [Google Scholar]
- 15.Min D-H, Mrksich M. Curr. Opin. Chem. Biol. 2004;8:554–558. doi: 10.1016/j.cbpa.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 16.a) Hanley L, Kornienko O, Ada ET, Fuoco E, Trevor JL. J. Mass Spectrom. 1999;34:705–723. doi: 10.1002/(SICI)1096-9888(199907)34:7<705::AID-JMS845>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]; b) Su J, Mrksich M. Angew. Chem. 2002;114:4909–4912. [Google Scholar]; Angew. Chem. Int. Ed. 2002;41:4715–4718. doi: 10.1002/anie.200290026. [DOI] [PubMed] [Google Scholar]; c) Mrksich M, Whitesides GM. Annu. Rev. Biophys. Biomol. Struct. 1996;25:55–78. doi: 10.1146/annurev.bb.25.060196.000415. [DOI] [PubMed] [Google Scholar]; d) Yeo W-S, Min D-H, Hsieh RW, Greene GL, Mrksich M. Angew. Chem. 2005;117:5616–5619. doi: 10.1002/anie.200501363. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:5480–5483. doi: 10.1002/anie.200501363. [DOI] [PubMed] [Google Scholar]
- 17.Hang HC, Bertozzi CR. Bioorg. Med. Chem. 2005;13:5021–5034. doi: 10.1016/j.bmc.2005.04.085. [DOI] [PubMed] [Google Scholar]
- 18.Ten Hagen KG, Fritz T, Tabak LA. Glycobiology. 2003;13:1R–16R. doi: 10.1093/glycob/cwg007. [DOI] [PubMed] [Google Scholar]
- 19.White T, Bennett EP, Takio K, Sørensen T, Bonding N, Clausen H. J. Biol. Chem. 1995;270:24156–24165. doi: 10.1074/jbc.270.41.24156. [DOI] [PubMed] [Google Scholar]
- 20.Bourgeaux V, Cadène M, Piller F, Piller V. ChemBioChem. 2007;8:37–40. doi: 10.1002/cbic.200600369. [DOI] [PubMed] [Google Scholar]
- 21.Gerken TA, Raman J, Fritz TA, Jamison O. J. Biol. Chem. 2006;281:32403–32416. doi: 10.1074/jbc.M605149200. [DOI] [PubMed] [Google Scholar]
- 22.Lahiri J, Isaacs L, Tien J, Whitesides GM. Anal. Chem. 1999;71:777–790. doi: 10.1021/ac980959t. [DOI] [PubMed] [Google Scholar]
- 23.Prime KL, Whitesides GM. J. Am. Chem. Soc. 1993;115:10714–10721. [Google Scholar]
- 24.Comer FI, Hart GW. Biochemistry. 2001;40:7845–7852. doi: 10.1021/bi0027480. [DOI] [PubMed] [Google Scholar]
- 25.Wilson IBH. J. Biol. Chem. 2002;277:21207–21212. doi: 10.1074/jbc.M201634200. [DOI] [PubMed] [Google Scholar]
- 26.Wandall HH, Hassan H, Mirgorodskaya E, Kristensen AK. J. Biol. Chem. 1997;272:23503–23514. doi: 10.1074/jbc.272.38.23503. [DOI] [PubMed] [Google Scholar]