

Abstract
The kinetochore, which consists of DNA sequence elements and structural proteins, is essential for high-fidelity chromosome transmission during cell division. In budding yeast, Sgt1 and Hsp90 help assemble the core kinetochore complex CBF3 by activating the CBF3 components Skp1 and Ctf13. In this study, we show that Sgt1 forms homodimers by performing in vitro and in vivo immunoprecipitation and analytical ultracentrifugation analyses. Analyses of the dimerization of Sgt1 deletion proteins showed that the Skp1-binding domain (amino acids 1–211) contains the Sgt1 homodimerization domain. Also, the Sgt1 mutant proteins that were unable to dimerize also did not bind Skp1, suggesting that Sgt1 dimerization is important for Sgt1-Skp1 binding. Restoring dimerization activity of a dimerization-deficient sgt1 mutant (sgt1-L31P) by using the CENP-B (centromere protein-B) dimerization domain suppressed the temperature sensitivity, the benomyl sensitivity, and the chromosome missegregation phenotype of sgt1-L31P. These results strongly suggest that Sgt1 dimerization is required for kinetochore assembly.
Spindle microtubules are coupled to the centromeric region of the chromosome by a structural protein complex called the kinetochore (1, 2). The kinetochore is thought to generate a signal that arrests cells during mitosis when it is not properly attached to microtubules, thereby preventing aberrant chromosome transmission to the daughter cells, which can lead to tumorigenesis (3, 4). The kinetochore of the budding yeast Saccharomyces cerevisiae has been characterized thoroughly, genetically and biochemically; thus, its molecular structure is the most well detailed to date. More than 70 different proteins comprise the budding yeast kinetochore, and several of those are conserved in mammals (2).
The budding yeast centromere DNA is a 125-bp region that contains three conserved regions, CDEI, CDEII, and CDEIII (5, 6). CDEI is bound by Cbf1 (7–9). CDEIII (25 bp) is essential for centromere function (10) and is the site where CBF3 binds to centromeric DNA. CBF3 contains four proteins: Ndc10, Cep3, Ctf13 (11–18), and Skp1 (17, 18), all of which are essential for viability. Mutations in any of the four CBF3 proteins abolish the ability of CDEIII to bind to CBF3 (19, 20). All of the described kinetochore proteins, except the CDEI-binding Cbf1, localize to kinetochores dependent on the CBF3 complex (2). Therefore, the CBF3 complex is the fundamental structure of the kinetochore, and the mechanism of CBF3 assembly is of major interest.
We previously isolated SGT1, the skp1-4 kinetochore-defective mutant dosage suppressor (21). Sgt1 and Skp1 activate Ctf13; thus, they are required for assembly of the CBF3 complex (21). The molecular chaperone Hsp90 is also required for the formation of the Skp1-Ctf13 complex (22). Sgt1 has two highly conserved motifs that are required for protein-protein interaction, the tetratricopeptide repeat (TPR)2 (21) and the CS (CHORD protein- and Sgt1-specific) motif. We and others (23–26) have found that both domains are important for the interaction with Hsp90. The Sgt1-Hsp90 interaction is required for the assembly of the core kinetochore complex; this interaction is an initial step in kinetochore assembly (24, 26, 27) that is conserved between yeast and humans (28, 29).
In this study, we further characterized the molecular mechanism of this assembly process. We found that Sgt1 forms dimers in vivo, and our results strongly suggest that Sgt1 dimerization is required for kinetochore assembly in budding yeast.
EXPERIMENTAL PROCEDURES
Yeast Strains and Medium—Supplemental Table I lists the genotypes of yeast strains used in this study. The medium for yeast growth and sporulation was described previously (30). Yeast transformation was performed according to the method described by Ito et al. (31). Strains that expressed tagged proteins were generated according to the procedure of Longtine et al. (32). Regions that encoded the Myc tags were inserted at the 3′ end of the endogenous locus.
Plasmid Construction and Primers—Supplemental Table II lists the plasmids used in this study. Details about their construction (33) and primer sequences are available upon request.
Antibodies—Anti-Skp1, anti-Sgt1, and anti-Hsp82 antibodies were described previously (21, 24, 34). Anti-hemagglutinin (HA) and anti-Myc (Roche Applied Science), anti-glutathione S-transferase (GST; Abcam), anti-FLAG (Sigma), and anti-His6 (Qiagen) antibodies were purchased from the indicated sources.
Protein Expression and Immunoprecipitation—Immunoprecipitation using yeast lysates was performed as described previously (24). Expression and purification of His6-Sgt1 and GST-Sgt1 proteins were performed according to the manufacturer's instructions, as described previously (24).
Analytical Ultracentrifugation—Experiments were carried out
in a ProteomeLab XL-I analytical ultracentrifuge with cells containing
sapphire or quartz windows and charcoal-filled Epon double-sector center
pieces (Beckman Coulter, Fullerton, CA). The software SEDNTERP was used to
convert the sedimentation coefficients to standard conditions (20 °C,
water as solvent, and zero concentration protein), and noted as
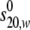 values, as well as for the calculation of
the density and viscosity of the buffer (50 mm Tris, 50
mm NaCl, 1 mm phenylmethylsulfonyl fluoride) and partial
specific volume and molecular mass of the protein
(35). Equilibrium as well as
velocity data were analyzed with SEDFIT and SEDPHAT software. Sedimentation
equilibrium was attained at 24 h at a temperature of 4 °C at increasing
rotor speeds of 16,000–30,000 rpm. Protein concentrations were
1.75–17.0 μm (120 μl), and absorbance distributions
were recorded at 280 nm. Modeling was performed at multiple rotor speeds using
a monomer-dimer self-association model
(36). For the sedimentation
velocity experiments, the loading volume was 360 μl (2.2 mg/ml), and the
protein was centrifuged at 20 °C at 60,000 rpm. Fringe displacement data
at time intervals of 1.0 min were collected with the Rayleigh interference
system for 12 h and analyzed using the models for continuous sedimentation
coefficient distribution (c(s)) as well as the hybrid
continuous/discrete distribution
(37–39).
The latter model was with two discrete species, one with a best fit apparent
molar mass of 41 kDa and an s value of 3.85 S and a second species
constrained to the molar mass of the dimer, 98.4 kDa, with a fixed s
value of 4.70 S.
values, as well as for the calculation of
the density and viscosity of the buffer (50 mm Tris, 50
mm NaCl, 1 mm phenylmethylsulfonyl fluoride) and partial
specific volume and molecular mass of the protein
(35). Equilibrium as well as
velocity data were analyzed with SEDFIT and SEDPHAT software. Sedimentation
equilibrium was attained at 24 h at a temperature of 4 °C at increasing
rotor speeds of 16,000–30,000 rpm. Protein concentrations were
1.75–17.0 μm (120 μl), and absorbance distributions
were recorded at 280 nm. Modeling was performed at multiple rotor speeds using
a monomer-dimer self-association model
(36). For the sedimentation
velocity experiments, the loading volume was 360 μl (2.2 mg/ml), and the
protein was centrifuged at 20 °C at 60,000 rpm. Fringe displacement data
at time intervals of 1.0 min were collected with the Rayleigh interference
system for 12 h and analyzed using the models for continuous sedimentation
coefficient distribution (c(s)) as well as the hybrid
continuous/discrete distribution
(37–39).
The latter model was with two discrete species, one with a best fit apparent
molar mass of 41 kDa and an s value of 3.85 S and a second species
constrained to the molar mass of the dimer, 98.4 kDa, with a fixed s
value of 4.70 S.
In Vitro Binding Assay Using the Hsp90 Inhibitor Geldanamycin (GA)—40 μl of rabbit reticulocyte lysate (Promega) was preincubated with either DMSO or geldanamycin for 30 min. Proteins were synthesized in the in vitro transcription and translation reaction of a 50-μl volume containing GA/DMSO-treated reticulocyte lysate, 0.02 mm methionine, and 800 ng of PCR-amplified DNA encoding FLAG-Sgt1 or HA-Sgt1 in the separate reactions and incubated at 30 °C for 90 min. HA-Sgt1 and FLAG-Sgt1 were mixed together with 300 μl of buffer A (50 mm Tris, pH 7.5, 50 mm NaCl, and 0.2% Triton X-100) and incubated at room temperature for 1 h. The reaction was immunoprecipitated with 30 μl of anti-HA cross-linked beads and incubated overnight at 4 °C, washed five times with Buffer A, and eluted in 30 μl of 2× gel loading buffer. Proteins were detected by Western blotting using tag-specific antibodies. HA-Skp1 used in the Skp1-Sgt1 binding reaction was expressed in baculovirus and was also pretreated with the same amount of DMSO or GA as used in the Sgt1-Sgt1 binding reaction.
RESULTS
Association of Sgt1 with Itself in Budding Yeast—In a two-hybrid analysis to further investigate the function of Sgt1, we discovered that Sgt1 interacts with itself (supplemental Fig. S1A). To confirm this interaction in vivo, we performed an immunoprecipitation assay using yeast strains expressing Sgt1-Myc and HA-Sgt1. HA-Sgt1 was detected specifically in precipitates of Sgt1-Myc but not in untagged Sgt1 control precipitations (Fig. 1A), and Sgt1-Myc was detected in precipitates of HA-Sgt1 but not in untagged control precipitations (supplemental Fig. S1B; data not shown). These results indicate that Sgt1 associates with itself in vivo.
FIGURE 1.

Sgt1 forms dimers. A, Sgt1 associates with Sgt1 in vivo. Lysates of yeast cells that expressed HA-Sgt1 (plasmid pB1354) and either Sgt1-Myc (Y1649, left) or untagged Sgt1 (YPH499, right) were subjected to immunoprecipitation (IP) with anti-Myc antibodies. Immunoblot analyses were performed with anti-HA or anti-Myc antibodies. T, total lysates; S, supernatant; P, precipitate. B, an analytical ultracentrifugation of Sgt1 is shown. Absorbance scans at 280 nm at equilibrium are plotted versus the distance from the axis of rotation Sgt1. Shown is protein at a concentration of 5μm centrifuged at 4 °C for at least 24 h at 16,000 rpm (black), 20,000 rpm (red), 24,000 rpm (blue), and 30,000 rpm (cyan). The solid lines represent the best fit of all the data sets to a monomer-dimer self-association model with a dissociation equilibrium constant of 20 nm. The root mean square deviation of the fit is 0.0040 absorbance units. C, the sedimentation velocity profiles (fringe displacement) of Sgt1 were fitted to a continuous sedimentation coefficient distribution model (c(s)) as well as a hybrid continuous/discrete distribution model (not shown). The root mean square deviation of the fit is 0.018 fringes.
Sgt1 Dimerization in Vitro—Next, we performed analytical ultracentrifugation by using bacterially expressed and purified His6-Sgt1. The monomer-dimer self-association model was used in the analysis of the sedimentation equilibrium data of Sgt1 (Fig. 1B). From these best fits, the dissociation equilibrium constant for Sgt1 was determined as 20 nm (strong dimerization). Thus, at an arbitrary concentration of 10 μm (500-fold higher in concentration than its dimer dissociation equilibrium constant of 20 nm), Sgt1 exists as a dimer in solution.
The sedimentation velocity data, c(s) distribution, of
Sgt1 show that it exists as a dimer in solution with a molar mass of ∼98.5
kDa (theoretical molar mass of 98,432 Da)
(Fig. 1C). The
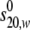 value is 4.75 S, larger than the maximum
theoretical value of 4.01 S for a monomer (data not shown). The frictional
ratio is 1.62, which indicates that this dimer is elongated and extended in
shape (data not shown). The second largest peak (s value of 3.85 S)
in the c(s) distribution does not appear to be the monomer
but a contaminant species with a molar mass of ∼41 kDa (data not shown).
The data could not be fitted to a rapid monomer-dimer self-association model.
Better fits were obtained with the hybrid continuous/discrete distribution
model where this 41-kDa contaminant species was taken into account and
comprised of no more than 10% of total protein. The existence of a dimer for
Sgt1 was confirmed with this model.
value is 4.75 S, larger than the maximum
theoretical value of 4.01 S for a monomer (data not shown). The frictional
ratio is 1.62, which indicates that this dimer is elongated and extended in
shape (data not shown). The second largest peak (s value of 3.85 S)
in the c(s) distribution does not appear to be the monomer
but a contaminant species with a molar mass of ∼41 kDa (data not shown).
The data could not be fitted to a rapid monomer-dimer self-association model.
Better fits were obtained with the hybrid continuous/discrete distribution
model where this 41-kDa contaminant species was taken into account and
comprised of no more than 10% of total protein. The existence of a dimer for
Sgt1 was confirmed with this model.
Binding of the N-terminal Domain of Sgt1 to Sgt1—Previous analyses of amino acid sequences revealed that Sgt1 has three conserved domains, viz. the TRP domain, the CS domain, and the SGS (Sgt1-specific sequence) domain (Fig. 2B). The TPR domain is located at the N-terminal end (amino acids 13–141) and contains TPR motifs that are homologous (27% similarity) to those of HOP (Sti1 in budding yeast), an Hsp90-Hsp70-organizing protein (21). The CS motif (amino acids 184–279) is homologous (18% similarity) to a domain in the co-chaperone p23 (25, 40). Previously, we reported (24) that in vitro, two of three repeats of the TPR domain and a short part (amino acids 184–211) of the CS domain are required for Sgt1 binding to Skp1 and that the full-length TPR domain and a short part (amino acids 184–211) of the CS domain are required for Sgt1 binding to Hsc82 (Fig. 2B).
FIGURE 2.

Sgt1-binding domain of Sgt1. A, the indicated mutant proteins were radiolabeled, mixed, and incubated for 1 h at 30 °C with an extract that contained unlabeled HA-Sgt1 (21, 24). HA-Sgt1 was immunoprecipitated (IP) by using anti-HA antibodies that had been conjugated to Sepharose. The immunoprecipitates were eluted and subjected to SDS-PAGE, and the radioactive bands were identified by autoradiography. HA-Sgt1 was detected by immunoblotting with anti-HA antibodies. T, total lysates (6% of the starting material); DS, deletion proteins. The numbers above the lanes indicate the positions of the N- and C-terminal amino acids (aa) in each Sgt1 deletion protein. B, an illustration of the Sgt1 deletion proteins used in the Sgt1 binding assays and a summary of the results of these assays are shown. The results of our previous study of Sgt1 binding to Skp1 and Hsc82 are shown for comparison only (24). Analyses of the Sgt1 binding ability of the Skp1-binding domain (Skp1-BD), Hsc82-binding domain (Hsc82-BD), and Sgt1-binding domain (Sgt1-BD) are shown. +, binding activity; –, no significant binding activity; +/–, weak binding activity; VR1, variable region 1; CS, CHORD protein- and Sgt1-specific; VR2, variable region 2; SGS, SGT1-specific. ND indicates analyses were not performed. C, the N-terminal domain of Sgt1 is sufficient for dimerization. Essentially the same experiments as shown in A were conducted, but HA-tagged mutant proteins were used in place of HA-tagged full-length Sgt1. HA-tagged Sgt1 mutant protein was expressed in the in vitro translation system but was not radiolabeled, whereas untagged Sgt1 mutant proteins were radiolabeled. HA-tagged Sgt1 was detected by immunoblotting with anti-HA antibodies. T, total lysates (6% of the starting material).
We performed immunoprecipitation experiments by using truncated Sgt1 proteins to identify the domains that participate in the Sgt1-Sgt1 association. Various deletion proteins (Fig. 2, A and B) were expressed and labeled with [35S]methionine by an in vitro translation system, and the reticulocyte lysates that contained the deletion proteins were mixed with a protein extract that contained HA-Sgt1 that had been expressed separately. HA-Sgt1 was immunoprecipitated by using Sepharose to which anti-HA antibodies were conjugated (Fig. 2A). Analysis of the binding activity of the deletion proteins revealed that the N-terminal region that contained the TPR domain (amino acids 1–182) bound to Sgt1 substantially (Fig. 2, A and B, and supplemental Fig. S1D). Deletion of amino acids 1–58 reduced binding to Sgt1 substantially (Fig. 2, A and B, and supplemental Fig. S1D). Thus, we conclude that the full-length TPR domain is required for Sgt1 binding to Sgt1 (Fig. 2B). The N-terminal region that contained the TPR domain (amino acids 1–182) bound to Sgt1 but not to Skp1 or Hsc82 (Fig. 2B). The finding that the Sgt1-binding domain overlaps the Skp1-binding and Hsp90-binding domains but is 29 amino acids shorter than both (Fig. 2B) suggests that Sgt1 first forms dimers and then binds to Skp1 and Hsp90. This hypothesis is consistent with the finding that Sgt1 can form dimers efficiently by itself.
Next, we examined whether these N-terminal polypeptides can form homodimers. The N-terminal region that consisted of amino acids 1–211 formed homodimers, but a shorter region (amino acids 1–182) did not (Fig. 2C). Also, the N-terminal region (amino acids 1–211) did not associate with the C-terminal region (amino acids 284–395) (Fig. 2C). These results indicate that the N-terminal end of Sgt1 binds to the N-terminal end of another Sgt1 protein.
Effect of Hsp90 on Sgt1 Dimerization—By using purified human Hsp90 protein, we previously showed that Hsp90 binding to Sgt1 is required for Sgt1 binding to Skp1 in vitro (24). Thus, we examined whether Hsp90 helps Sgt1 dimerization or dissociation.
In the present study, we used MBP-fused yeast Hsp90 protein (MBP-Hsp82) (26) and confirmed the previous results. Indeed, MBP-Hsp82 stimulated Sgt1 binding to GST-Skp1 in vitro (Fig. 3A). Using the same proteins, we examined whether Hsp90 stimulates the formation or dissociation of Sgt1 dimers. We found that MBP-Hsp82 moderately stimulates (but does not inhibit) the association between His6-Sgt1 and GST-Sgt1 (Fig. 3B). Under these conditions, the molecular ratio of Skp1 to Sgt1 is ∼1:2 (1:2.18), supporting the model that the Sgt1 dimer binds to Skp1.
FIGURE 3.

Effect of Hsp90 on Sgt1 Dimerization. A, recombinant MBP-Hsp90 induces Sgt1-Skp1 binding. The indicated amounts of MBP-Hsp82 were added to ∼300 ng of His-Sgt1 and GST-Skp1 and incubated at 30 °C for 1 h before immunoprecipitation (IP) with anti-His antibodies. Input refers to total lysates (6% of starting material); Pellet refers to the immunoprecipitate. B, increased levels of Hsp82 stimulate Sgt1-Sgt1 binding moderately. The experiment is essentially the same as the one described in A, except that GST-Sgt1 was used in place of GST-Skp1. His-Sgt1 or GST-Sgt1 was detected by anti-His or anti-GST antibodies. C, Hsp90 is required for Sgt1-Skp1 binding but not Sgt1-Sgt1 binding in vitro. Upper, HA-Skp1-FLAG-Sgt1 binding was monitored in the presence of the Hsp90 inhibitor geldanamycin. In each reaction, equal amounts of HA-Skp1 and FLAG-Sgt1 proteins pretreated with DMSO or GA were immunoprecipitated with anti-HA antibody. Proteins in the immunoprecipitates were detected by tag-specific antibodies. Lower, similar to the experiment in the upper panel, except that HA-Sgt1 was used instead of HA-Skp1 in the Sgt1-Sgt1 binding assay. D, Sgt1-Sgt1 association is unaltered in hsp82 mutants, but Sgt1-Skp1 binding is reduced. Lysates were collected for the mutant strain hsp82-T101I (strain Y1653) that expressed HA-Sgt1 and Myc-Sgt1. Before lysis, the cells were grown either at 25 °C only or at 25 °C and shifted to 37 °C for 3 h. Lysates were subjected to immunoprecipitation with anti-Myc antibodies. Bound fractions were analyzed by using anti-HA, anti-Myc, and anti-Skp1 antibodies.
In the in vitro translation system using reticulocyte lysates (presumably including abundant Hsp90), we found that GA, an Hsp90 inhibitor, substantially inhibits Sgt1-Skp1 association but not Sgt1 dimerization (Fig. 3C). These results strongly suggest that Hsp90 is required for binding of Sgt1 to Skp1 but not Sgt1 itself.
Next, we examined the role of Hsp90 in Sgt1 dimerization in vivo. In hsp82 temperature-sensitive mutant cells at the nonpermissive temperature (37 °C), Sgt1 binding to Skp1 was substantially reduced, but Sgt1 binding to Sgt1 was not significantly affected (Fig. 3D). Note that the association of Sgt1 and Skp1 was not altered in wild-type strains at 37 °C (supplemental Fig. S1E, left) and that the Hsp82 mutant protein was not reduced at the nonpermissive temperature (supplemental Fig. S1E, right). These results indicate that Hsp82 is required for Sgt1 binding to Skp1 but not for generating Sgt1 dimers in vivo.
Importance of the Three Repeats in the TPR to Sgt1 Dimerization and Skp1 Binding—The sgt1-3 cells, which undergo arrest in G2/M phases at the nonpermissive temperature, exhibit a chromosome missegregation phenotype (21). The CBF3 complex fails to form in sgt1-3 cells, and the Sgt1-3 protein cannot bind to Skp1 (21). A functionally different sgt1 mutant called sgt1-5 undergoes arrest in the G1 phase at the nonpermissive temperature and is deficient in SCF (Skp1-Cullin-F-box) ubiquitin ligase activity and activated adenylyl cyclase (21, 25). We found that Sgt1-3 protein cannot bind to Sgt1-3 protein in vivo (Fig. 4A), although Sgt1-5 protein can bind to Sgt1-5 protein (Fig. 4A). Three missense mutations are present in sgt1-3: L31P, F99L, and N213I. The first two are in the TPR domain (Fig. 4B). Previously, Lingelbach and Kaplan (26) showed that the N213I mutation does not contribute to the kinetochore defects in sgt1-3 mutants. Therefore, we tested whether the L31P or F99L mutant protein can form dimers. Interestingly, the L31P mutant protein did not bind to Sgt1 or Skp1 (Fig. 4C), and the F99L mutant protein bound to Sgt1 and Skp1 weakly (Fig. 4C). The other kinetochore-defective alleles, sgt1-7 and sgt1-12, have missense mutations; L117P and Q191K occur in sgt1-7, and A74T and I151V occur in sgt1-12. To address the importance of all three repeats in the TPR domain (Fig. 4B), we examined whether the A67Y, L117P, or A74T mutant protein can form homodimers (note that Ala-67 and Ala-74 are conserved in Sti1, a typical TPR protein) (Fig. 4B). The dimerization of the A67Y mutant protein and that of the A74T mutant protein were reduced, and that of the L117P mutant protein was substantially reduced (Fig. 4C).
FIGURE 4.

The TPR domain of Sgt1 is important for its kinetochore function. A, the N-terminal region of Sgt1 that contains the TPR domain is important for Sgt1-Sgt1 and Sgt1-Skp1 interactions. HA-Sgt1, HA-sgt1-3, or HA-sgt1-5 was expressed in Sgt1-Myc, sgt1-3-Myc, or sgt1-5-Myc cells (Y1650, Y1651, and Y1652, respectively). Cells were either grown at 25 °C only or at 25 °C and shifted to 37 °C for 3 h before harvesting. The lysates were subjected to immunoprecipitation with anti-Myc antibodies conjugated to Sepharose. Anti-HA or anti-Myc antibodies were used to detect HA-tagged or Myc-tagged proteins, respectively, in the immunoprecipitates (IP). Skp1 was detected by anti-Skp1 antibodies. B, shown is an alignment of TPR motifs in Sgt1 with those in Sti1, a typical TPR protein (27). Identical residues are in black boxes; similar residues are in gray boxes; and the indicated residues, except for Ala-67, are those whose significance for the kinetochore function was described in our previous reports (21, 24). The starting amino acid position of each residue is shown in parentheses. C, all three TPR motifs of the Sgt1 TPR domain are important for kinetochore function. Strains (Y1662–1666 and Y1668) in which SGT1 was deleted were designed to express pRS414-HA-Sgt1 (plasmid pB180) and pRS415-Myc-Sgt1 (plasmid pB1367) or the indicated point mutations in pRS414-HA (plasmids pB1361–1365) and pRS415-Myc (plasmids pB1368–1372). Lysates were subjected to immunoprecipitation with anti-Myc antibodies conjugated to Sepharose (lower panels). Error bars indicate the S.D. of two independent results. The signals in the upper panels were quantified, and binding activity was defined as the ratio of the amount of coprecipitated protein to the amount of input protein. Specifically, the binding activity of wild-type Sgt1 was defined as 1, and the binding activity of each mutant was normalized to that value.
Catlett and Kaplan (27) reported that the second repeat in the TPR domain is not necessary for binding to Skp1 because the A67Y mutant protein is able to bind to Skp1 in vitro. However, we found that the A67Y mutant protein is not able to associate efficiently with itself or with Skp1 in vivo (Fig. 4C). These results strongly suggest that all three TPR repeats in the TPR domain are important for Sgt1 dimerization and Skp1 binding.
Role of Dimerization in the Restoration of Defects in the sgt1-3 Mutant—To directly confirm the importance of the Sgt1-Sgt1 association for the essential function of Sgt1, we fused the 59-amino acid dimerization domain of human CENP-B (41) to the N-terminal end of the L31P mutant protein. The CENP-B dimerization domain (DD) is sufficient to form dimers (41). CENP-B is not conserved in budding yeast; thus, its dimerization domain is not relevant to kinetochore function in budding yeast. We confirmed by immunoprecipitation that the CENP-B-DD-L31P mutant protein forms dimers in vivo (Fig. 5A). Indeed, restoring dimerization activity by using CENP-B-DD suppressed the temperature sensitivity of sgt1-L31P (Fig. 5B), the benomyl sensitivity of sgt1-L31P (Fig. 5C), and the chromosome missegregation phenotype of sgt1-L31P (Fig. 5D). These results strongly support the hypothesis that Sgt1 dimerization is required for the protein's function in kinetochore assembly.
FIGURE 5.

Sgt1 dimerization is required for the kinetochore function. A, fusion of the CENP-B-DD with an Sgt1 mutant protein results in partial recovery of a lost phenotype. The 59 C-terminal amino acids of human CENP-B, in which the dimerization domain resides, were fused with Sgt1 (CENP-B-DD-Sgt1) and with Sgt1-L31P (CENP-B-DD-Sgt1-L31P). Both HA- and Myc-tagged constructs for each (wild-type and mutant) were expressed in sgt1Δ cells (Y1677 and Y1678, respectively). Untagged (without CENP-B-DD), HA-tagged, and Myc-tagged constructs were used for each (wild-type and mutant) and were expressed in sgt1Δ cells (Y1662 and Y1663, respectively). Cell lysate from all four strains were subjected to immunoprecipitation (IP) with anti-Myc antibodies conjugated to Sepharose. Anti-HA and anti-Myc antibodies were used to detect HA- and Myc-tagged CENP-B-DD-Sgt1, CENP-B-DD-Sgt1-L31P, Sgt1, and Sgt1-L31P. Skp1 was detected with anti-Skp1 antibodies. The asterisk indicates a nonspecific band. HA-Sgt1 and HA-Sgt1-L31P protein in Input lanes were very close to the nonspecific band. B, cells that expressed CENP-B-DD fused with Sgt1 (Y1675) or Sgt1-L31P (Y1676) were streaked on yeast extract-peptone-dextrose plates. The plates were also inoculated with a strain that expressed wild-type Sgt1 (Y1655) and a strain that expressed sgt1-L31P without CENP-B-DD (Y1656). Plates were incubated at 25 °C and 37 °C. Pictures were taken after 3 days of growth. C, the indicated strains (the same as in B) were grown on yeast extract-peptone-dextrose plates containing 15 mg/ml benomyl or DMSO only. The numbers of cells that were spotted onto each plate (left to right) were ∼5 × 104, 1 × 104, 2 × 103, and 4 × 102. The plates were incubated at 30 °C for 3 days. D, shown are the results of a colony color-sectoring assay to analyze the chromosome missegregation phenotype of cells expressing Sgt1-L31P (Y1670) and CENP-B-DD-Sgt1-L31P (Y1680).
DISCUSSION
In this study, we have shown that Sgt1 forms homodimers. Analyses of Sgt1 domains revealed that the N-terminal domain, which includes TPR motifs, binds to Sgt1 itself. Further analyses suggested that the dimerization of Sgt1 is important for kinetochore assembly. On the basis of this work, we have developed a model in which CBF assembly is regulated by Sgt1 and Hsp90.
Updated Model of CBF3 Assembly—Recombinant purified Sgt1 forms dimers efficiently in vitro. The dimerization domain is a little smaller than the Skp1-binding and Hsp90-binding domains. These results suggest that Sgt1 forms homodimers before it binds to other proteins in vivo.
By activating Ctf13, Sgt1, together with Skp1, is required for the formation of the CBF3 complex (21). Hsp90 is also required for the formation of the Skp1-Ctf13 complex (22). Previously, we and others (24, 26) showed that the Sgt1-Hsp90 complex is required for the formation of the CBF3 complex. Here, our results suggest that Sgt1 first forms dimers. Next, Sgt1 binds to Hsc82 to form the proper structure for binding to Skp1, and by binding to Skp1, Sgt1 activates the Skp1-Ctf13 complex for the subsequent formation of the CBF3 complex (42). Skp1 can form a complex with Sgt1 and an F-box protein (e.g. Ctf13) (21). Because Sgt1 is not in the CBF3 complex (21), Sgt1 is expected to dissociate from the complex after assembly.
Necessity of the TPR Domain in Sgt1 Dimerization—Of the three conserved Sgt1 domains, we found that the N-terminal TPR domain is required for dimerization. Our in vivo analyses of sgt1 mutants that carry mutations in the TPR domain strongly suggested that all three TPR motifs are important for Sgt1 dimerization and Skp1 binding. Catlett and Kaplan (27) showed that, like wild-type protein, the Sgt1-A67Y protein binds to GST-Skp1 in vitro, and they concluded that the second TPR is not important for Skp1 binding. However, we found that in vivo, Sgt1-A67Y does not bind to itself very efficiently or to Skp1 at all. We believe that this difference in binding is explained by the difference between in vitro and in vivo conditions. Consistent with our result showing that Sgt1-A67Y does not bind to Skp1, a mutation at another conserved residue in the second TPR (A74T) reduced dimerization and abolished Skp1 binding. These results suggest that, at least in vivo, the second TPR motif also contributes to Skp1 binding.
Hsp90 Is Not Essential for Generating Sgt1 Dimers in Vivo—Although we found that Hsp90 stimulates Sgt1 dimerization moderately in vitro, Sgt1 forms dimers efficiently in hsp90 mutant lysates in which Sgt1 does not substantially bind to Skp1. Also, in the in vitro translation system using reticulocyte lysates, geldanamycin inhibits binding of Sgt1 to Skp1 but does not inhibit Sgt1 dimerization. Therefore, it is likely that Hsp90 stimulates the Sgt1 dimer to bind to Skp1. However, we cannot exclude the possibility of the existence of a redundant pathway that stimulates Sgt1 dimerization when Hsp90 fails in vivo. Further studies are required to address this issue.
Kinetochore Function of Human SGT1—Human SGT1A can rescue the yeast sgt1-null mutant (21), a result that suggests that the function of Sgt1 is conserved between humans and yeast. SGT1 associates with HSP90 in human and yeast cells (28, 43). Interestingly, Steensgaard et al. (29) and Niikura et al. (28) found that CENP-I, CENP-F, and Hec1, but not CENP-C, are absent from kinetochores in human cells depleted of SGT1 or HSP90 or treated with 17-(allylamino)-17-demethoxygeldanamycin (an HSP90 inhibitor). This absence suggests that the human SGT1-HSP90 complex is crucial for the assembly of mammalian kinetochores. Therefore, the kinetochore function of Sgt1 is evolutionally conserved between humans and yeast.
We initially detected the human SGT1-SGT1 interaction in vitro by immunoprecipitation, although it was weak (data not shown). However, Nyarko et al. (44) recently showed that human SGT1 does not form dimers efficiently by gel-filtration chromatography. Therefore, we further performed analytical ultracentrifuge experiments to determine the strength of dimerization. A monomer-dimer self-association model predicted dissociation equilibrium constants of 173 μm for human SGT1A (data not shown) and 20 nm for yeast Sgt1 (Fig. 1, B and C). Thus, we conclude that human SGT1A exists mainly as a monomer and that yeast Sgt1 exists mainly as a dimer in solution. These results are consistent with the results of Nyarko et al.
Supplementary Material
Acknowledgments
We thank V. Measday and R. Kitagawa for helpful comments, members of the Kitagawa laboratory for stimulating conversation and advice, K. B. Kaplan and S. Lindquist for generous gifts of reagents, the Hartwell Center for protein purification, and A. McArthur and V. Shanker for editing this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM68418 and Cancer Center Support Grant CA21765 from NCI. This work was also supported by the American Lebanese Syrian Association Charities. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains “Experimental Procedures,” Fig. S1, and supplemental Tables I and II.
Footnotes
The abbreviations used are: TPR, tetratricopeptide repeat; HA, hemagglutinin; GST, glutathione S-transferase; GA, geldanamycin; MBP, maltose-binding protein; DD, dimerization domain; DMSO, dimethyl sulfoxide.
References
- 1.Cheeseman, I. M., and Desai, A. (2008) Nat. Rev. Mol. Cell Biol. 9 33–46 [DOI] [PubMed] [Google Scholar]
- 2.Westermann, S., Drubin, D. G., and Barnes, G. (2007) Annu. Rev. Biochem. 76 563–591 [DOI] [PubMed] [Google Scholar]
- 3.Sudakin, V., and Yen, T. J. (2007) BioDrugs 21 225–233 [DOI] [PubMed] [Google Scholar]
- 4.Weaver, B. A., Silk, A. D., Montagna, C., Verdier-Pinard, P., and Cleveland, D. W. (2007) Cancer Cell 11 25–36 [DOI] [PubMed] [Google Scholar]
- 5.Fitzgerald-Hayes, M., Clarke, L., and Carbon, J. (1982) Cell 29 235–244 [DOI] [PubMed] [Google Scholar]
- 6.Hieter, P., Pridmore, D., Hegemann, J. H., Thomas, M., Davis, R. W., and Philippsen, P. (1985) Cell 42 913–921 [DOI] [PubMed] [Google Scholar]
- 7.Baker, R. E., and Masison, D. C. (1990) Mol. Cell. Biol. 10 2458–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai, M., and Davis, R. W. (1990) Cell 61 437–446 [DOI] [PubMed] [Google Scholar]
- 9.Cai, M. J., and Davis, R. W. (1989) Mol. Cell. Biol. 9 2544–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hegemann, J. H., Shero, J. H., Cottarel, G., Philippsen, P., and Hieter, P. (1988) Mol. Cell. Biol. 8 2523–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lechner, J., and Carbon, J. (1991) Cell 64 717–725 [DOI] [PubMed] [Google Scholar]
- 12.Doheny, K. F., Sorger, P. K., Hyman, A. A., Tugendreich, S., Spencer, F., and Hieter, P. (1993) Cell 73 761–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goh, P. Y., and Kilmartin, J. V. (1993) J. Cell Biol. 121 503–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang, W., Lechner, J., and Carbon, J. (1993) J. Cell Biol. 121 513–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lechner, J. (1994) EMBO J. 13 5203–5211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strunnikov, A. V., Kingsbury, J., and Koshland, D. (1995) J. Cell Biol. 128 749–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Connelly, C., and Hieter, P. (1996) Cell 86 275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stemmann, O., and Lechner, J. (1996) EMBO J. 15 3611–3620 [PMC free article] [PubMed] [Google Scholar]
- 19.Sorger, P. K., Doheny, K. F., Hieter, P., Kopski, K. M., Huffaker, T. C., and Hyman, A. A. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 12026–12030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan, K. B., Hyman, A. A., and Sorger, P. K. (1997) Cell 91 491–500 [DOI] [PubMed] [Google Scholar]
- 21.Kitagawa, K., Skowyra, D., Elledge, S. J., Harper, J. W., and Hieter, P. (1999) Mol. Cell 4 21–33 [DOI] [PubMed] [Google Scholar]
- 22.Stemmann, O., Neidig, A., Kocher, T., Wilm, M., and Lechner, J. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 8585–8590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Azevedo, C., Sadanandom, A., Kitagawa, K., Freialdenhoven, A., Shirasu, K., and Schulze-Lefert, P. (2002) Science 295 2073–2076 [DOI] [PubMed] [Google Scholar]
- 24.Bansal, P. K., Abdulle, R., and Kitagawa, K. (2004) Mol. Cell. Biol. 24 8069–8079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubacq, C., Guerois, R., Courbeyrette, R., Kitagawa, K., and Mann, C. (2002) Eukaryot. Cell 1 568–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lingelbach, L. B., and Kaplan, K. B. (2004) Mol. Cell. Biol. 24 8938–8950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Catlett, M. G., and Kaplan, K. B. (2006) J. Biol. Chem. 281 33739–33748 [DOI] [PubMed] [Google Scholar]
- 28.Niikura, Y., Ohta, S., Vandenbeldt, K. J., Abdulle, R., McEwen, B. F., and Kitagawa, K. (2006) Oncogene 25 4133–4146 [DOI] [PubMed] [Google Scholar]
- 29.Steensgaard, P., Garre, M., Muradore, I., Transidico, P., Nigg, E. A., Kitagawa, K., Earnshaw, W. C., Faretta, M., and Musacchio, A. (2004) EMBO Rep. 5 626–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rose, M. D., Winston, F., and Hieter, P. (1990) Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press, New York
- 31.Ito, H., Fukuda, Y., Murata, K., and Kimura, A. (1983) J. Bacteriol. 153 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Longtine, M. S., McKenzie, A., III, Demarini, D. J., Shah, N. G., Wach, A., Brachat, A., Philippsen, P., and Pringle, J. R. (1998) Yeast 14 953–961 [DOI] [PubMed] [Google Scholar]
- 33.Kitagawa, K., and Abdulle, R. (2002) BioTechniques 33 288–294 [DOI] [PubMed] [Google Scholar]
- 34.Kitagawa, K., Abdulle, R., Bansal, P. K., Cagney, G., Fields, S., and Hieter, P. (2003) Mol. Cell 11 1201–1213 [DOI] [PubMed] [Google Scholar]
- 35.Laue, T. M., Shah, B. D., Ridgeway, T. M., and Pelletier, S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding, S. E., Rowe, A. J., and Horton, J. C., eds) pp. 90–125 The Royal Society of Chemistry, Cambridge
- 36.Vistica, J., Dam, J., Balbo, A., Yikilmaz, E., Mariuzza, R. A., Rouault, T. A., and Schuck, P. (2004) Anal. Biochem. 326 234–256 [DOI] [PubMed] [Google Scholar]
- 37.Schuck, P. (2000) Biophys. J. 78 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuck, P. (2005) in Analytical Ultracentrifugation: Techniques and Methods (Scott, D. J., Harding, S. E., and Rowe, A. J., eds) pp. 26–50 The Royal Society of Chemistry, Cambridge
- 39.Schuck, P., Perugini, M. A., Gonzales, N. R., Howlett, G. J., and Schubert, D. (2002) Biophys. J. 82 1096–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia-Ranea, J. A., Mirey, G., Camonis, J., and Valencia, A. (2002) FEBS Lett. 529 162–167 [DOI] [PubMed] [Google Scholar]
- 41.Kitagawa, K., Masumoto, H., Ikeda, M., and Okazaki, T. (1995) Mol. Cell. Biol. 15 1602–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Russell, I. D., Grancell, A. S., and Sorger, P. K. (1999) J. Cell Biol. 145 933–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee, Y. T., Jacob, J., Michowski, W., Nowotny, M., Kuznicki, J., and Chazin, W. J. (2004) J. Biol. Chem. 279 16511–16517 [DOI] [PubMed] [Google Scholar]
- 44.Nyarko, A., Mosbahi, K., Rowe, A. J., Leech, A., Boter, M., Shirasu, K., and Kleanthous, C. (2007) Biochemistry 46 11331–11341 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.