

Abstract
Although centrally acting opioid analgesics produce profound antinociception under basal conditions, the antinociceptive properties of peripherally restricted opioid analgesics are generally only detectable after inflammation or injection of inflammatory mediators. Despite considerable research, the cellular mechanisms regulating the functional competence of peripheral opioid receptor systems for inhibition of nociception remain unclear. Recent work has demonstrated that brief pre-treatment (priming) with bradykinin, arachidonic acid, protease-activated receptor-2 agonists, or direct activators of protein kinase C (PKC) are capable of inducing the functional competence of the opioid receptor system in cultures of primary sensory neurons in vitro. Here we report that the peripheral delta opioid receptor system also requires PKC-dependent priming to inhibit prostaglandin E2 (PGE2)-induced thermal allodynia in the rat. Peripheral hindpaw injection of [D-Pen2,5]-enkephalin (DPDPE), a selective delta opioid receptor agonist, did not alter PGE2-induced thermal allodynia. However, following priming (15 min) with bradykinin or arachidonic acid, DPDPE produced a significant reduction in allodynia that was antagonist reversible, peripherally restricted, and exhibited a typical dose-response relationship. Furthermore, the bradykinin priming effect was blocked by the PKC inhibitors, bisindolylmaleimide I and chelerythrine. Collectively, these data support prior in vitro findings that, although present on primary sensory neurons, peripheral opioid receptor systems are functionally inactive under basal conditions and require activation of a PKC- and arachidonic acid-dependent signaling pathway to develop functional competence in vivo.
Keywords: DPDPE, hindpaw, prostaglandin, bradykinin, allodynia
1. Introduction
Centrally acting opioids are highly effective at reducing nociceptive behaviors under either basal conditions or after injury. However, drugs that act in the central nervous system (CNS) often trigger potentially serious adverse side effects and opioids are no exception. In contrast to the dramatic antinociceptive effects of centrally acting opioids, peripherally restricted opioids usually have little-to-no detectable effects on basal nociceptive thresholds (for reviews see Cabot, 2001; Przewlocki and Przewlocka, 2001; Stein et al., 2001). Interestingly, although some variability has been reported (Ibrahim et al., 2005), peripherally restricted opioids can have profound efficacy for inhibiting hyperalgesia/allodynia in inflamed tissues (for review, see Stein et al., 2003; 1989; Joris et al., 1987; Ferreira and Nakamura, 1979). Furthermore, peripheral antihyperalgesic/antiallodynic effects of opioids have been reported in models of inflammation (eg., carrageenan, complete Freund’s adjuvant, formalin), nerve injury (e.g., chronic constriction injury) and tissue sensitization (e.g., local injection of prostaglandin E2, bradykinin or capsaicin) (Ko et al., 2000; Catheline et al., 1996; Keita et al., 1995; Hong and Abbott, 1995; Levine and Taiwo, 1989; Joris et al., 1987; Ferreira and Nakamura, 1979). However, whereas numerous animal studies demonstrate nearly 80–100% efficacy in reversing hyperalgesia/allodynia, this is not the case in clinical trials. Several clinical trials have not detected a significant peripheral component to opioid analgesia, and one systematic review of the clinical literature estimated that the overall efficacy of peripheral opioid analgesia is “mild” for reducing clinical pain (see Gupta et al., 2001). Thus, there is a large difference in efficacy estimates between animal and clinical studies on peripheral opioids. Consequently a better understanding of the mechanisms mediating the development of inflammation-induced functional competence of the opioid receptor system is required.
Peripherally selective opioids may offer reduced risk of CNS-mediated adverse effects, however, comparatively little is known about the cellular mechanisms mediating the development of functional competence of peripheral opioid receptor systems for inhibiting nociception. Recent work has demonstrated that opioid receptor systems on primary sensory neurons in vitro require a pretreatment, or priming stimulus, such as bradykinin, protease-activated receptor agonists or arachidonic acid, to inhibit neuropeptide release and adenylyl cyclase activity (Berg et al., 2007a, 2007b; Patwardhan et al., 2006, 2005). Furthermore, induction of opioid receptor system competence is mediated via a cyclooxygenase-dependent arachidonic acid metabolite that is downstream from PKC. However, it is not known whether the development of opioid receptor system competence in vivo requires activation of these cellular signaling pathways. Accordingly, in the present study, we evaluated whether bradykinin and arachidonic acid are effective for priming opioid receptor systems using an in vivo model of thermal allodynia, and further, whether this effect is mediated by activation of PKC signaling pathways.
2. Materials and Methods
2.1. Animals
Experiments were performed on male Sprague-Dawley rats (175–200g; Charles River, Wilmington, MA). A 12 hr light/dark cycle was used with all testing occurring in the light phase. Animals were housed for 1 week before the experiment with food and water available ad libitum. The animal study protocol was approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio and conformed to the International Association for the Study of Pain and US federal guidelines.
2.2. Materials
Prostaglandin E2 (PGE2) and arachidonic acid were purchased from Cayman Chemicals (Ann Arbor, MI). Bisindolylmaleimide I (BIS) was purchased from Calbiochem (San Diego, CA). Bradykinin acetate, [D-Pen2,5]-enkephalin (DPDPE), [D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin (DAMGO), D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP), naltrindole hydrochloride, and chelerythrine chloride were purchased from Sigma-Aldrich (St. Louis, MO). Stock solutions were prepared in ethanol (PGE2 and arachidonic acid), dimethylsulfoxide (BIS), sterile water (DAMGO, CTOP, DPDPE, naltrindole, and chelerythrine), or saline (bradykinin). All stock solutions were diluted in saline prior to injection (BIS was diluted in saline containing 5% Tween). Drugs were administered via intraplantar (i.pl.) injection at a final volume of 50μl (BIS injections were 100μl).
2.3. Behavioral Testing
Paw withdrawal latency (PWL) to a thermal stimulus was measured with a plantar test apparatus (Hargreaves et al., 1988). Briefly, rats were placed in plastic boxes with a glass floor. After a 30 min habituation period, the plantar surface of the hindpaw was exposed to a beam of radiant heat through the glass floor, causing a gradual increase in the temperature of the hindpaw of the animal (Dirig et al., 1997). The PWL was automatically determined by a photoelectric cell. The rate of increase in temperature of the glass floor was adjusted so that baseline PWL values were close to 10 s; cut-off time was 20 s. Measurements were taken in duplicate at least 30 s apart and the average was used for statistical analysis. Observers were blinded to the treatment allocation.
2.4. Time Course of Effects
To evaluate the magnitude and duration of thermal allodynia to PGE2 and bradykinin, animals were injected with vehicle, bradykinin (25 μg), or PGE2 (0.3 μg) and PWLs were measured every 5 min for 20 min.
2.5. Priming Requirement
To determine if priming is required for functional competence of the peripheral delta opioid receptor system, animals were pretreated with vehicle, bradykinin (25 μg), or arachidonic acid (35 μg) 15 min prior to co-injection of PGE2 (0.3 μg) and either vehicle or DPDPE (20 μg). PWLs were measured 5 and 10 min after pretreatment, and every 5 min for 20 min after co-injection. DPDPE was also evaluated with and without bradykinin priming (25 μg) at 0.2, 2 and 20 μg to assess a dose-response relationship for DPDPE.
2.6. Peripherally Restricted
To verify that the effect of DPDPE is mediated in the periphery, animals were pretreated with bradykinin (25 μg) 15 min prior to co-injection of PGE2 (0.3 μg) and either vehicle or DPDPE (20 μg) in the ipsilateral hindpaw and either vehicle or DPDPE (20 μg) in the contralateral hindpaw. PWLs were measured on the ipsilateral hindpaw 5 and 10 min after pretreatment, and every 5 min for 20 min after co-injection.
2.7. Pharmacology
To determine whether the effects of DPDPE were mediated via delta opioid receptors, animals were pretreated with bradykinin (25 μg) and the selective delta opioid receptor antagonist naltrindole (40 μg) or bradykinin (25 μg) alone 15 min prior to co-injection of PGE2 (0.3 μg) and either vehicle or DPDPE (20 μg). PWLs were measured 5 and 10 min after pretreatment, and every 5 min for 20 min after co-injection. In a separate experiment, animals were pretreated with bradykinin (25 μg) and the selective mu opioid receptor antagonist CTOP (10 μg) or bradykinin (25 μg) alone 15 min prior to co-injection of PGE2 (0.3 μg) and either vehicle, DPDPE (20 μg), or the mu opioid receptor agonist DAMGO (8 μg). PWLs were again measured 5 and 10 min after pretreatment, and every 5 min for 20 min after co-injection.
To determine whether the priming effect of bradykinin on the peripheral delta opioid receptor system was PKC-dependent, animals were pretreated with the PKC inhibitors chelerythrine (5 μg) or BIS (25μg) 30 min prior to priming with vehicle or bradykinin (25 μg). Animals were co-injected with PGE2 (0.3 μg) and either vehicle or DPDPE (20 μg) 15 min post-priming as before. PWLs were measured 5 and 10 min after both PKC-inhibitor injection and priming pretreatments, and every 5 min for 20 min after co-injection.
2.8. Statistical Analysis
Results are expressed as mean ± S.E.M. of 5–9 animals per group and reflect changes from pre-injection baseline PWLs. Statistical significance was determined by one- or two-way ANOVA, where appropriate, with Bonferroni. P<0.05 was considered statistically significant.
3. Results
Both PGE2 (0.3 μg) and bradykinin (25 μg) produced significant thermal allodynia (Fig. 1). The peak magnitude of PGE2-induced thermal allodynia was significantly smaller (P<0.01) than bradykinin-induced thermal allodynia. The bradykinin effect was transient, returning to baseline by 15 min after injection. The thermal allodynia produced by PGE2 was sustained for the full 20 minutes of the experiment (P<0.001).
Fig. 1. Time course of bradykinin- and PGE2-induced thermal allodynia.
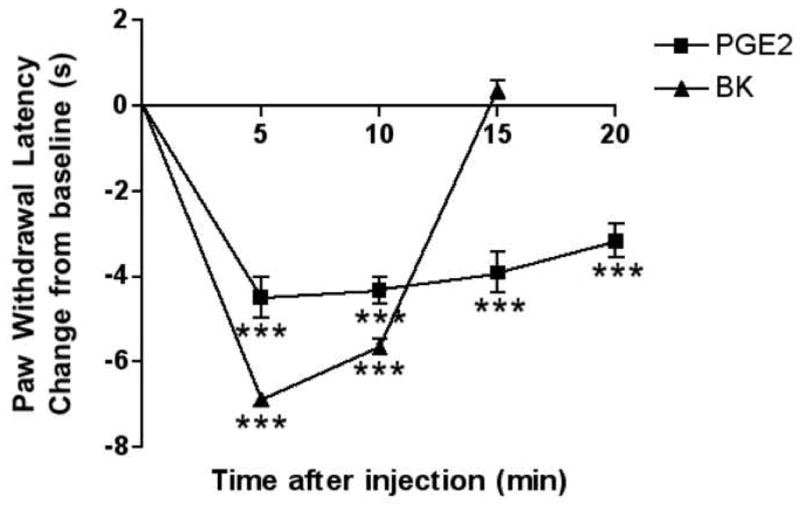
Bradykinin (BK; 25 μg) or PGE2 (0.3 μg) was injected i.pl. in 50 μl saline. PWLs were measured at 5 min intervals for at least 15 min post-injection. Baseline PWLs were 9.75 ± 0.40 s. Data are expressed as mean ± S.E.M. of 5–9 animals per group (Note: Error bars for some data points are within the size of the symbol). ***, P<0.001 vs. vehicle treated controls (not shown). All observations were collected by observers blinded to treatment allocation.
Injection of DPDPE (20 μg) into the hindpaw did not alter PGE2-induced thermal allodynia when injected after vehicle pretreatment (Fig. 2). However, 15 min after injection of bradykinin (25 μg) into the hindpaw, DPDPE (20 μg) completely reversed PGE2-induced thermal allodynia, producing withdrawal latencies greater than baseline levels (Fig. 2). Similarly, DPDPE (20 μg) also blocked PGE2-induced thermal allodynia following priming with arachidonic acid (35 μg), which did not affect the response to PGE2 alone (Fig. 2). Fig. 3 shows the dose-response relationship for DPDPE in bradykinin-primed animals.
Fig. 2. Functional competence of the peripheral delta opioid receptor system requires priming with bradykinin or arachidonic acid.

Separate groups of animals were injected with vehicle, bradykinin (BK; 25 μg), or arachidonic acid (AA; 35μg) 15 min prior to the co-injection of PGE2 (0.3 μg) and DPDPE (20 μg) or vehicle. PWLs were measured at 5 min intervals for at least 20 min after the second injection. Baseline PWLs were 8.83 ± 0.23 s. Data are expressed as mean ± S.E.M. PWL measured 10 min after the second injection of 5–9 animals per group. ***, *, P<0.001, 0.05 vs. first three groups or vs. AA/Veh/PGE2, respectively.
Fig. 3. Dose-response relationship for DPDPE to reduce PGE2-induced thermal allodynia.
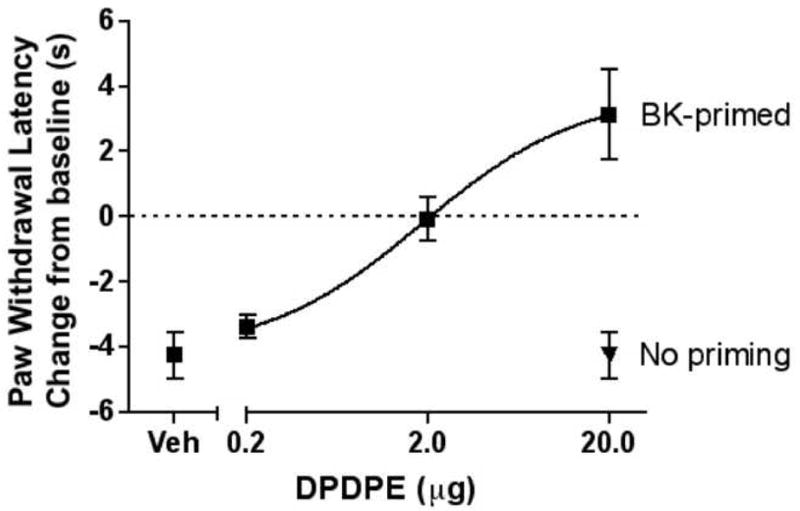
Animals were pretreated with bradykinin (BK; 25 μg) or vehicle 15 min prior to the co-injection of PGE2 (0.3 μg) and DPDPE (0.2, 2, or 20 μg). PWLs were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 8.61 ± 0.27 s. Data are represented as mean ± S.E.M. PWL measured 10 min after the last injection of 5–9 animals per group. For comparison, animals receiving bradykinin pretreatment prior to the co-injection of vehicle and PGE2 (0.3 μg) are shown on the lower left (“Veh”), and animals not receiving bradykinin pretreatment prior to the co-injection of DPDPE (20 μg) and PGE2 (0.3 μg) are shown on the lower right (“No priming”). ED50=1.84 μg.
In bradykinin-primed (25 μg) animals, neither vehicle injected ipsilaterally nor DPDPE (20 μg) injected contralaterally altered PGE2-induced thermal allodynia (Fig. 4). However, DPDPE (20 μg) injected ipsilaterally completely reversed PGE2-induced thermal allodynia to withdrawal latencies above baseline levels (Fig. 4).
Fig. 4. Antinociception of DPDPE is restricted to the periphery.

15 min after ipsilateral bradykinin priming (BK; 25 μg), animals received single injections into both the ipsilateral and contralateral hindpaws. All animals received PGE2 (0.3 μg) in the ipsilateral hindpaw (“Ipsi”), co-injected with DPDPE (20 μg) or vehicle, and either DPDPE (20 μg) or vehicle in the contralateral hindpaw (“Contra”). PWLs of the ipsilateral hindpaw were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 10.39 ± 0.52 s. Data are represented as mean ± S.E.M. PWL measured 10 min after the last injection of 5–9 animals per group. ***, P<0.001 vs. both other groups.
Naltrindole pretreatment (40 μg) did not affect PGE2-induced thermal allodynia in bradykinin-primed (25 μg) animals, but reversed the anti-allodynic effect of DPDPE (20 μg; Fig. 5). Chelerythrine and BIS pretreatment also had no effect on PGE2-induced thermal allodynia in the absence of bradykinin priming, but both chelerythrine and BIS reversed the effect seen with DPDPE (20 μg) in bradykinin-primed (25 μg) animals (Fig. 5). In bradykinin-primed (25 μg) animals, peripheral administration of the mu opioid selective agonist, DAMGO (8 μg), also reversed PGE2-induced thermal allodynia. The mu opioid selective antagonist, CTOP (10 μg), completely blocked the anti-allodynic effect of DAMGO (8 μg) but had no effect on DPDPE (20 μg; Fig. 6).
Fig. 5. The anti-allodynic effect of DPDPE is mediated by delta opioid receptors and is PKC-dependent.

Animals received either bradykinin (BK; 25 μg) or bradykinin with naltrindole (NAL; 40 μg) 15 min prior to the co-injection of PGE2 (0.3 μg) and DPDPE (20 μg) or vehicle (first four bars). In another experiment, separate groups of animals were pretreated with either chelerythrine (CHEL; 5 μg) or BIS (25 μg; 100 μl) 30 min prior to treatment with either vehicle or bradykinin (BK; 25 μg); 15 min after vehicle or bradykinin, animals were co-injected with PGE2 (0.3 μg) and DPDPE (20 μg; last four bars). PWLs were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 10.50 ± 0.31 s. Data are represented as mean ± S.E.M. PWL measured 10 min after the last injection of 5–9 animals per group. ***, P<0.001 vs. all other groups.
Fig. 6. The anti-allodynic effect of DPDPE is not mediated by mu opioid receptors.

Animals received either bradykinin (BK; 25 μg) or bradykinin with CTOP (10 μg) 15 min prior to the co-injection of PGE2 (0.3 μg) and DAMGO (8 μg), DPDPE (20 μg), or vehicle. PWLs were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 10.27 ± 0.56 s. Data are represented as mean ± S.E.M. PWL measured 10 min after the last injection of 5–9 animals per group. ***, P<0.001 upper three groups vs. lower three groups. n.s., not significant.
4. Discussion
This study demonstrates that the in vivo peripheral delta opioid receptor system does not function to reduce PGE2-mediated thermal allodynia unless a priming stimulus (such as bradykinin or arachidonic acid) is first applied to the tissue. This finding is also consistent with the in vitro culture models of primary sensory neurons for both mu- (Berg et al., 2007a; 2007b) and delta-opioid receptors (Patwardhan et al., 2006; 2005) where opioid receptor activation does not inhibit neuropeptide release or adenylyl cyclase activity unless primed with bradykinin, PAR-2 agonists, arachidonic acid or PKC activators. Similarly, the majority of studies (for reviews see Cabot, 2001; Przewlocki and Przewlocka, 2001; Stein et al., 2001), but not all (Ibrahim et al., 2005), indicate peripherally administered opioids have no appreciable antinociceptive effects, but can, under appropriate conditions such as inflammation, exert profound antihyperalgesic/antiallodynic effects.
Following bradykinin priming, the antiallodynic effect of DPDPE did not occur when DPDPE was injected into the paw contralateral to the hindpaw receiving the bradykinin priming and PGE2 injection, indicating that, under these experimental conditions, the effect of DPDPE was localized to the ipsilateral inflamed hindpaw. The DPDPE effect was reversed by the delta opioid receptor antagonist naltrindole and not by the mu opioid receptor antagonist CTOP, suggesting mediation by peripheral delta receptors in the hindpaw. Furthermore, the efficacy of DPDPE was substantial, as PGE2-induced thermal allodynia was blocked completely.
PKC has been demonstrated to be important in the regulation of nociceptor function (Blaukat, 2003; Souza, 2002; Cesare et al., 1999) and activation of PKC signaling pathways is required for priming of the peripheral opioid receptor system in vitro by bradykinin (Patwardhan et al., 2005; Berg et al., 2007a). Our findings here are consistent with these in vitro results since two structurally distinct PKC inhibitors, chelerythrine and BIS, reversed DPDPE-mediated antinociception following bradykinin priming. Since PKC is activated in response to bradykinin receptor stimulation (Leeb-Lundberg et al., 2005), it is likely that BIS and chelerythrine blocked the priming effect of bradykinin.
Recently, it has been shown that the opioid receptor system on peripheral sensory neurons may be functionally primed not only by administration of bradykinin, but also by exogenously applied arachidonic acid, and that arachidonic acid, produced downstream from PKC, mediates the priming effect of bradykinin (Berg et al., 2007a; Patwardhan et al., 2005). Here we also found that arachidonic acid can substitute for bradykinin priming to induce functional competence of the peripheral delta opioid receptor system in vivo. Taken together, these studies suggest that opioid receptor system functional competence in vivo can be produced by activation of Gαq-coupled receptors and is dependent upon activation of PKC and production of arachidonic acid. Further, these data demonstrate strong congruence with those obtained with primary cultures of sensory neurons, suggesting that the cell culture model may be useful in delineating the mechanisms of regulation of opioid receptor system functional competence.
In addition to activation of Gq/11-mediated signaling pathways, functional competence of opioid receptor systems may be induced by other mechanisms. For example, in trigeminal and nodose neurons pretreated with forskolin, adenosine A1 and mu opioid receptors are responsive to inhibit CGRP release and voltage-dependent cation channels (Carruthers et al. 2001; Ingram and Williams, 1994). These data suggest that perhaps cAMP produced via activation of adenylyl cyclase may also act as a priming stimulus. Furthermore, following pre-treatment with capsaicin, which activates TRPV1 channels, in dorsal root ganglion neurons or animals in vivo, mu opioid receptors were functional to inhibit TRPV1 currents and thermal allodynia, respectively (Endres-Becker et al., 2007), suggesting that perhaps increases in intracellular calcium as a result of TRPV1 activation (for review see Caterina and Julius, 2001), may also promote competence.
The changes within the opioid receptor system that underlie the enhanced responsiveness following a priming stimulus are not known. Such changes may include increased receptor density on the cell surface, increased affinity of agonist for the receptor or enhanced coupling of receptor to G proteins. Previous work with primary cultures of rat trigeminal ganglion suggests that priming with bradykinin increases the ability of MOR agonists to stimulate GTP[γ35S] binding (Berg et al., 2007a), which could result from any of the above mechanisms. Identification of the mechanisms responsible for induction of opioid receptor system competence may allow for new adjuvant treatments for improved pharmacotherapy with peripherally selective opioids.
Understanding the mechanisms underlying development of functional competence of the peripheral delta opioid receptor system by bradykinin- or arachidonic acid-mediated priming may have fundamental significance for treatment of peripheral pain disorders. Peripherally restricted opioid analgesics have the advantage of reduced centrally mediated adverse effects, however, in clinical studies the analgesic efficacy of peripherally applied opioids is variable and some studies have not detected a significant peripheral component to opioid analgesia. A better understanding of the cellular mechanisms by which opioid receptor system functional competence is produced and maintained will aid in the development of improved pharmacotherapy for peripheral pain conditions.
Supplementary Material
Figure S1. Functional competence of the peripheral delta opioid receptor system requires priming with BK or AA. Separate groups of animals were injected with either saline (V), BK (25 μg), or AA (35μg) 15 min prior to the co-injection of PGE2 (0.3 μg) and DPDPE (20 μg; D+P) or vehicle (V+P). PWLs were measured at 5 min intervals for at least 20 min after the second injection. Baseline PWLs were 8.83 ± 0.23 sec. Data are expressed as mean ± SEM PWL after the second injection of 5–9 animals per group. ***, *, p<0.001, 0.05 vs. first three groups or vs. AA, V+P, respectively.
Figure S2. Dose-response relationship for DPDPE to reduce PGE2-induced thermal allodynia. Animals were pretreated with BK (25 μg) or vehicle 15 min prior to the co-injection of PGE2 (0.3 μg) and DPDPE (0.2, 2, or 20 μg; D+P) or vehicle (V+P). PWLs were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 8.61 ± 0.27 sec. Data are expressed as mean ± SEM PWL after the second injection of 5–9 animals per group. ED50=1.84 μg. ***, *, p<0.001, 0.05 vs. lower three groups on graph.
Figure S3. Antinociception of DPDPE is restricted to the periphery. 15 min after ipsilateral BK priming (25 μg), animals received single injections into both the ipsilateral and contralateral hindpaws. All animals received PGE2 (0.3 μg) in the ipsilateral hindpaw (“Ipsi”), co-injected with DPDPE (20 μg; D+P) or vehicle (V+P), and either DPDPE (20 μg) or vehicle (V) in the contralateral hindpaw (“Contra”). PWLs of the ipsilateral hindpaw were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 10.39 ± 0.52 sec. Data are expressed as mean ± SEM PWL after the second injection of 5–9 animals per group. ***, p<0.001 vs. both other groups.
Figure S4. The anti-allodynic effect of DPDPE is mediated by delta opioid receptors and is PKC-dependent. Animals received either BK (25 μg) or BK with NAL (40 μg; BK+NAL) 15 min prior to the co-injection of PGE2 (0.3 μg) and DPDPE (20 μg; D+P) or vehicle (V+P). In another experiment, separate groups of animals were pretreated with either CHEL (5 μg) or BIS (25 μg; 100 μl) 30 min prior to treatment with either vehicle (V) or BK (25 μg); 15 min after vehicle or BK, animals were co-injected with PGE2 (0.3 μg) and DPDPE (20 μg; D+P). PWLs were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 10.50 ± 0.31 sec. Data are represented as mean ± SEM PWL after the last injection of 5–9 animals per group. ***, p<0.001 vs. all other groups.
Figure S5. The anti-allodynic effect of DPDPE is not mediated by mu opioid receptors. Animals received either BK (25 μg) or BK with CTOP (10 μg; BK+CTOP) 15 min prior to the co-injection of PGE2 (0.3 μg) and DAMGO (8 μg; GO+P), DPDPE (20 μg; D+P), or vehicle (V+P). PWLs were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 10.27 ± 0.56 sec. Data are represented as mean ± SEM PWL after the last injection of 5–9 animals per group. ***, p<0.001 upper three groups on graph vs. lower three groups on graph.
Acknowledgments
The authors would like to thank Dharshini Amarneethi for excellent technical assistance. This work was supported by USPHS grant P01DA016719, a Research Support Grant award from the Oral and Maxillofacial Surgery Foundation, the Texas Advanced Research Program under Grant No. 3659-0023, and COSTAR Training Grant NIDCR T32DE14318 (M.P.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berg KA, Patwardhan AM, Sanchez TA, Silva YM, Hargreaves KM, Clarke WP. Rapid modulation of μ-opioid receptor signaling in primary sensory neurons. J Pharmacol Exp Ther. 2007a;321:839–847. doi: 10.1124/jpet.106.116681. [DOI] [PubMed] [Google Scholar]
- Berg KA, Zardeneta G, Hargreaves KM, Clarke WP, Milam SB. Integrins regulate opioid receptor signaling in trigeminal ganglion neurons. Neurosci. 2007b;144:889–897. doi: 10.1016/j.neuroscience.2006.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaukat A. Structure and signaling pathways of kinin receptors. Andrologia. 2003;35:17–23. doi: 10.1046/j.1439-0272.2003.00533.x. [DOI] [PubMed] [Google Scholar]
- Cabot PJ. Immune-derived opioids and peripheral antinociception. Clin Exp Pharmacol Physiol. 2001;28:230–232. doi: 10.1046/j.1440-1681.2001.03425.x. [DOI] [PubMed] [Google Scholar]
- Carruthers AM, Sellers LA, Jenkins DW, Jarvie EM, Fenuik W, Humphrey PPA. Adenosine A1 receptor-mediated inhibition of protein kinase A-induced calcitonin gene-related peptide release from rat trigeminal neurons. Mol Pharmacol. 2001;59:1533–41. doi: 10.1124/mol.59.6.1533. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Ann Rev Neurosci. 2001;24:487–517. doi: 10.1146/annurev.neuro.24.1.487. [DOI] [PubMed] [Google Scholar]
- Catheline G, Kayser V, Guilbaud G. Further evidence for a peripheral component in the enhanced antinociceptive effect of systemic morphine in mononeuropathic rats: involvement of kappa-, but not delta-opioid receptors. Eur J Pharmacol. 1996;315:135–143. doi: 10.1016/s0014-2999(96)00629-2. [DOI] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-ε in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Dirig DM, Salami A, Rathbun ML, Ozaki GT, Yaksh TL. Characterization of variables defining hindpaw withdrawal latency evoked by radiant thermal stimuli. J Neurosci Meth. 1997;76:183–191. doi: 10.1016/s0165-0270(97)00097-6. [DOI] [PubMed] [Google Scholar]
- Endres-Becker J, Heppenstall PA, Mousa SA, Labuz D, Oksche A, Schafer M, Stein C, Zollner C. μ-Opioid receptor activation modulates transient receptor potential vanilloid 1 (TRPV1) currents in sensory neurons in a model of inflammatory pain. Mol Pharmacol. 2007;71:12–8. doi: 10.1124/mol.106.026740. [DOI] [PubMed] [Google Scholar]
- Ferreira SH, Nakamura M. Prostaglandin hyperalgesia: the peripheral analgesic activity of morphine, enkephalins and opioid antagonists. Prostaglandins. 1979;18:191–200. doi: 10.1016/0090-6980(79)90104-7. [DOI] [PubMed] [Google Scholar]
- Gupta A, Bodin L, Holmstrom B, Berggren L. A systematic review of the peripheral analgesic effects of intraarticular morphine. Anesth Analg. 2001;93:761–770. doi: 10.1097/00000539-200109000-00042. [DOI] [PubMed] [Google Scholar]
- Hargreaves KM, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hong Y, Abbott FV. Peripheral opioid modulation of pain and inflammation in the formalin test. Eur J Pharmacol. 1995;277:21–28. doi: 10.1016/0014-2999(95)00045-m. [DOI] [PubMed] [Google Scholar]
- Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A, Davar G, Makriyannis A, Vanderah TW, Mata HP, Malan TP. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci USA. 2005;102:3093–3098. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram SL, Williams JT. Opioid inhibition of Ih via adenylyl cyclase. Neuron. 1994;13:179–86. doi: 10.1016/0896-6273(94)90468-5. [DOI] [PubMed] [Google Scholar]
- Joris JL, Dubner R, Hargreaves KM. Opioid analgesia at peripheral sites: a target for opioids released during stress and inflammation? Anesth Analg. 1987;66:1277–1281. [PubMed] [Google Scholar]
- Keita H, Kayser V, Guilbaud G. Antinociceptive effect of a kappa-opioid receptor agonist that minimally crosses the blood-brain barrier (ICI 204448) in a rat model of mononeuropathy. Eur J Pharmacol. 1995;277:275–280. doi: 10.1016/0014-2999(95)00122-2. [DOI] [PubMed] [Google Scholar]
- Ko MC, Tuchman JE, Johnson MD, Wiesenauer K, Woods JH. Local administration of mu or kappa opioid agonists attenuates capsaicin-induced thermal hyperalgesia via peripheral opioid receptors in rats. Psychopharmacol. 2000;148:180–185. doi: 10.1007/s002130050040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb-Lundberg LM, Marceau F, Muller-Esterl W, Pettibone DJ, Zuraw BL. International Union of Pharmacology. XLV. Classification of the Kinin Receptor Family: from Molecular Mechanisms to Pathophysiological Consequences. Pharmacol Rev. 2005;57:27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- Levine JD, Taiwo YO. Involvement of the mu-opiate receptor in peripheral analgesia. Neurosci. 1989;32:571–575. doi: 10.1016/0306-4522(89)90279-0. [DOI] [PubMed] [Google Scholar]
- Patwardhan AM, Berg KA, Akopian AN, Jeske NA, Gamper N, Clarke WP, Hargreaves KM. Bradykinin-induced functional competence and trafficking of the δ-opioid receptor in trigeminal nociceptors. J Neurosci. 2005;25:8825–8832. doi: 10.1523/JNEUROSCI.0160-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan AM, Diogenes A, Berg KA, Fehrenbacher JC, Clarke WP, Akopian AN, Hargreaves KM. PAR-2 agonists activate trigeminal nociceptors and induce functional competence in the delta opioid receptor. Pain. 2006;125:114–124. doi: 10.1016/j.pain.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Przewlocki R, Przewlocka B. Opioids in chronic pain. Eur J Pharmacol. 2001;429:79–91. doi: 10.1016/s0014-2999(01)01308-5. [DOI] [PubMed] [Google Scholar]
- Souza ALS, Moreira FA, Almeida KR, Bertollo CM, Costa KA, Coelho MM. In vivo evidence for a role of protein kinase C in peripheral nociceptive processing. Br J Pharmacol. 2002;135:239–247. doi: 10.1038/sj.bjp.0704434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Machelska H, Binder W, Schafer M. Peripheral opioid analgesia. Cuur Opin Pharmacol. 2001;1:62–65. doi: 10.1016/s1471-4892(01)00005-4. [DOI] [PubMed] [Google Scholar]
- Stein C, Millan MJ, Shippenberg TS, Peter K, Herz A. Peripheral opioid receptors mediating antinociception in inflammation. Evidence for involvement of mu, delta, and kappa receptors. J Pharmacol Exp Ther. 1989;248:1269–1275. [PubMed] [Google Scholar]
- Stein C, Shafer M, Machelska H. Attacking pain at its source: new perspectives on opioids. Nat Med. 2003;9:1003–1008. doi: 10.1038/nm908. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Functional competence of the peripheral delta opioid receptor system requires priming with BK or AA. Separate groups of animals were injected with either saline (V), BK (25 μg), or AA (35μg) 15 min prior to the co-injection of PGE2 (0.3 μg) and DPDPE (20 μg; D+P) or vehicle (V+P). PWLs were measured at 5 min intervals for at least 20 min after the second injection. Baseline PWLs were 8.83 ± 0.23 sec. Data are expressed as mean ± SEM PWL after the second injection of 5–9 animals per group. ***, *, p<0.001, 0.05 vs. first three groups or vs. AA, V+P, respectively.
Figure S2. Dose-response relationship for DPDPE to reduce PGE2-induced thermal allodynia. Animals were pretreated with BK (25 μg) or vehicle 15 min prior to the co-injection of PGE2 (0.3 μg) and DPDPE (0.2, 2, or 20 μg; D+P) or vehicle (V+P). PWLs were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 8.61 ± 0.27 sec. Data are expressed as mean ± SEM PWL after the second injection of 5–9 animals per group. ED50=1.84 μg. ***, *, p<0.001, 0.05 vs. lower three groups on graph.
Figure S3. Antinociception of DPDPE is restricted to the periphery. 15 min after ipsilateral BK priming (25 μg), animals received single injections into both the ipsilateral and contralateral hindpaws. All animals received PGE2 (0.3 μg) in the ipsilateral hindpaw (“Ipsi”), co-injected with DPDPE (20 μg; D+P) or vehicle (V+P), and either DPDPE (20 μg) or vehicle (V) in the contralateral hindpaw (“Contra”). PWLs of the ipsilateral hindpaw were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 10.39 ± 0.52 sec. Data are expressed as mean ± SEM PWL after the second injection of 5–9 animals per group. ***, p<0.001 vs. both other groups.
Figure S4. The anti-allodynic effect of DPDPE is mediated by delta opioid receptors and is PKC-dependent. Animals received either BK (25 μg) or BK with NAL (40 μg; BK+NAL) 15 min prior to the co-injection of PGE2 (0.3 μg) and DPDPE (20 μg; D+P) or vehicle (V+P). In another experiment, separate groups of animals were pretreated with either CHEL (5 μg) or BIS (25 μg; 100 μl) 30 min prior to treatment with either vehicle (V) or BK (25 μg); 15 min after vehicle or BK, animals were co-injected with PGE2 (0.3 μg) and DPDPE (20 μg; D+P). PWLs were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 10.50 ± 0.31 sec. Data are represented as mean ± SEM PWL after the last injection of 5–9 animals per group. ***, p<0.001 vs. all other groups.
Figure S5. The anti-allodynic effect of DPDPE is not mediated by mu opioid receptors. Animals received either BK (25 μg) or BK with CTOP (10 μg; BK+CTOP) 15 min prior to the co-injection of PGE2 (0.3 μg) and DAMGO (8 μg; GO+P), DPDPE (20 μg; D+P), or vehicle (V+P). PWLs were measured at 5 min intervals for at least 20 min after the last injection. Baseline PWLs were 10.27 ± 0.56 sec. Data are represented as mean ± SEM PWL after the last injection of 5–9 animals per group. ***, p<0.001 upper three groups on graph vs. lower three groups on graph.