

Abstract
The phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway is highly conserved throughout evolution and regulates cell size and survival and cell cycle progression. It regulates the latter by stimulating procession through G1 and the G1/S phase transition. Entry into S phase requires an abundant supply of purine nucleotides, but the effect of the PI3K/Akt pathway on purine synthesis has not been studied. We now show that the PI3K/Akt cassette regulates both de novo and salvage purine nucleotide synthesis in insulin-responsive mouse mesenchymal cells. We found that serum and insulin stimulated de novo purine synthesis in serum-starved cells largely through PI3K/Akt signaling, and pharmacologic and genetic inhibition of PI3K/Akt reduced de novo synthesis by 75% in logarithmically growing cells. PI3K/Akt regulated early steps of de novo synthesis by modulating phosphoribosylpyrophosphate production by the non-oxidative pentose phosphate pathway and late steps by modulating activity of the bifunctional enzyme aminoimidazole-carboxamide ribonucleotide transformylase IMP cyclohydrolase, an enzyme not previously known to be regulated. The effects of PI3K/Akt on purine nucleotide salvage were likely through regulating phosphoribosylpyrophosphate availability. These studies define a new mechanism whereby the PI3K/Akt cassette functions as a master regulator of cellular metabolism and a key player in oncogenesis.
Insulin and a variety of other growth factors activate the PI3K3/Akt (protein kinase B) pathway. Activated Akt regulates several intracellular processes including protein synthesis, glucose metabolism, and cell cycle progression (1, 2). Frequently regulation occurs at more than one step. (i) Akt increases protein synthesis by activating mTOR, which regulates the activities of S6 kinase-1 and 4E-BP1, two translational regulators (3). (ii) Akt regulates glucose metabolism by inducing translocation of glucose transporters to the cell surface, by activating hexokinase, and by inhibiting glycogen synthase kinase-3 (4). (iii) Akt stimulates the cell cycle by phosphorylating, and thereby inhibiting, the cyclin-dependent kinase inhibitors p21Cip/WAF1 and p27Kip1 and the FOXO transcription factors and through phosphorylation of glycogen synthase kinase-3, which regulates the G1 cyclins, cyclins D and E (2, 5).
The de novo synthesis of purines consists of 10 sequential steps, starting with phosphoribosylpyrophosphate (PRPP) and ending with IMP; the latter is converted to AMP or GMP (see Fig. 1). PRPP amidotransferase catalyzes the first committed step of the pathway (see Fig. 1, reaction 2) and is subject to feedback inhibition by purine nucleotides; therefore, it has been considered a major point of pathway regulation (6). We showed previously that production of ribose 5-phosphate, the immediate precursor of PRPP and an end product of the pentose phosphate pathway, also contributes to the regulation of purine synthesis (7). Salvage purine nucleotide synthesis involves hypoxanthine phosphoribosyltransferase, which catalyzes the conversion of hypoxanthine or guanine to IMP, a PRPP-requiring reaction (see Fig. 1, reaction 6).
FIGURE 1.

Schematic diagram of de novo and salvage purine nucleotide synthesis with points of regulation by Akt. Glucose (Glu) can be converted to ribose 5-phosphate (Rib-5-P) via either the oxidative or non-oxidative pentose phosphate pathway (PPP). Under normal conditions, most ribose 5-phosphate used in purine synthesis comes from the non-oxidative pathway. Ribose 5-phosphate is converted by PRPP synthetase to PRPP (reaction 1), which is converted to phosphoribosylamine (PRA) by PRPP amidotransferase, the first committed step of the de novo pathway (reaction 2). Glycine combines with phosphoribosylamine to produce glycinamide ribonucleotide (GAR), which is combined with formate to produce FGAR (reaction 3); the latter is converted to AICAR in five subsequent steps (shown as a dashed arrow with the first step, reaction 4, inhibited by azaserine). AICAR and formate are converted by the bifunctional enzyme ATIC (reaction 5) to IMP, which can then be converted to either AMP or GMP. The major routes of AMP and GMP catabolism differ. AMP is converted to hypoxanthine (Hx) via adenosine (Ado) and inosine (Ino), and GMP is converted to guanine (Gua); in both cases ribose 1-phosphate (Rib-1-P) is generated. The ribose 1-phosphate can undergo isomerization to ribose 5-phosphate and then be converted to PRPP, and hypoxanthine and guanine can be converted to IMP by hypoxanthine phosphoribosyltransferase (reaction 6). PRPP production from glucose is shown in green, the de novo purine pathway is shown in pink, purine nucleotide catabolism and the purine salvage pathway are shown in yellow, and points of regulation by PI3K/Akt are shown in red. Glu-6-P, glucose 6-phosphate; 10f-THF, 10-formyltetrahydrofolate.
Although no previous studies have assessed whether the PI3K/Akt cassette regulates purine synthesis, several lines of evidence suggest that this may indeed be the case. First, the intracellular ATP concentration is decreased in mouse embryo fibroblasts that lack Akt1 and Akt2 (8). Second, treating keratinocytes with epidermal or keratinocyte growth factor, both of which activate PI3K and Akt, increases expression of several purine synthetic enzymes (9). Third, the intracellular ATP concentration is 3 times higher in Rat1a fibroblasts that overexpress myristoylated, constitutively active Akt compared with control cells (8, 10). Although the concentrations of ADP and AMP were not reported, their intracellular concentrations are considerably less than ATP, and thus the increased ATP likely arose from increased purine synthesis rather than from increased ADP and AMP phosphorylation. Finally in the IL-3-dependent hematopoietic cell line FL5.12, activity of the oxidative pentose phosphate pathway is higher in cells expressing myristoylated Akt than in control cells and does not decrease on removing IL-3 as occurs in control cells (4). Similarly increased pentose phosphate pathway activity on antigen receptor cross-linking of B lymphocytes is attenuated by LY294002, a PI3K inhibitor (11). Thus, it seemed possible that PI3K/Akt regulate purine synthesis.
EXPERIMENTAL PROCEDURES
Materials—Antibodies against Akt and phospho-Akt were from Cell Signaling Technology (Danvers, MA), and an anti-tubulin antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). An expression vector encoding human Akt1 with a myristoylation signal sequence was from J. R. Woodgett (Mount Sinai Hospital, Toronto, Canada) and human ATIC was produced as described previously (12). LY294002, LY303511, and wortmannin were from Calbiochem; 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICA-riboside) and AICA-riboside monophosphate (AICAR) were from Sigma-Aldrich; 5-formyltetrahydrofolate was from Schircks Laboratories (Jona, Switzerland); and AG 50 resin was from Bio-Rad. LY294002 and LY303511 were dissolved in dimethyl sulfoxide (DMSO) and when added to cells yielded a final DMSO concentration of 0.2%; cells not treated with either drug also received 0.2% DMSO, referred to as vehicle. [14C]Formate, [8-14C]adenine, [8-14C]hypoxanthine, [1-14C]glucose, and [6-14C]glucose were from Moravek (Brea, CA).
Cell Culture and DNA Transfection—C2C12 mouse mesenchymal cells were from the American Tissue Culture Collection (ATCC), and tuberin-deficient mouse embryo fibroblasts were provided by D. J. Kwiatkowski (13); both cell types were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Human erythroleukemia (HEL) cells from the ATCC were cultured in RPMI 1640 medium supplemented with 10% FBS. In some experiments, cells were transfected 48 h prior to use with a cDNA plasmid or siRNA using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol.
To reduce endogenous Akt1, two siRNAs targeting different Akt1 sequences were used: Akt1 siRNA1 was from Dharmacon, and Akt1 siRNA2 had the sequence UGCCCUUCUACAACCAGGA. An siRNA targeting green fluorescent protein was used as a control.
Measurement of Rates of de Novo Purine Synthesis—Rates of de novo purine synthesis were measured as described previously by following [14C]formate incorporation into all cellular purine nucleotides, i.e. the soluble purine nucleotide pool as well as purines incorporated into newly synthesized DNA and RNA (14). Briefly cells were plated in 12-well culture dishes at 5 × 105 cells/well in DMEM supplemented with 10% FBS. About 18 h later, the cells were washed with phosphate-buffered saline, placed in 1 ml of fresh DMEM supplemented with 10% dialyzed FBS, and incubated for 2 h with 0.2% DMSO, 20 μm LY294002, 20 μm LY303511, or 100 nm wortmannin. In experiments where cells were serum-starved, they were incubated for 14 h in DMEM containing 0.1% FBS; some cells received either 10% FBS (serum-stimulated) or 10 nm insulin for 2 h in the absence or presence of 20 μm LY294002. After the 2-h incubation with drugs, [14C]formate (10 μCi; specific activity, 57 mCi/mmol) was added for 2 h, and then the cells were extracted in situ in 0.4 n perchloric acid. Cell lysates were boiled for 70 min to release purine bases from DNA, RNA, and purine nucleotides, and then the samples were cooled on ice and centrifuged for 5 min at 800 × g. The clarified lysates were loaded onto 0.5 × 5-cm AG 50 columns, which were washed with 0.1 n HCl to remove unreacted [14C]formate; purine bases were eluted from the columns in 6 n HCl. Radioactivity in the column eluates was measured by liquid scintillation counting, and the data are expressed as [14C]formate incorporation into purines/h/106 cells. As shown in supplemental Fig. S1, the assay was linear with time to at least 3 h. In addition, the assay was linear with cell number to at least 1 × 106 cells/well, and all subsequent assays using radioactive labels were linear with time and cell number.
To measure [14C]formate incorporation into adenine and guanine separately, the AG 50 column eluates were dried under vacuum, resuspended in 50 μl of water, and spotted on cellulose acetate thin layer chromatography (TLC) plates. The plates were developed in butanol/acetonitrile/water (65:20:20), spots corresponding to adenine and guanine were cut out, and radioactivity was measured by liquid scintillation counting.
In some experiments, de novo purine synthesis was measured following glycine incorporation into purines (15). Briefly [14C]glycine (10 μCi; specific activity, 54 mCi/mmol) was added to cells instead of [14C]formate, 2 h later the cells were lysed in perchloric acid, and the lysates were boiled as described above. After cooling the lysates on ice, KHCO3 was added to neutralize the perchloric acid, and insoluble potassium perchlorate was removed by centrifugation. The supernatants were dried, and adenine and guanine in the samples were separated from unreacted [14C]glycine by TLC as described above. The data are expressed as [14C]glycine incorporation into purines/h/106 cells.
Measurement of PRPP Availability—Incorporation of [8-14C]adenine into adenine nucleotides by adenine phosphoribosyltransferase requires intracellular PRPP and can be used to measure cellular PRPP availability (7). Cells were plated as described for measuring de novo purine synthesis. After a 3-h incubation with the indicated drugs, [8-14C]adenine (0.2 μCi; specific activity, 56 mCi/mmol) was added to the cells, and 30 min later cells were washed with ice-cold phosphate-buffered saline and lysed in water. The cell lysates were placed on 2 × 2-cm squares of diethylaminoethyl cellulose paper (DE-81, Whatman), which were washed four times in 1 mm ammonium formate to remove unincorporated [8-14C]adenine. Radioactivity in adenine nucleotides, which remain bound to the cellulose squares, was measured by liquid scintillation counting.
Measurement of Carbon Flow through the Pentose Phosphate Pathway—Carbon flow through the non-oxidative branch of the pentose phosphate pathway was measured by following [6-14C]glucose incorporation into ATP as described previously (16). Briefly C2C12 cells were incubated as described for measuring rates of de novo purine synthesis except [6-14C]glucose (10 μCi; specific activity, 45 mCi/mmol) replaced the formate label. The cells were extracted in 0.4 n perchloric acid, the extracts were neutralized with KHCO3, and the extracts were fractionated by high performance liquid chromatography (HPLC) on a strong anion exchange column eluted with a linear gradient from 10 mm NH4PO4, pH 4.0 to 480 mm NH4PO4, pH 4.0 over 75 min. Fractions containing ATP were collected, and radioactivity was quantified by liquid scintillation counting. Although this method potentially includes carbon flow through the oxidative branch of the pentose phosphate pathway, it predominantly measures flow through the non-oxidative branch because the latter provides the majority of ribose 5-phosphate used in purine synthesis (16–19).
Carbon flow through the oxidative branch of the pentose phosphate pathway was measured by following CO2 release from [1-14C]glucose as described previously (16). Briefly 1 × 106 HEL cells were incubated in 16 × 100-mm glass tubes containing 1 ml of RPMI 1640 medium supplemented with 25 mm HEPES and 10% dialyzed FBS. The tubes were capped tightly with a rubber septum that held a plastic center well (Kontes Glass Co.) containing a fluted piece of Whatman No. 1 filter paper saturated with 1 n NaOH. After a 2-h equilibration period with or without 20 μm LY294002 or LY303511, 10 μCi of [1-14C]glucose (specific activity, 55 mCi/mmol) was added to the cells by piercing a Hamilton syringe through the rubber septum. At the end of the incubation, the center well was transferred to a scintillation vial, and radioactivity trapped in the NaOH was measured by liquid scintillation counting. Because these experiments must be performed in a sealed system, they were performed on suspension (HEL) cells in capped tubes rather than on surface-adherent cells.
Measurement of PRPP Synthetase and ATIC Activity—Cells incubated for 3 h in 0.2% DMSO or 20 μm LY294002 were extracted by three 10-s bursts of sonication on ice, and the extracts were centrifuged for 10 min at 10,000 × g. For measuring PRPP synthetase activity, the extract buffer and assay conditions were as described by Torres et al. (20). For measuring ATIC activity, cells were extracted in 50 mm Tris-HCl, pH 7.4, 25 mm KCl, and 5 mm β-mercaptoethanol. The assay contained 10-formyltetrahydrofolate synthesized from 5-formyltetrahydrofolate as described by Rabinowitz (21) and was performed as described by Mueller and Benkovic (22) with the following changes. (i) 1 mm β-glycerophosphate and 1 mm sodium pyrophosphate were included to inhibit nonspecific phosphatases, and 2 mm α,β-methylene adenosine diphosphate (AOPCP) was included to inhibit 5′-nucleotidase activity (23). (ii) The IMP product was separated from the AICAR substrate (and AOPCP) by HPLC on a C18 reverse phase column eluted with a linear gradient from 20 mm K2PO4, pH 5.5 to 60% aqueous methanol. The assay measures both AICAR transformylase and IMP cyclohydrolase activity. Protein in the cell extracts was measured by the method of Bradford (24), and enzyme activities were measured at two time points.
Measurement of Intracellular Purine Nucleotides—Intracellular purine nucleotides were measured as described previously (14). Briefly about 10 × 106 C2C12 cells were extracted rapidly in ice-cold 0.4 n perchloric acid, precipitated macromolecules were removed by centrifugation, and the lysates were neutralized with KHCO3. Purine nucleotides in the samples were analyzed by HPLC on a strong anion exchange column using the same system as described for following [6-14C]glucose incorporation into ATP.
Measurement of the Early and Late Steps of the de Novo Purine Synthesis Pathway—Rates of the early steps of the de novo purine synthesis pathway were measured by following [14C]formate incorporation into formylglycinamide ribonucleotide (FGAR) as described previously (14). Briefly cells were incubated with a 10 μm concentration of the glutamine analog azaserine; this azaserine concentration inhibits formylglycinamidine ribonucleotide synthase (Fig. 1, reaction 4) but has minimal effect on other glutamine-requiring processes, such as protein synthesis (14, 25, 26). After a 45-min incubation with azaserine, [14C]formate was added, and 2 h later cells were washed with ice-cold phosphate-buffered saline and extracted in acetonitrile/water (50:50). After centrifugation to remove precipitated proteins, supernatants were spotted on cellulose acetate TLC plates and developed in a butanol/acetone/water/acetic acid/ammonium hydroxide (35:35:22.5:15:2.5) system. The plates were exposed to x-ray film, and FGAR was identified on the autoradiographs based on its Rf value in the TLC system and its presence only in azaserine-treated cells (14, 27). Corresponding areas on the TLC plates were cut out, and radioactivity in FGAR was measured by liquid scintillation counting.
Rates of the last two steps of the de novo purine synthesis pathway were measured as described for measuring total de novo purine synthesis except 10 μm azaserine and 200 μm AICA-riboside were added to the cells (14, 26). The azaserine was present to inhibit the early steps of the de novo pathway, thereby making purine synthesis dependent on the exogenously added AICA-riboside.
Measurement of Purine Synthesis by the Salvage Pathway—Purine synthesis by the salvage pathway was assessed by measuring [8-14C]hypoxanthine incorporation into purine nucleotides as described previously (14). The assay was the same as described for measuring de novo purine synthesis except the cells were incubated with 10 μCi of [8-14C]hypoxanthine (specific activity, 47 mCi/mmol).
Western Blots—Western blots were performed as described previously using chemiluminescence detection (28). Cells were extracted in situ in hot SDS sample buffer and applied to 8% polyacrylamide gels. Resolved proteins were transferred to polyvinylidene difluoride membranes, which were incubated with the appropriate primary antibody and a horseradish peroxidase-tagged secondary antibody.
Data Presentation and Analysis—All data presented in bar and line graphs are the mean ± S.E. of at least three independent experiments performed in duplicate. Statistical analyses were performed using Prism 5 software (GraphPad, Carlsbad, CA). Student's t test was used for paired comparisons, and a one-way analysis of variance with a Dunnett's or Bonferroni post-test analysis was used for multiple comparisons with the control group or comparisons across experimental conditions, respectively.
RESULTS
Effect of Akt Inhibition on de Novo Purine Synthesis—To determine whether the PI3K/Akt cassette regulates de novo purine synthesis, we treated C2C12 cells with LY294002 for 2 h and found that the drug reduced [14C]formate incorporation into purines by >75% over the ensuing 2 h (Fig. 2a). Rates of purine synthesis were reduced during the earliest time interval that could be measured, i.e. the first 30 min after adding the formate label, and remained reduced for at least 3 h (supplemental Fig. S1; all subsequent measurements with the formate label were performed at 2 h, well within the linear range of the assay). LY303511, which is structurally similar to LY294002 but does not inhibit PI3K (29), had no significant effect on purine synthesis (Fig. 2a), whereas wortmannin, another PI3K inhibitor, also severely reduced purine synthesis (data not shown). Starving cells for serum for 15 h reduced purine synthesis, and adding back serum or insulin returned purine synthesis rates toward those of control, non-starved cells; under all three conditions, LY294002 significantly inhibited rates of purine synthesis (Fig. 2a). We confirmed that LY294002 inhibited Akt activation by assessing Akt phosphorylation on serine 473 and threonine 308 (Fig. 2a). LY303511 had no effect on Akt phosphorylation, and serum starvation partially reduced Akt phosphorylation (Fig. 2a). The marked reduction of purine synthesis in serum-starved cells in the presence of some residual Akt activity suggests that serum regulates purine synthesis in part through non-PI3K/Akt mechanisms; further reduction in purine synthesis by LY294002 in serum-starved cells is consistent with this hypothesis. However, that LY294002 largely prevented serum and insulin from restoring purine synthesis in serum-starved cells indicates that PI3K/Akt signaling is critical to serum and insulin regulation of purine synthesis.
FIGURE 2.

Effect of PI3K inhibitors or Akt1 siRNA on de novo purine synthesis. a–c, C2C12 cells were incubated for 2 h with either 0.2% DMSO (white bars, No add), 20 μm LY294002 in DMSO (LY2; black bars), or 20 μm LY303511 in DMSO (LY3; gray bars); some cells received 100μm folic acid (FA; b, vertical striped bar) or 100 μm 5-formyltetrahydrofolate (5-fTHF; c, horizontal striped bar). In a, serum-starved cells were incubated for 14 h in DMEM containing 0.1% FBS and then received either phosphate-buffered saline (Serum-starved), 10% serum (Serum-stimulated), or 10 nm insulin at the same time as receiving LY294002 or LY303511. After the 2-h incubation with drugs, rates of de novo purine synthesis were measured for 2 h by incubating cells with either 10 μCi of [14C]formate (a and b) or 10 μCi of [14C]glycine (c). Below the bar graph of a is a Western blot for total Akt and Akt phosphorylated on Ser-473 (pSer473) and Thr-308 (pThr308). Experimental conditions for the blots are the same as for the corresponding bars: lane 1, control; lane 2, control plus LY294002; lane 3, control plus LY303511; lane 4, serum-starved; lane 5, serum-starved plus LY294002; lane 6, serum-starved plus serum; lane 7, serum-starved plus serum and LY294002; lane 8, serum-starved plus insulin; and lane 9, serum-starved plus insulin and LY294002. d, C2C12 cells were transfected with either a control siRNA directed against green fluorescent protein (GFP) or one of two different Akt1 siRNAs (Akt1 siRNA1 in the main figure and Akt1 siRNA2 in the inset). In the main figure, cells additionally were transfected with either vector DNA or a myristoylated (Myr) human Akt1 cDNA resistant to the effects of the Akt1 siRNA. After 48 h, rates of de novo purine synthesis were measured using [14C]formate as described in a and b. Below the bar graphs are Western blots for Akt and tubulin; experimental conditions for the blots are the same as for the corresponding bars. e, Tsc2-deficient (white bar) and matched (black bar) mouse embryo fibroblasts were incubated with 0.2% DMSO (No add), 20μm LY294002, or 20μm LY303511 for 2 h, and then rates of purine synthesis were measured as described in a. In a–c and e, asterisks indicate p < 0.05 when comparing LY294002-treated cells with non-LY294002-treated cells. Asterisks in d indicate p < 0.05 when comparing cells transfected with Akt1 siRNAs with cells transfected with green fluorescent protein siRNA. Data in the bar graphs are the mean ± S.E. (error bars) of three independent experiments performed in duplicate.
Because formate is incorporated into purines via 10-formyltetrahydrofolate (Fig. 1, reactions 3 and 5), decreased tetrahydrofolate availability would decrease apparent rates of purine synthesis using a [14C]formate label. However, LY294002 reduced purine synthesis to a similar extent in folic acid-supplemented cells as in non-supplemented cells (Fig. 2b). Further evidence that LY294002 directly inhibited purine synthesis was that the drug inhibited [14C]glycine incorporation into purines to a similar extent as [14C]formate incorporation (Fig. 2c). The inactive analog LY303511 had no significant effect on [14C]glycine incorporation into purines, and supplementing cells with 5-formyltetrahydrofolate, which is in rapid equilibrium with 10-formyl-tetrahydrofolate, did not attenuate the effect of LY294002 (Fig. 2c); the latter experiment confirmed that LY294002 was not acting by reducing 10-formyltetrahydrofolate availability.
To determine whether regulation of purine synthesis by the PI3K/Akt cassette occurred distal to PI3K, we reduced Akt1 expression in C2C12 cells using an siRNA approach (none of the commercially available Akt inhibitors significantly reduced Akt activity over the relatively short time period of our experiments). Two different Akt1 siRNAs significantly reduced purine synthesis, whereas a control siRNA had no effect (Fig. 2d; the control siRNA was targeted to green fluorescent protein; rates of purine synthesis with this siRNA were similar to those in untreated cells shown in Fig. 2, a and b). Neither Akt1 siRNA reduced purine synthesis to the same extent as LY294002, which is likely related to the siRNAs only partially reducing Akt protein and Akt activation (Fig. 2d, Western blots; only Akt protein is shown). Expressing myristoylated human Akt1 in C2C12 cells treated with Akt1 siRNA1 returned rates of purine synthesis to those found in cells transfected with control siRNA (Fig. 2d; these experiments could not be performed with Akt1 siRNA2 because the latter was directed to a sequence common to the mouse and human genes). Expressing myristoylated Akt1 in otherwise untreated cells increased rates of purine synthesis, but the increase did not reach statistical significance. Because LY294002 more effectively inhibited Akt activation than Akt1 siRNAs, subsequent experiments were performed with LY294002 using LY303511 as a negative control.
To examine whether PI3K/Akt regulate purine synthesis via the mTOR/S6 kinase pathway, we performed two separate sets of experiments. First, we studied tuberin (Tsc2)-deficient mouse embryo fibroblasts; tuberin regulates mTOR activity by serving as a Rheb GTPase and is inhibited when phosphorylated by Akt (13). We found that LY294002 reduced purine synthesis to the same extent in tuberin-deficient cells as in sister cells containing Tsc2 (Fig. 2e). Tsc2-deficient cells are resistant to the effects of Akt on the mTOR/S6 kinase pathway and demonstrate constitutive S6 kinase activation (13). Second, we showed that the mTOR inhibitor rapamycin only modestly reduced total purine synthesis in C2C12 cells and had no effect on the early or late steps of de novo purine synthesis (shown later in Fig. 4, a and b). Thus, Akt appeared to regulate purine synthesis independently of the mTOR/S6 kinase pathway.
FIGURE 4.

Effect of PI3K inhibitors on early and late steps of de novo purine synthesis. C2C12 cells were treated for 2 h with 0.2% DMSO (Vehicle; white bars), 20 μm LY294002 (LY2; black bars), 20 μm LY303511 (LY3; gray bars), or 10 nm rapamycin (diagonal striped bars). During the last 45 min of the 2-h incubation, the cells received 10 μm azaserine (a and b) and 100 μm AICA-riboside (b). [14C]Formate incorporation into FGAR (a) and purine nucleotides (b) was then measured over 2 h. Asterisks indicate p < 0.05 when comparing LY294002-treated cells with vehicle-treated cells. Data are the mean ± S.E. (error bars) of three independent experiments performed in duplicate.
Effect of Akt Inhibition on PRPP Availability—Because ribose 5-phosphate and PRPP production can be limiting for purine synthesis (7, 30) and expression of a constitutively active Akt increases carbon flow through the oxidative pentose phosphate pathway (4), we hypothesized that PI3K/Akt could regulate purine synthesis by regulating PRPP availability. We measured PRPP availability by incubating cells with [14C]adenine, which is converted to AMP by adenine phosphoribosyltransferase, a reaction requiring PRPP (7). LY294002 decreased PRPP availability in C2C12 cells by 31%, whereas LY303511 had no effect (Fig. 3a). Serum starvation reduced PRPP availability albeit not to the extent it reduced purine synthesis, and replenishing cells with serum or insulin increased PRPP toward control levels (Fig. 3a). LY294002 further reduced PRPP availability under serum-starved conditions and largely prevented the effects of restoring serum or insulin (Fig. 3a). Treating cells with rapamycin had no significant effect on PRPP availability under any of the four conditions tested. These data suggest that some of the effects of Akt and serum on purine synthesis are from a decrease in PRPP availability.
FIGURE 3.

Effect of PI3K inhibitors on PRPP availability and the pentose phosphate pathway. a and b, C2C12 cells were treated for 3 h with either 0.2% DMSO (Vehicle; white bars in a and open triangles in b), 20 μm LY294002 (black bars and filled triangles), or 20 μm LY303511 (gray bar). Cells were serum-starved, serum-stimulated, and treated with insulin as described in the legend to Fig. 2. Cellular PRPP availability was assessed by measuring [8-14C]adenine incorporation into adenine nucleotides over 30 min. The culture medium contained 25 mm glucose in a (DMEM with high glucose) and the indicated glucose concentrations in b. c, C2C12 cells were incubated for 2 h with 0.2% DMSO (Vehicle; white bar), 20 μm LY294002 (LY2; black bar), or 20 μm LY303511 (LY3; white bar), and then [6-14C]glucose was added to the cells for 2 h. The cells were extracted, the extracts were fractionated by high performance liquid chromatography, and radioactivity in ATP-containing fractions was measured. d, HEL cells were incubated for 2 h with 0.2% DMSO (Vehicle; white bar), 20 μm LY294002 (black bar), or 20μm LY303511 (white bar) in a tightly capped tube fitted with a center well containing NaOH. [1-14C]Glucose was added, and CO2 was collected in the NaOH over 2 h. Asterisks in a and d indicate p < 0.05 when comparing LY294002-treated cells with vehicle-treated cells. Data in the bar graphs are the mean ± S.E. (error bars) of three independent experiments performed in duplicate.
To investigate the basis for decreased PRPP availability in LY294002-treated C2C12 cells, we performed three separate experiments. First, we measured PRPP availability at several glucose concentrations and found a similar reduction in PRPP at each concentration (Fig. 3b). These data suggest that decreased PRPP in LY294002-treated cells was not from regulation of glucose transport because a more profound effect on PRPP availability would have been expected at lower glucose concentrations. Second, we measured carbon flow through the pentose phosphate pathway. We and others have shown that the non-oxidative branch of the pathway supplies most of the ribose 5-phosphate used in purine nucleotide synthesis (16–19), and we found that LY294002 reduced carbon flow through this pathway by 39% (Fig. 3c). LY294002 had no effect on carbon flow through the oxidative branch of the pathway in HEL cells, whereas it inhibited purine synthesis to a similar extent in these cells as in C2C12 cells (Fig. 3d). The lack of an effect of LY294002 on carbon flow through the oxidative pentose phosphate pathway is further evidence that the effect of Akt on PRPP production was not from decreased glucose transport or glucose phosphorylation. Third, we measured the activity of PRPP synthetase, a multimeric enzyme whose activity is regulated by the intracellular phosphate concentration and PRPP synthetase-associated protein PAP 39 (31). We found that PRPP synthetase activity was the same in control and LY294002-treated cells: 2.2 ± 0.3 and 2.4 ± 0.4 nmol/min/mg of protein, respectively (mean ± S.D. of three independent experiments). Thus, reduced flow through the non-oxidative pentose phosphate pathway appeared to be the basis for reduced PRPP availability in Akt-inhibited cells.
Effect of Akt Inhibition on Early and Late Steps of de Novo Purine Synthesis—The early steps of de novo purine synthesis can be measured by following [14C]formate incorporation into FGAR in the presence of 10 μm azaserine; at this concentration, azaserine inhibits formylglycinamidine ribonucleotide synthase (Fig. 1, reaction 4) but has minimal effect on other glutamine-requiring processes (14, 25, 26). LY294002 decreased FGAR synthesis by ∼40% in C2C12 cells, whereas LY303511 and rapamycin had no significant effect (Fig. 4a). Because PRPP amidotransferase, which catalyzes the first committed step of the de novo pathway (Fig. 1, reaction 2), is inhibited by purine nucleotides, it seemed possible that Akt could inhibit FGAR synthesis by altering nucleotide levels. However, LY294002 had no effect on the intracellular concentrations of ATP, ADP, AMP, GTP, GDP, or GMP when present for up to 9 h (supplemental Table S1; only the data at 3 h are shown, but similar results were seen at 6 and 9 h of LY294002 treatment). The intracellular concentrations of purine nucleotides did decrease after 15 h of LY24002 exposure at which time they fell by ∼50% (supplemental Fig. S2). Because LY294002 decreased FGAR synthesis to a similar extent as it reduced PRPP availability, these data suggest that Akt inhibits the early steps of purine synthesis by decreasing PRPP availability.
The last two steps of de novo purine synthesis, which are catalyzed by the bifunctional enzyme ATIC (Fig. 1, reaction 5), can be measured by following [14C]formate incorporation into purine nucleotides in the presence of azaserine and AICA-riboside. Azaserine renders the assay dependent on exogenously supplied AICA-riboside, which is converted to AICAR by adenosine kinase. LY294002 inhibited AICAR-dependent purine synthesis in C2C12 cells by 50%, whereas LY303511 and rapamycin had no effect (Fig. 4b).
To study the mechanism of the decrease in AICAR-dependent purine synthesis, we measured ATIC activity in cell extracts and observed a 20% decrease in enzyme activity in C2C12 cells that had been treated with LY294002: 3.71 ± 0.38 versus 2.94 ± 0.33 nmol/min/mg of protein in control and LY294002-treated cells, respectively (mean ± S.E. of five independent experiments; p = 0.0015 for the difference between control and LY294002-treated cells). Enzyme activity was unchanged in extracts of cells treated with LY303511, and adding LY294002 to either purified ATIC or cell extracts had no effect. In other experiments, expressing human ATIC in C2C12 cells increased rates of de novo purine synthesis, indicating that endogenous ATIC was limiting to purine synthesis; Sidi and Mitchell (32) reached a similar conclusion after finding increased AICAR (ZMP), and ZDP, and ZTP in erythrocytes from Lesch-Nyhan patients.
Effect of Akt Inhibition on Adenine and Guanine Nucleotide Synthesis—IMP, the product of the de novo purine pathway, is converted to either AMP or GMP (Fig. 1). We found that LY294002 reduced adenylate synthesis by 50% and guanylate synthesis by 75%, whereas LY303511 had no effect on synthesis of either nucleotide (Fig. 5, a and b). Similar results were found for AICA-riboside-dependent AMP and GMP synthesis, eliminating the effects of Akt inhibition on PRPP availability and providing a more focused evaluation of AMP and GMP synthesis. We previously found preferential reduction in guanylate synthesis during other states of reduced purine nucleotide synthesis (14). Thus, it is possible that under states of reduced IMP synthesis a shift to adenylates occurs, constituting a protective cellular mechanism to preserve ATP.
FIGURE 5.

Effect of PI3K inhibitor on adenine and guanine nucleotide synthesis and on purine nucleotide synthesis by the salvage pathway. C2C12 cells were treated for 2 h with either 0.2% DMSO (Vehicle; white bars), 20 μm LY294002 (LY2; black bars), or 20 μm LY303511 (LY3; gray bars). [14C]Formate incorporation into adenine (a) and guanine nucleotides (b) or [8-14C]hypoxanthine incorporation into purine nucleotides (c) was then measured over a 2-h period. Asterisks indicate p < 0.05 when comparing LY294002-treated cells with vehicle-treated cells. Data are the mean ± S.E. (error bars) of three independent experiments performed in duplicate.
Effect of Akt Inhibition on Salvage Purine Nucleotide Synthesis—To determine whether the PI3K/Akt cassette regulates purine nucleotide synthesis from the salvage pathway (Fig. 1, reaction 6), we measured [8-14C]hypoxanthine incorporation into purine nucleotides. This assay assesses the activity of hypoxanthine phosphoribosyltransferase, the major enzyme of purine salvage. LY294002 reduced salvage synthesis by almost 50%, whereas LY303511 had no effect (Fig. 5c). The basis for the decrease in salvage pathway synthesis was likely from decreased PRPP availability because the salvage pathway requires this substrate, and the reduction in salvage synthesis was similar to the reduction in PRPP availability.
DISCUSSION
We have found that the PI3K/Akt cassette regulates the synthesis of purine nucleotides via both the de novo and salvage pathways. Akt is known to positively regulate glucose, protein, and lipid metabolism, and we now show that it regulates the synthesis of yet another major class of macromolecules. The regulation of purine synthesis by Akt is consistent with its positive effects on cell growth and proliferation, which require a concomitant increase in ATP and GTP production. We found that Akt regulates de novo purine synthesis at two separate steps: PRPP production and ATIC-catalyzed conversion of AICAR to IMP. This dual effect is analogous to Akt regulation of glucose metabolism and protein synthesis at several levels (1, 33). In addition to being required for purine synthesis, PRPP is required for pyrimidine and pyridine (NAD/NADP) synthesis. Thus, by regulating PRPP production, Akt can regulate multiple cellular processes, contributing to its role as a master cellular regulator (2).
ATIC would seem an unlikely candidate for regulation because it is the last enzyme in the de novo pathway. However, because ATIC activity appears to be limiting to purine synthesis, small changes in enzyme activity could have profound effects on purine synthesis; thus, the 20% decrease in ATIC activity we found in Akt-inhibited cells could be the basis for the 50% decrease in AICAR-dependent purine synthesis. Moreover AICAR would be expected to accumulate in cells with decreased ATIC activity as occurs in patients with genetic ATIC deficiency (34). AICAR is a potent activator of AMP-activated protein kinase (35); the latter inhibits anabolic and activates catabolic pathways, decreasing protein, carbohydrate, and lipid synthesis and inhibiting cell growth and proliferation (35, 36). Thus, decreased Akt activation during growth factor deprivation could both directly (through decreased phosphorylation of Tsc2 and other substrates) and indirectly (through AMP-activated protein kinase activation) reduce macromolecule synthesis, and the two mechanisms could have a potentially synergistic effect.
During catabolism of purine nucleotides, GMP is degraded to guanine, and AMP is degraded to hypoxanthine via inosine (Fig. 1). Guanine and hypoxanthine are converted to IMP by hypoxanthine phosphoribosyltransferase (Fig. 1, reaction 6), and in the presence of an adequate supply of these purine bases, cells derive purine nucleotides via this salvage pathway (37). Because hypoxanthine phosphoribosyltransferase-mediated salvage activity was significantly reduced in Akt-inhibited cells, purine bases will be lost from the cell under states of low Akt activation. When combined with low rates of de novo purine synthesis, the cell will become depleted of purine nucleotides. Thus, the combination of a purine synthesis inhibitor, such as methotrexate, with a PI3K or Akt inhibitor could potentially be beneficial in treating cancer and other proliferative diseases requiring an abundant supply of purines.
We measured PRPP availability using [14C]adenine because the
Km of adenine phosphoribosyltransferase for PRPP is about
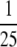 that of the
Km of hypoxanthine phosphoribosyltransferase for PRPP
(38,
39). Thus, assessing adenine
incorporation into purine nucleotides should be more sensitive to changes in
the intracellular PRPP concentration than measuring hypoxanthine or guanine
incorporation into nucleotides. Accurate assessment of the purine salvage
pathway required measuring hypoxanthine incorporation into purine nucleotides
because hypoxanthine phosphoribosyltransferase is the major enzyme of purine
salvage.
that of the
Km of hypoxanthine phosphoribosyltransferase for PRPP
(38,
39). Thus, assessing adenine
incorporation into purine nucleotides should be more sensitive to changes in
the intracellular PRPP concentration than measuring hypoxanthine or guanine
incorporation into nucleotides. Accurate assessment of the purine salvage
pathway required measuring hypoxanthine incorporation into purine nucleotides
because hypoxanthine phosphoribosyltransferase is the major enzyme of purine
salvage.
In conclusion, the PI3K/Akt cassette regulates de novo and salvage purine nucleotide synthesis by regulating PRPP availability and ATIC activity. These newly discovered activities of the PI3K/Akt cassette can profoundly influence cellular metabolism and cell growth and proliferation.
Supplementary Material
Acknowledgments
We thank J. R. Woodgett and D. J. Kwiatkowski for the Akt1 plasmid with a myristoylation signal sequence and Tsc2-deficient mouse embryo fibroblasts, respectively.
This work was supported, in whole or in part, by National Institutes of Health Grants NS58030 (to G. R. B.) and AI42266 (to I. A. W. and S. C.) and Training Grant DK69263 (to W. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2 and Table S1.
Footnotes
The abbreviations used are: PI3K, phosphatidylinositol 3-kinase; AICA-riboside, 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside; AICAR, AICA-riboside monophosphate, also known as ZMP; ATIC, AICAR transformylase IMP cyclohydrolase; DMEM, Dulbecco's modified Eagle's medium; DMSO, dimethyl sulfoxide; FBS, fetal bovine serum; FGAR, formylglycinamide ribonucleotide; HPLC, high performance liquid chromatography; HEL, human erythroleukemia; PRPP, phosphoribosylpyrophosphate; TLC, thin layer chromatography; mTOR, mammalian target of rapamycin; siRNA, small interfering RNA; AOPCP, α,β-methylene adenosine diphosphate; ZDP, AICA-riboside diphosphate; ZTP, AICA-riboside triphosphate.
References
- 1.Kandel, E. S., and Hay, N. (1999) Exp. Cell Res. 253 210–229 [DOI] [PubMed] [Google Scholar]
- 2.Manning, B. D., and Cantley, L. C. (2007) Cell 129 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isotani, S., Hara, K., Tokunaga, C., Inoue, H., Avruch, J., and Yonezawa, K. (1999) J. Biol. Chem. 274 34493–34498 [DOI] [PubMed] [Google Scholar]
- 4.Rathmell, J. C., Fox, C. J., Plas, D. R., Hammerman, P. S., Cinalli, R. M., and Thompson, C. B. (2003) Mol. Cell. Biol. 23 7315–7328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang, J., and Slingerland, J. M. (2003) Cell Cycle 2 339–345 [PubMed] [Google Scholar]
- 6.Holmes, E. W., Wyngaarden, J. B., and Kelley, W. N. (1973) J. Biol. Chem. 248 6035–6040 [PubMed] [Google Scholar]
- 7.Pilz, R. B., Willis, R. C., and Boss, G. R. (1984) J. Biol. Chem. 259 2927–2935 [PubMed] [Google Scholar]
- 8.Hahn-Windgassen, A., Nogueira, V., Chen, C. C., Skeen, J. E., Sonenberg, N., and Hay, N. (2005) J. Biol. Chem. 280 32081–32089 [DOI] [PubMed] [Google Scholar]
- 9.Gassmann, M. G., Stanzel, A., and Werner, S. (1999) Oncogene 18 6667–6676 [DOI] [PubMed] [Google Scholar]
- 10.Gottlob, K., Majewski, N., Kennedy, S., Kandel, E., Robey, R. B., and Hay, N. (2001) Genes Dev. 15 1406–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doughty, C. A., Bleiman, B. F., Wagner, D. J., Dufort, F. J., Mataraza, J. M., Roberts, M. F., and Chiles, T. C. (2006) Blood 107 4458–4465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolan, D. W., Greasley, S. E., Beardsley, G. P., and Wilson, I. A. (2002) Biochemistry 41 15505–15513 [DOI] [PubMed] [Google Scholar]
- 13.Zhang, H., Cicchetti, G., Onda, H., Koon, H. B., Asrican, K., Bajraszewski, N., Vazquez, F., Carpenter, C. L., and Kwiatkowski, D. J. (2003) J. Clin. Investig. 112 1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boss, G. R., and Erbe, R. W. (1982) J. Biol. Chem. 257 4242–4247 [PubMed] [Google Scholar]
- 15.Martin, D. W., Jr., and Owen, N. T. (1972) J. Biol. Chem. 247 5477–5485 [PubMed] [Google Scholar]
- 16.Boss, G. R., and Pilz, R. B. (1985) J. Biol. Chem. 260 6054–6059 [PubMed] [Google Scholar]
- 17.Raivio, K. O., Lazar, C. S., Krumholz, H. R., and Becker, M. A. (1981) Biochim. Biophys. Acta 678 51–57 [DOI] [PubMed] [Google Scholar]
- 18.Boros, L. G., Puigjaner, J., Cascante, M., Lee, W. N., Brandes, J. L., Bassilian, S., Yusuf, F. I., Williams, R. D., Muscarella, P., Melvin, W. S., and Schirmer, W. J. (1997) Cancer Res. 57 4242–4248 [PubMed] [Google Scholar]
- 19.Boros, L. G., Bassilian, S., Lim, S., and Lee, W. N. (2001) Pancreas 22 1–7 [DOI] [PubMed] [Google Scholar]
- 20.Torres, R. J., Mateos, F. A., Puig, J. G., and Becker, M. A. (1996) Clin. Chim. Acta 245 105–112 [DOI] [PubMed] [Google Scholar]
- 21.Rabinowitz, J. C. (1963) Methods Enzymol. 6 814–815 [Google Scholar]
- 22.Mueller, W. T., and Benkovic, S. J. (1981) Biochemistry 20 337–344 [DOI] [PubMed] [Google Scholar]
- 23.Boss, G. R., Thompson, L. F., Spiegelberg, H. L., Pichler, W. J., and Seegmiller, J. E. (1980) J. Immunol. 125 679–682 [PubMed] [Google Scholar]
- 24.Bradford, M. M. (1976) Anal. Biochem. 72 248–254 [DOI] [PubMed] [Google Scholar]
- 25.Henderson, J. F. (1962) J. Biol. Chem. 237 2631–2635 [PubMed] [Google Scholar]
- 26.Boss, G. R. (1987) Biochem. J. 242 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyle, J. A., Raivio, K. O., Becker, M. A., and Seegmiller, J. E. (1972) Biochim. Biophys. Acta 269 179–183 [DOI] [PubMed] [Google Scholar]
- 28.Scheele, J. S., Pilz, R. B., Quilliam, L. A., and Boss, G. R. (1994) J. Biol. Chem. 269 18599–18606 [PubMed] [Google Scholar]
- 29.Vlahos, C. J., Matter, W. F., Hui, K. Y., and Brown, R. F. (1994) J. Biol. Chem. 269 5241–5248 [PubMed] [Google Scholar]
- 30.Boss, G. R. (1984) J. Biol. Chem. 259 2936–2941 [PubMed] [Google Scholar]
- 31.Becker, M. A. (2001) Prog. Nucleic Acids Res. Mol. Biol. 69 115–148 [DOI] [PubMed] [Google Scholar]
- 32.Sidi, Y., and Mitchell, B. S. (1985) J. Clin. Investig. 76 2416–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coffer, P. J., Jin, J., and Woodgett, J. R. (1998) Biochem. J. 335 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marie, S., Heron, B., Bitoun, P., Timmerman, T., Van Den Berghe, G., and Vincent, M. F. (2004) Am. J. Hum. Genet. 74 1276–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henin, N., Vincent, M. F., Gruber, H. E., and Van Den Berghe, G. (1995) FASEB J. 9 541–546 [DOI] [PubMed] [Google Scholar]
- 36.Hardie, D. G. (2007) Nat. Rev. Mol. Cell Biol. 8 774–785 [DOI] [PubMed] [Google Scholar]
- 37.Hershfield, M. S., and Seegmiller, J. E. (1976) J. Biol. Chem. 251 7348–7354 [PubMed] [Google Scholar]
- 38.Krenitsky, T. A., Papaioannou, R., and Elion, G. B. (1969) J. Biol. Chem. 244 1263–1270 [PubMed] [Google Scholar]
- 39.Henderson, J. F., Miller, H. R., Kelley, W. N., Rosenbloom, F. M., and Seegmiller, J. E. (1968) Can. J. Biochem. 46 703–706 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.