

Abstract
Late infantile neuronal ceroid lipofuscinosis is a fatal childhood neurological disorder caused by a deficiency in the lysosomal protease tripeptidyl-peptidase 1 (TPP1). TPP1 represents the only known mammalian member of the S53 family of serine proteases, a group characterized by a subtilisin-like fold, a Ser-Glu-Asp catalytic triad, and an acidic pH optimum. TPP1 is synthesized as an inactive proenzyme (pro-TPP1) that is proteolytically processed into the active enzyme after exposure to low pH in vitro or targeting to the lysosome in vivo. In this study, we describe an endoglycosidase H-deglycosylated form of TPP1 containing four Asn-linked N-acetylglucosamines that is indistinguishable from fully glycosylated TPP1 in terms of autocatalytic processing of the proform and enzymatic properties of the mature protease. The crystal structure of deglycosylated pro-TPP1 was determined at 1.85 Å resolution. A large 151-residue C-shaped prodomain makes extensive contacts as it wraps around the surface of the catalytic domain with the two domains connected by a 24-residue flexible linker that passes through the substrate-binding groove. The proenzyme structure reveals suboptimal catalytic triad geometry with its propiece linker partially blocking the substrate-binding site, which together serve to prevent premature activation of the protease. Finally, we have identified numerous processing intermediates and propose a structural model that explains the pathway for TPP1 activation in vitro. These data provide new insights into TPP1 function and represent a valuable resource for constructing improved TPP1 variants for treatment of late infantile neuronal ceroid lipofuscinosis.
Late infantile neuronal ceroid lipofuscinosis (LINCL)3 (OMIM number 204500) is a neurodegenerative lysosomal storage disease of childhood that presents typically between the ages of 2 and 4 years with the onset of seizures. Disease progression is reflected by blindness, dementia, mental retardation, and an increase in the severity of seizures. LINCL is always fatal, and the life span of patients is typically 6-15 years. LINCL is caused by mutations in TPP1 (previously named CLN2, for ceroid lipofuscinosis neuronal type 2 gene) (1), which normally encodes a lysosomal protease, tripeptidyl-peptidase 1 (TPP1, EC 3.4.14.9) (2, 3).
There is currently no treatment of demonstrated efficacy for LINCL, but promising progress is being made in some directions. Proof-of-principle for virus-mediated gene therapy has been established in a mouse model of LINCL, with a significant improvement in disease phenotype achieved with the use of adeno-associated virus vectors expressing TPP1 (4-7). Affected children have also been treated with adeno-associated virus vectors, although it is too soon to determine whether significant clinical benefits have been achieved in these early trials (8). Enzyme replacement therapy, an approach that has proven successful in a number of other lysosomal storage diseases, has also been investigated in LINCL. Purified recombinant human TPP1 that contains the mannose 6-phosphate lysosomal targeting modification can be taken up by LINCL fibroblasts where it degrades storage material (9), and the protein has been introduced into the cerebrospinal fluid of the LINCL mouse model via intraventricular injection, resulting in significant uptake into the brain and some correction of neuropathology (10).
For therapeutic approaches that rely upon replacing a mutant gene product with a functional protein via recombinant methods, e.g. gene and enzyme replacement therapy, a thorough understanding of the biological and biophysical properties of the protein in question are essential for success. Thus, for LINCL, considerable effort has been directed toward the investigation of TPP1, and as a result, this is a well characterized enzyme at the functional and molecular levels (reviewed in Refs. 11, 12). TPP1 encodes a 563-residue preproprotein with a cleavable N-terminal 19-residue signal sequence. The proenzyme (residues 20-563) is a soluble monomer that undergoes proteolytic cleavage in the lysosome, converting the zymogen to an active, mature protease (residues 196-563) (1). Studies on purified pro-TPP1 demonstrate that maturation is autocatalytic in vitro (13, 14) but may involve other proteases in vivo (15). TPP1 is glycosylated, and its N-linked oligosaccharides have been implicated in maturation, activity, targeting, and stability of the processed enzyme (16, 17).
TPP1 is a serine protease (14) that possesses two catalytic functions as follows: a primary tripeptidyl exopeptidase activity with a pH optimum of ∼5.0 that catalyzes the sequential release of tripeptides from the unsubstituted N termini of substrates (18), and a much weaker endoproteolytic activity with a pH optimum of ∼3.0 (19). TPP1 exhibits broad substrate specificity (20) and is the only mammalian member of the S53 sedolisin family (reviewed in Ref. 21), which includes a number of unusual bacterial serine peptidases (22). High resolution crystal structures of both free and inhibitor-bound complexes have been determined for three bacterial members of this family (sedolisin (23-26), kumamolisin (27, 28), and kumamolisin-As (29, 30)), and for one (kumamolisin), the structure of a mutant, inactive precursor form has also been obtained (28). These proteins share a common subtilisin-like fold, an octahedrally coordinated calcium-binding site, and an active site that contains an unusual Ser-Glu-Asp (SED) catalytic triad, rather than the Ser-His-Asp (SHD) triad of subtilisin (31, 32). Chemical modification studies of TPP1 have revealed that Ser475 is the active site nucleophile (14). Modeling studies suggest that Glu272 and Asp276 complete the catalytic triad and that Asp360 is homologous to the conserved Asn in the subtilisin family in its role in stabilization of the oxyanion of the tetrahedral intermediate during catalysis (33). Site-directed mutagenesis studies are consistent with these conclusions (14, 34).
A detailed understanding of the tertiary structure of TPP1 may have implications for developing or improving therapeutic strategies. First, a high resolution model would provide the basis for targeted protein engineering efforts to design TPP1 derivatives with increased half-life prior to and/or upon delivery to the lysosome. Successful creation of a longer lived TPP1 molecule could significantly enhance gene or enzyme replacement approaches to LINCL. Second, a structural model for TPP1 could be valuable in designing derivatives tagged with protein transduction domains to facilitate crossing of the blood-brain barrier for delivery to the central nervous system from the bloodstream. In this study, we present the crystal structure of the proform of human TPP1 at 1.85 Å resolution. This model provides novel insights into the structural basis for the pH-induced auto-activation of the proform of TPP1. A structure of glycosylated pro-TPP1 has been independently determined, displaying features similar to those of deglycosylated TPP1.4
EXPERIMENTAL PROCEDURES
Protein Production—Production and purification of recombinant human TPP1 from a Chinese hamster ovary (CHO) expression system were conducted using modifications of methods described previously (9). Cells were cultured in 1050-cm2 roller bottles for 9 days with serum-free Dulbecco's modified Eagle's/F-12 medium containing 1 μg/ml kifunensin. Conditioned media were harvested and adjusted to contain 2.5 m NaCl as well as 20 mm Tris, pH 8.0, and 20 μm CaCl2 (TC Buffer). All subsequent steps were conducted at 4 °C unless noted otherwise. The solution was clarified by centrifugation and filtration and then applied to a butyl-Sepharose 4 fast-flow column (GE Healthcare). After washing with 2.5 m NaCl in TC Buffer, the column was eluted with a 7.5-column volume linear gradient of 2.5 to 0 m NaCl in TC Buffer. Fractions containing most of the pro-TPP1 were concentrated and equilibrated with 20 mm NaCl/TC Buffer by diafiltration. The solution was filtered, loaded onto a Mono Q12 column (Bio-Rad), and washed with 4 column volumes of 20 mm NaCl in TC Buffer. The column was eluted with 10 column volumes of a linear gradient of 20 to 515 mm NaCl in TC Buffer. Peak fractions containing pro-TPP1 were adjusted to contain 100 mm sodium citrate, pH 5.5. Protein was deglycosylated by incubation with Endo Hf (New England Biolabs, 26,500 units per mg of pro-TPP1) at 25 °C for 15 h followed by 1 h at 37 °C. The solution was buffer exchanged and repurified by Mono Q12 chromatography as described above. Fractions containing pro-TPP1 were concentrated and applied to a Superdex 75 column equilibrated in 150 mm NaCl in TC Buffer to separate pro-TPP1 monomer from a minor amount of aggregated material.
A similar procedure was used to isolate glycosylated pro-TPP1, omitting kifunensin from the cell culture media and the Endo H digestion and second Mono Q12 chromatography steps. Hexahistidine-tagged human pro-TPP1 was produced using an insect cell system and purified as described previously (14) except that all column buffers contained 20 μm CaCl2, and a final Superdex 75 gel filtration step was performed as described above.
TPP1 Activation Kinetics—TPP1 proenzyme was diluted to the desired concentration using 0.1% Triton X-100, 150 mm NaCl, 20 mm Tris, pH 8.0. For experiments using hexahistidine TPP1, the protein was preincubated with 1 mm E-64 to inhibit an insect cell thiol protease contaminant that was present in trace amounts in some preparations. Proenzyme (20 μl) was mixed with 180 μl of 0.1% Triton X-100, 150 mm NaCl, and 100 mm sodium formate adjusted with HCl to the indicated pH. The reaction was incubated at 25 °C for the indicated time, and the activation process was terminated by diluting 1 part sample into 9 parts ice-cold 0.1% Triton X-100, 150 mm NaCl, 500 mm sodium acetate, pH 5.0, frozen on dry ice, and stored at -80 °C. The amount of enzymatically active TPP1 (total final TPP1 concentration typically 1.5 nm) was measured using the previously described kinetic fluorescence assay (35) using 200 μm Ala-Ala-Phe-AMC substrate in 0.1% Triton X-100, 150 mm NaCl, 500 mm sodium acetate, pH 5.0. Data for activation conducted at pH 4.0 were fit to a one-phase exponential association model using built in functions in Prism version 5.01 (GraphPad software). The activation reactions at pH 2.5 to 3.5 were fit to an irreversible consecutive reaction model where the inactive TPP1 zymogen (Z) is converted to an intermediate (I) with partial activity, which is then converted into a fully active mature form (M) as shown in Reaction 1,
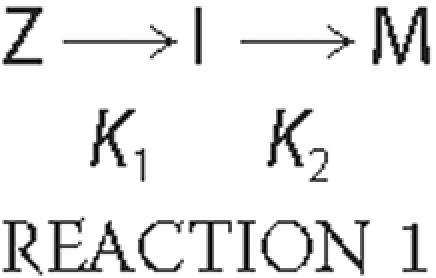
Using this model, one can calculate the activity at any given time from Equation 1,
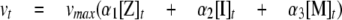 |
(Eq.1) |
Here vt denotes the observed TPP1 enzymatic activity at any given time; vmax denotes the enzymatic activity of the preparation when existing as the fully activated form; α denotes the relative enzymatic activity of the different forms, and for any given time [Z]t + [I]t + [M]t is a constant that corresponds to the total added TPP1, with all existing as Z at time = 0. Data were fit using the integrated rate equations given in Ref. 36 using Prism version 5.01, with the following constraints: no activity for the zymogen (α1 = 0), partial activity for the intermediate form (0 ≤ α2 ≤ 1), and maximal activity for the mature form (α3 = 1).
Protein Chemistry—Pro-TPP1 was activated as described above. For matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS), reactions were terminated by mixing with an equal volume of ice-cold solution containing 0.5% trifluoroacetic acid, 50% acetonitrile, mixed with sinapinic acid matrix, and analyzed using an Applied Biosystems/MDS-Sciex 4800 mass spectrometer operated in the linear mode. For SDS-PAGE analysis, reactions were terminated using reducing sample buffer and fractionated on 10% NuPAGE BisTris gels using MOPS running buffer (Invitrogen). Gels were either stained using Sypro Ruby (Molecular Probes) or transferred to membranes, stained with Coomassie Blue, and relevant regions of the membrane excised and sequenced as described previously (9, 14).
Crystallization of Pro-TPP1—Purified Endo H-deglycosylated pro-TPP1 was dialyzed into TC buffer containing 100 mm NaCl, concentrated to 7 mg/ml using a 30,000 centrifugal device (Millipore), and filtered through 0.22-μm pore size cellulose acetate filters (Corning Costar). Initial crystallization conditions were identified by high throughput screening at the Hauptman Woodward Medical Research Institute, Buffalo, NY. In the optimized condition, crystals of pro-TPP1 ranging in size between 50 × 100 × 100 μm and 50 × 100 × 300 μm appeared in ∼3 days when set up and grown at 4 °C using hanging drop diffusion by mixing 1 μl of pro-TPP1 solution at 5 mg/ml and 1 μl of reservoir solution containing 0.1 m citrate, 4-7% polyethylene glycol 6000, pH 5.0.
Data Collection, Structure Determination, and Refinement—Data for two native, one mercury-derivatized, and one lead-derivatized crystals were collected to 1.85 Å at X4C at the National Synchrotron Light Source, Brookhaven National Laboratory, Upton, NY. The lead and mercury derivatives were obtained by soaking the crystals overnight at 4 °C in reservoir solutions containing heavy atom salts at concentrations of 2.5 mm Pb(NO3)2 and 0.5 mm C2H5HgCl. Cryoprotection was achieved with passage of the crystals held in cryoloops through 1-μl drops of 50% ethylene glycol for 1 s and flash-cooling in liquid N2 stream. For all crystals, 180° of data with 1° oscillation angle were collected. All four data sets were processed and scaled in the primitive orthorhombic P212121 space group using DENZO and SCALEPACK (37) (Table 1 and supplemental Table S1). Initial attempts for structure solution with molecular replacement using a homology model of TPP1 (33) or homologous structures (24, 27, 28) did not yield reliable solutions. Furthermore, attempts for solving the structure by multiple isomorphous replacement using multiple heavy atom derivatives were unsuccessful because of lack of isomorphism between the datasets (supplemental Table S1). Subsequently, the crystal structure was solved by single wavelength anomalous dispersion (SAD) technique using the mercury dataset. Four mercury sites were obtained from the anomalous differences using SHELX programs (38) with the HKL2MAP software (39). The sites were used for SAD phasing at 2.5 Å resolution through Solve/Resolve (40, 41) as implemented in the software package Phenix (42). Taking advantage of the anisomorphism among the data sets, the SAD phases were improved and extended to 1.9 Å resolution through multiple crystal form averaging four data sets (two native, mercury-soaked and lead-soaked) using DMmulti (43) integrated into CCP4 version 6.0 package (44). The initial model consisting of 511 of the 544 residues was obtained by automated chain tracing with the ARP/wARP routine (45) using the averaged phases. The remaining residues were modeled following iterative cycles of maximum likelihood and isotropic temperature refinement with Refmac 5.1.2.4 (46), and manual model building using Coot (47). The Translation/Libration/Screw (TLS) motion determination server (48-50) was used to determine the optimal number of TLS groups (five groups used) and to generate parameters that were used in the final stages of refinement in refmac 5.1.2.4 (46). Initial models for solvent molecules were obtained from the Hic-up server (51). Solvent molecules with Fourier difference peaks greater than 3σ were included. The final model, refined to 1.85 Å with Rwork and Rfree of 0.179 and 0.205, respectively, includes all 544 residues, 1 bound Ca2+ ion, 4 N-acetyl-d-glucosamine (GlcNAc) groups, 4 ethylene glycol molecules, and 237 water oxygen atoms (Table 1). The structure was validated using the MOLPROBITY server (52), which reported 97.05% of the residues in the Ramachandran favored regions. Loop regions Thr191-Leu196 and Glu395-Thr400 were modeled using a combination of composite omit and difference Fourier maps calculated with map algorithms within CNS version 1.2 (53), and side chain conformations for residues in the aforesaid loops as well as for residues Lys101, Glu232, Gln264, Arg268, and Arg297 were modeled as the most common rotamer, because of disordered electron density corresponding to these regions.
TABLE 1.
Data collection and refinement statistics
Values corresponding to highest resolution shells are shown in parentheses.

Structural Analyses—Superposition of structural models was accomplished using LSQKAB (54) from the CCP4 program suite version 6.0 (44). Structural comparisons were made using the Secondary Structure Matching server (55), and overall root mean square deviation (r.m.s.d.) values were obtained using the r.m.s.d. calculation algorithm within CNS version 1.2 (53). The protein-protein interaction server (56, 57), the iMOLTALK server (58), and the WHATIF web interface (59) were used for analyses of interdomain and lattice contacts. Buried surface areas were calculated using CNS version 1.2 (53), and all structural figures were generated using PyMOL version 1.0 (60).
Electrostatic Potential Calculations—Analysis of pH dependence on interdomain binding energy in pro-TPP1 was accomplished using the Adaptive Poisson-Boltzmann Solver version 0.5.1 (APBS) (61). The PDB2PQR server (62, 63) implementing the PARSE forcefield and the PROPKA program (64) to assign specific protonation states at different pH conditions (pH 2.5-8.0) was used to convert the pro-TPP1 protein data bank (PDB) coordinates to three sets of PQR files, one for the intact zymogen (residues 20-563) (ZpH), one for the propiece (residues 20-195) (PpH), and the third for the mature domain (residues 196-563) (MpH) for a given pH condition. These pH-specific PQR files were used as input for electrostatic binding energy calculations at that pH using a linearized Poisson-Boltzmann equation (PB) using cubic B-spline discretization of the charge distributions. PB calculations were performed at 298 K with a dielectric constant of 78.0 for water and 2.0 for the protein interior. The ion concentrations were set to 0.15 m with ionic radius of 2.0 Å. Care was taken to remove self-interaction energies by using exactly the same grid spacing, length and center, determined by the psize.py script provided in the APBS distribution. The electrostatic binding energy at a given pH (BEpH) is calculated by APBS as shown in Equation 2,
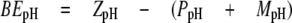 |
(Eq.2) |
RESULTS AND DISCUSSION
Production of Deglycosylated TPP1—Initial attempts to obtain suitable crystals from previously characterized glycosylated recombinant human pro-TPP1 produced in insect cells (14) and in CHO cells (9) were unsuccessful (data not shown).5 Given that heterogeneity in glycosylation may have been a complicating factor, we tried different methods to deglycosylate the protein produced from CHO cells. In our hands, peptide:N-glycosidase F was not completely effective in removing oligosaccharides unless the protein was denatured, suggesting that steric hindrance prevented cleavage of the GlcNAc-Asn bond. Endo H, which cleaves between the first two Asn-linked Glc-NAc residues, was partially effective in deglycosylating the native molecule, but some N-linked glycans were retained, reflecting the presence of oligosaccharides that had been processed to Endo H-resistant forms (15, 65). We therefore cultured the CHO cells in the presence of the endoplasmic reticulum mannosidase I inhibitor kifunensin, which prevents further processing of N-linked glycans to Endo H-resistant forms (66). When treated with Endo H, pro-TPP1 produced in this manner yielded a single species by SDS-PAGE analysis, corresponding to a deglycosylated form in which four single Asn-linked Glc-NAc monosaccharides remain, as confirmed by the crystal structure (supplemental Fig. S1 and see below).
Properties of Deglycosylated TPP1—Glycosylation has been reported to be critical for the folding, processing, and/or stability of TPP1 in cultured cells (17, 67). In this study, it was therefore important to compare the functional properties of deglycosylated TPP1 prepared for structural studies and the previously well characterized glycosylated native protein.
Purified pro-TPP1 is a zymogen that undergoes low pH-triggered autoactivation and processing (9, 13, 14, 16, 68). The kinetics for autoactivation of deglycosylated pro-TPP1 were essentially identical to those for glycosylated protein produced from untreated CHO cells or a hexahistidine-tagged protein produced from insect cells (Fig. 1). Rates were greatly accelerated at lower pH values and showed biphasic kinetics at pH 3.5 and below (Fig. 1).
FIGURE 1.

Autoactivation of pro-TPP1. Highly purified human recombinant TPP1 preparations (filled circles, hexahistidine-tagged pro-TPP1 from an insect cell expression system; open circles, fully glycosylated pro-TPP1 from CHO cells; and open squares, Endo H-deglycosylated pro-TPP1 from kifunensin-treated CHO cells) were incubated at the indicated pH for the specified time before stopping the reaction and measuring TPP1 activity as described under “Experimental Procedures.” Data for each experiment were normalized to plateau values, all of which were comparable. The curves shown are fit to the reaction curves for the hexahistidine-tagged pro-TPP1 preparation as described under “Experimental Procedures.”
We also measured the activity of preactivated glycosylated and deglycosylated TPP1 using two different fluorogenic substrates, Ala-Ala-Phe-AMC and Arg-Nle-Nle-AMC. Initial rates were determined as a function of substrate concentration and pH. The kcat values (Vmax/protein concentration) for the deglycosylated and glycosylated proteins were similar (Fig. 2, A and B). Although it appeared that the deglycosylated protein had slightly higher activity, the difference fell within the variation that we typically observe with independent preparations of glycosylated TPP1. The Michaelis constant (Km) (Fig. 2, A and B) and substrate specificity constant (kcat/Km) (Fig. 2C) of the glycosylated and deglycosylated protein were essentially indistinguishable. Our results do not rule out a role for glycosylation in folding or trafficking of pro-TPP1, nor do they address the role of the terminal Asn-linked GlcNAc that remains after Endo H-deglycosylation. However, they do demonstrate that Endo H-deglycosylated TPP1 is equivalent in terms of autocatalytic and enzymatic function to fully glycosylated TPP1.
FIGURE 2.

Enzymatic properties of glycosylated and Endo H-deglycosylated TPP1. CHO cell-produced enzyme preparations described in Fig. 1 were incubated at pH 3.5 for 2 h at 37 °C. Enzyme activity measurements were conducted using eight different concentrations of either Ala-Ala-Phe-AMC (800 to 6.25 μm) or Arg-Nle-Nle-AMC (400 to 3.12 μm) substrate. Final reactions contained 150 mm NaCl, 0.1% Triton X-100, and either 100 mm sodium formate, pH 2.5, or sodium acetate (all other pH values) adjusted to the indicated pH using HCl. Kinetic constants were calculated using the built-in Michaelis-Menten curve fitting functions in Prism version 5.01. Open symbols denote deglycosylated protein; filled symbols denote glycosylated protein. A and B, circles, kcat; squares, Km. C, circles, Ala-Ala-Phe-AMC substrate; squares, Arg-Nle-Nle-AMC substrate.
Crystal Structure of Pro-TPP1 Reveals a Conserved Subtilisinlike Fold—We have determined the crystal structure of the inactive precursor of Endo H-deglycosylated form of TPP1 at 1.85 Å resolution (supplemental Fig. S1 and Fig. 3). The data processed in the P212121 space group with one molecule in the asymmetric unit with a Mathhews coefficient and solvent content of 2.37 Å3/Da and 48.0%, respectively. The structure is markedly similar to the glycosylated form of pro-TPP1 determined at 2.35 Å resolution in the P21212 space group with two molecules in the asymmetric unit (PDB ID 3EE6). Superposition of Cα coordinates of deglycosylated pro-TPP1 with those of the two chains of glycosylated pro-TPP1 yield r.m.s.d. values of 0.61 and 0.66 Å, respectively, with differences in positioning of a few solvent exposed loops and conformations of a couple of active site residues (as discussed below). Pro-TPP1 exhibits a characteristic bi-domain precursor structure with a C-shaped prodomain (residues Ser20-Phe169) tightly packed against a more globular catalytic domain (residues Leu196-Pro563) connected by a 24-residue linker (Gly172-Gly195). The propiece has an antiparallel-α/antiparallel-β fold and the catalytic domain has a typical subtilisin-like fold.
FIGURE 3.

Overall structure of pro-TPP1. Stereo view of pro-TPP1 backbone depicted in coil representation with the propiece shown in cyan and blue and the catalytic domain in gold and red, for α helices and β strands, respectively, and the linker loop shown in magenta. Side chains of active site catalytic triad (Ser475, Glu272, and Asp276) and oxyanion hole (Asp360) residues are shown in CPK mode. Glycosylation sites with residual GlcNAc moieties (circles, Asn210, 1; Asn286, 2; Asn313, 3; and Asn443, 4) and disulfide bridges (squares, Cys111-Cys122, 1; Cys365-Cys526, 2; and Cys522-Cys537, 3) are depicted in stick mode. A bound Ca2+ ion is shown as a green sphere.
The N-terminal portion (residues 172-179) of the propiece linker is well ordered and threads through the substrate-binding groove making strong short distance contacts (one hydrogen bond, one salt bridge, and three hydrophobic interactions) with the catalytic domain. In contrast, the remaining portion of the linker (residues 180-195) loops out of the interface making only minimal long range van der Waal contacts. The conformation of this portion of the linker is stabilized by a few strong lattice contacts involving residues 180-190 as discussed below. Interestingly, the well structured nature of the N-terminal portion of the linker is supported by identical conformations and its involvement in interdomain contacts in both glycosylated and deglycosylated structures. The flexible nature of the C-terminal portion of the linker is emphasized by its involvement in lattice contacts in the deglycosylated structure and by its absence in the glycosylated structure, possibly because of disorder. Furthermore, a TLS motion analysis (48-50) for evidence of thermal motion detects significant flexibility in the linker region as compared with the rest of the protein (supplemental Fig. S2). Such flexibility may be important in positioning the linker for autocatalytic cleavage during zymogen activation (see below).
The structure has marked similarities with those of other S53 family members (24, 27, 28, 30) in terms of the overall fold, barring minor differences in the presence or absence of a few secondary structural elements in both the propiece and mature domain and orientation of the linker (supplemental Figs. S3 and S4). Superpositions of Cα coordinates of corresponding regions of pro-TPP1 with propiece of prokumamolisin (PDB ID 1T1E) (28), and mature enzymes of sedolisin (PDB ID 1GA4) (24), kumamolisin (PDB ID 1GTG) (27), and kumamolisin-As (PDB ID 1SN7) (30) yield r.m.s.d. values of 2.17, 2.07, 1.81, and 1.92 Å, respectively. Compared with prokumamolisin, the propiece and mature domain in pro-TPP1 are more compact with 550 Å2 more buried surface area than the former, whereas the respective linkers are oriented at an angle of ∼30° (supplemental Fig. S4). The total buried surface area between the propiece and mature domain in pro-TPP1 is 2200 A2 with 22 direct and 10 water-mediated hydrogen bonds, 3 salt bridges, and 6 hydrophobic interactions (supplemental Fig. S5 and supplemental Table S2).
Key features of the S53 family members, including Cα positions of catalytic residues forming the charge transfer complex (viz. Ser475, Glu272, Asp276, and Asp360), two cis-peptide bonds, and the calcium-binding site are conserved. Three cis-Pro residues are found in pro-TPP1 (Pro379, Pro450, and Pro563), of which the former two are completely conserved in the bacterial homologs (supplemental Fig. S3). The structural role and binding partners of the octahedrally coordinated calcium ion, including the side chain carboxyl groups of Asp517 and Asp453, backbone carbonyl groups of Val518, Gly539, and Gly541, and a bound water molecule, are conserved within the S53 family. In addition, three disulfide linkages unique to pro-TPP1 (viz. Cys111-Cys122, Cys365-Cys526, and Cys522-Cys537) stabilize the fold. The lone Cys227 is completely buried within the core of the mature domain and is within hydrogen bonding distance of the side chain carboxyl of Asp276 (see below).
Endo H treatment leaves one GlcNAc moiety at each N-linked glycosylation site. Mutagenesis data indicate that all five potential glycosylation sites of TPP1 (viz. Asn210, Asn222, Asn286, Asn313, and Asn443) are utilized (67). We find clear evidence for single GlcNAc moieties at all sites except for Asn222, a surface residue, which is clearly not modified in our structure. The structure of the glycosylated form of pro-TPP1 also shows that Asn222 is not utilized (PDB ID 3EE6). It is formally possible that this site is heterogeneously glycosylated and that crystallization conditions have selected out the four-sugar glycosylated form from a population of four- and five-sugar forms. In our structure, the GlcNAc moiety linked to Asn286 participates in lattice contacts with a symmetry-related molecule (supplemental Fig. S6 and supplemental Table S3).
Active Site of Pro-TPP1 and Maintenance of the Zymogen in the Inactive State—The active site in pro-TPP1 contains an SED (Ser475-Glu272-Asp276) catalytic triad, similar to all other members of the S53 family of serine peptidases and contrasting with the SHD catalytic triad of canonical serine peptidases. The replacement of the His residue with the acidic residue Glu in the S53 family allows the active site serine to function in nucleophilic attack of substrate at acidic pH (14), with the Glu abstracting the hydroxyl proton from the serine (14, 23-26, 34). In addition, there is a second conserved Asp residue (Asp360), designated here as D′, in place of a conserved Asn in the subtilisin family. The role of the latter is to bind and stabilize the oxyanion of the tetrahedral intermediate of the peptidase reaction by hydrogen bond interactions, as described by NMR analyses and high resolution crystal structures of members of the subtilisin family bound to inhibitors acting as transition-state analogs that mimic the tetrahedral intermediate (reviewed in Ref. 72). The catalytic efficiency in proteolytic enzymes is significantly increased by transition-state stabilization of the tetrahedral intermediate in the reaction. Computational studies on kumamolisin-As suggest that the D′ residue has multiple roles in catalyzing peptide bond hydrolysis, serving as a general acid to protonate the substrate during formation of the tetrahedral intermediate and subsequently functioning as a general base, extracting a proton from the tetrahedral intermediate during conversion to the acyl enzyme (73, 74). In TPP1, these roles are likely to be performed by Asp360. The D′ residue is conserved in the S53 family, suggesting a common proton transfer mechanism that distinguishes these enzymes from canonical serine proteases.
The crystal structure of pro-TPP1 reveals a number of features that help maintain the zymogen in an inactive state, with the active site assuming a geometry that is suboptimal for catalysis. Low pH-mediated pro-TPP1 zymogen maturation is likely to be associated with different positioning of key elements at the active site as well as different protonation states of active site carboxylates that might favor conformations poised for catalysis.
Although the Cα positions of key catalytic residues, including SEDD′ are superimposable with those of corresponding residues in all previously determined structures of bacterial homologs (24, 27, 28, 30), the side chain conformations and hydrogen bond network at the active site in pro-TPP1 are different (Fig. 4). As revealed by several high resolution crystal structures of free and inhibitor-bound mature enzymes of sedolisin, kumamolisin, and kumamolisin-As, the geometry of an active site poised for catalysis is characterized by a charge transfer complex involving the key SED residues with hydrogen bonds between one of the side chain carboxyl oxygens of the Glu with the hydroxyl of the Ser and the other with one of the carboxyl oxygens of the Asp. In contrast, in pro-TPP1, side chains of both Glu272 and Asp276 are directed away from each other and from the active site Ser475. Such alternate rotameric conformations of these catalytic residues allow hydrogen bonds distinct from those characteristic of a charge transfer complex (Fig. 4). PROPKA predictions (64) yield pKa values of 3.6 and 6.4 for Glu272 and Asp276 in the mature enzyme, respectively (supplemental Table S4). From this it follows that such rotameric conformations for the Glu and the Asp, as observed in pro-TPP1, might arise from electrostatic charge repulsions and could likely be favored at neutral pH, at which both of these residues are predicted to be deprotonated and negatively charged. However, at the crystallization pH condition, pH 5.0, only Glu272 and not Asp276 is predicted to be deprotonated. The active site Glu is also found in dual conformations in some inhibitor complexes in structures of homologs crystallized at higher pH (25). In addition, the conformations of Glu272 and Asp276 in the glycosylated form of pro-TPP1 are similar to those found in mature enzymes of bacterial homologs. These results suggest that the acidic residues at the active site most likely exist in equilibrium between alternate rotameric states and that different conditions favoring different pH-driven protonation states increase the probability of one state over another.
FIGURE 4.

Comparison of the active site of pro-TPP1 zymogen with that of mature kumamolisin. Backbones are shown as ribbon representations with featured side chains in ball-and-stick mode, bound water molecules depicted as red spheres, and hydrogen bonds shown as dashed lines with distances in Å. A, stereo view of TPP1 showing catalytic residues in a conformation that is recalcitrant to activity. In TPP1, Glu272 (E), Asp276 (D), and Ser475 (S) form the catalytic triad, and Asp360 (D′) forms part of the stabilizing oxyanion hole. Ser324 (S′) is completely conserved in the S53 family. Backbone carbonyl groups are shown only for the Ser and S′ residues. Side chain oxygen atoms are shown in red and backbone carbonyl oxygens in yellow. Both Glu272 and Asp276 side chains are oriented away from Ser475. The position of Glu272 is stabilized by a hydrogen bond to Gln233. A bound water molecule (1) occupies the position expected for one of the carboxyl oxygens of Glu272, if the active site were poised for activity. One of the carboxyl oxygens of Asp276 forms a hydrogen bond to a water molecule (2), and the other is within weak hydrogen bond distance to the sulfhydryl of unpaired Cys277. The side chain hydroxyls of Ser475 and Ser324 participate in a strong hydrogen bond. A third water molecule (3) makes a strong hydrogen bond with the Ser475 hydroxyl and is also within hydrogen bond distance of the Asp360 carboxyl constituting the oxyanion hole, close to where the carbonyl oxygen of the scissile peptide bond is expected to be positioned. B, comparison of catalytic machinery of pro-TPP1 with that in mature kumamolisin. Least square superpositions of Cα coordinates of the pro-TPP1 catalytic domain with those of all available structures of S53 family members were performed and yielded similar results. Superposition of TPP1 with mature kumamolisin (PDB ID 1GTL) (27) is shown with ribbon depictions in gray and green, respectively. Although the corresponding oxyanion D′ residues in TPP1 and kumamolisin are in similar conformations, the corresponding catalytic Glu and Asp residues are both in different conformations. Furthermore, the S′ residue in TPP1 (Ser324) is in a different rotameric conformation, and its Cα position is displaced by 1.5 Å from the corresponding S′ (Ser316) in kumamolisin, resulting in different hydrogen bond schemes with the active site Ser.
Furthermore, in both structures of the glycosylated and Endo H-deglycosylated forms of pro-TPP1, the side chain hydroxyl group of the active site Ser475 is also involved in a strong hydrogen bond with the side chain hydroxyl of Ser324 at 2.6 Å. Interestingly, this Ser (designated here as S′) is completely conserved within the S53 family (supplemental Fig. S3). It is noteworthy that in the crystal structures of mature enzymes of the bacterial homologs, the region of the protein containing the conserved S′ residue exhibits a different conformation and consequently a distinct hydrogen bond geometry with the active site Ser (Fig. 4). For instance, in mature kumamolisin, the S′ residue Ser316 is in a different rotameric conformation, and its Cα position is displaced by 1.5 Å away from that of the pro-TPP1 S′ Ser324. This geometry supports a hydrogen bond between the backbone carbonyl of the active site Ser466 and the side chain hydroxyl of Ser316 at 2.8 Å (backbone-side chain bond) and another between the side chain hydroxyl of the active site Ser466 and the backbone carbonyl of Ser316 at 3.5 Å (side chain-backbone bond), in place of a strong side chain-side chain hydrogen bond in pro-TPP1. With lack of mutational data on this residue, its contribution to the catalytic machinery is not certain but is worth consideration because of its complete conservation. In pro-TPP1, Ser324 is located at the tip of the mβ4 strand. The differences in positioning of structural elements of this region compared with those in mature enzymes are likely propagated as a result of interdomain contacts involving residues C-terminal to Ser324, viz. within the mβ4-mβ5 loop and the mβ5 strand (region III in supplemental Fig. S5).
The side chain conformation of the D′ residue Asp360 constituting the oxyanion hole is indistinguishable from the corresponding residue in homologous structures except in prokumamolisin where the corresponding Asp352 is in a different rotameric conformation. In addition, a bound water molecule (water 3) occupies a similar position as the backbone carbonyl of the scissile peptide bond of a substrate as observed upon superposition of the pro-TPP1 catalytic domain with an inhibitor bound homologous structure (27). This water makes strong hydrogen bonds with the side chain oxygens of active site Ser475 and Asp360 (Fig. 4).
Zymogens are commonly kept in their inactive states by occlusion of their active sites by propeptides that function as competitive inhibitors, occupying the substrate-binding site in a nonhydrolyzable configuration (reviewed in Ref. 75). To test whether the pro-TPP1 linker can function in this role, we docked a model hexapeptide substrate to the catalytic domain in which the side chain conformations of Glu272 and Asp276 were reoriented to assumed active conformations, based on the structures of mature enzymes of bacterial homologs. This hexapeptide was readily accommodated in the absence of the propiece linker. However, when the linker was superimposed on the docked model, the P3 and P3′ residues overlap with linker residues Pro178 and Arg185, respectively, suggesting steric occlusion. The docking study further suggests that the linker residue Arg183 would hinder approach of the substrate to the S2′ and S3′ subsites (Fig. 5).
FIGURE 5.

Model of peptide substrate in the TPP1 active site. The model was constructed by docking a hexapeptide substrate, AAFFAA, with a fixed position of the carbonyl oxygen of the P1 residue (Phe) relative to the catalytic domain of TPP1, based on the prokumamolisin structure (PDB ID 1T1E) (28), onto the catalytic domain of TPP1 in which the side chains of active site residues Glu272 and Asp276 were reoriented in the presumed active conformation as found in mature bacterial homologs. A ribbon diagram and a transparent surface rendition of the catalytic domain are shown in gray, a ball-and-stick depiction of the modeled hexapeptide substrate in yellow, and the superimposed linker with side chains of featured residues shown in ball-and-stick mode in cyan. Overlapping residues are shown in dotted surface. This model is an approximation because the positions of the mα3 helix with Glu272 and Asp276 as well as mβ4 with Ser324 together with the mβ4-mβ5 loop with Asp327 are likely to differ in the mature enzyme (Fig. 4). The N terminus of the tripeptide substrate is likely to interact with the Asp327 side chain carboxyl groups. The predicted protonation state of Asp327 (see text) is consistent with a role in binding to an unsubstituted positively charged N terminus of the P3 residue of a putative substrate, thus providing a possible structural basis for the tripeptidyl peptidase activity. In addition, in the absence of crystal lattice contacts, positively charged Arg183 of the linker is likely to participate in electrostatic interactions with negatively charged Asp360 of the oxyanion hole, potentially restricting its role in general acid and base catalysis.
In addition, a putative salt bridge between the positively charged Arg183 and negatively charged Asp360 is also likely to render a nonfunctional oxyanion hole. This is consistent with the pKa of 4.7 predicted for Asp360 in the zymogen form (supplemental Table S4). A corresponding salt bridge between Arg169 and the oxyanion hole Asp352 observed in prokumamolisin has been implicated in substrate-mediated inactivation (28). However, in pro-TPP1 this salt bridge is absent (>5.0 Å), presumably because of crystal lattice interactions involving the linker region C-terminal to Arg183 (supplemental Fig. S6).
Structural Basis for Tripeptidyl Peptidase Activity—The substrate-binding subsite and the active site pocket are likely to undergo subtle changes in the mature enzyme with different rotameric conformations of catalytic triad and neighboring residues (such as Ser324, see above). Nevertheless, a docked model of a hexapeptide substrate AAFFAA revealed that the Asp327 side chain is a likely candidate for binding to the positively charged unsubstituted N termini of P3 residues of substrates, thus facilitating release of tripeptides after catalysis (Fig. 5). Asp327 is completely conserved in all mammalian TPP1 proteins but not among the bacterial homologs (supplemental Fig. S3). Interestingly, in the latter, a neighboring Trp side chain blocks this region. In pro-TPP1, the Asp327 carboxyl is 5.3 Å away from the N terminus of the modeled hexapeptide, although this is likely to change in the mature domain. In addition, pKa predictions using the PROPKA program (64) indicate that the pKa of Asp327 is 6.2 when it is buried as part of pro-TPP1, whereas it is 3.9 when exposed in the mature enzyme. From this it follows that at pH <3.9 it is likely to be protonated when its binding to the positively charged N terminus of a substrate is suboptimal. This correlates with the Km profile of the enzyme, which reveals a substantial increase in Km below pH 4.0 (Fig. 2). Such a role for Asp327 was also suggested from the previously predicted homology model for TPP1 (33), supported by site-directed mutagenesis (34).
Model for Autoproteolytic Endopeptidase Cleavage of Pro-TPP1—MALDI-TOF-mass spectrometric (MS) analysis revealed differences in the size of the liberated propeptide at different pH values (Fig. 6A). This indicates that the zymogen can utilize three different endopeptidase cleavage sites (Fig. 6B) as follows: 1) Ser181-Leu182 site within pH range 4.0-4.5; 2) Gln189-Val190 site within pH range 3.0-4.0; and 3) Asp168-Phe169 site within pH range 2.5-4.0. Cleavages at sites 1 and 2 have been reported previously (13). Calculations using APBS electrostatic analysis software (61) with pro-TPP1 domains revealed that the binding energy between the propiece and mature domain is more positive (i.e. interactions are less stable) with lower pH (Fig. 6C). A similar profile is also observed for calculations done using prokumamolisin coordinates (data not shown). This phenomenon of pH-induced effects on binding is intuitive. Key salt bridge interactions as well as hydrogen bonds contributed by ionizable residues may be disrupted at low pH where acidic residues are likely to become protonated. This conclusion is supported by a recent report that the extent of inhibition of TPP1 activity by the prodomain decreases with lower pH and is negligible at pH 3.5 (68). In addition, complex formation between kumamolisin-As E78H/D164N mutant mature domain and wild-type propiece was observed to be less at lower pH (29). These results are consistent with a pH-induced change in binding between the propiece and the mature domain. In pro-TPP1, the binding interface has many electrostatic bonds and ionizable residues with pKa values within the physiological pH range (Fig. 6B and supplemental Fig. S6 and supplemental Tables S2 and S4). Predictions of pKa values of ionizable residues at the interdomain interface reveal that these residues cluster into groups that map to specific regions of the binding interface (supplemental Fig. S6 and supplemental Table S4). These regions are associated with distinct ranges of pKa values, suggesting pH-specific disruption of domain contacts.
FIGURE 6.

Model for autoproteolytic endopeptidase cleavage during pH-dependent pro-TPP1 zymogen maturation. A, MS analysis of autoproteolytic cleavage products. Endo H-deglycosylated pro-TPP1 was incubated at either 25 °C for 0.5 min (pH 2.5 and 3.0), 6.3 min (pH 3.5), 13 min (pH 4.0), or at 37 °C for 18 h (pH 4.5). Indicated m/z are those of the predicted TPP1 fragments spanning residues 20-168 (16640), 20-181 (18024), and 20-189 (19029). Satellite peaks likely represent matrix adducts. B, endopeptidase cleavage sites and positions of ionizable residues at the interdomain interface. The pro-TPP1 backbone is shown with the catalytic domain in orange, the prodomain in cyan, and the propiece linker in magenta. The endopeptidase cleavage sites (1, Ser181-Leu182; 2, Gln189-Val190; and 3, Asp168-Phe169) and active site residues (Ser475, Glu272, Asp276, and Asp360) are depicted in stick mode. The Cα positions of ionizable interface residues with pKa values predicted to be within the relevant physiological pH range for TPP1 (supplemental Table S4) are shown as spheres: 1, Glu232; 2, Glu299; 3, Glu302; 4, Glu24; 5, Asp26; 6, Asp168; 7; Asp118; 8, Glu343; and 9, Asp70. Details of these interactions are shown in supplemental Fig. S4 and supplemental Table S2. C, titration curve of binding energies of the propiece and mature domain of pro-TPP1. Binding energies were calculated as described under “Experimental Procedures.” D, model for pro-TPP1 zymogen maturation. A schematic depiction of pro-TPP1 is shown with colors, and endopeptidase cleavage sites and interdomain contacts involving ionizable residues are indicated as in B. The active site cleft is depicted as a triangle within the catalytic domain. The geometry of one side of the cleft (top half of triangle) defined by the acidic residues is subject to pH-driven protonation states, whereas the structure of the other side (lower half of triangle) defined by Ser475 and Ser324 is subject to restructuring by propagation of long range prodomain interactions. At higher pH, the prodomain and mature domains are tightly bound, and the active site catalytic machinery is preformed, and the active site exists in equilibrium between two states, one suboptimal for activity (hatched triangle with mouth closed) and one poised for activity (white triangle with open mouth). As the pH is lowered, changes in protonation states progressively disrupt interdomain interactions and affect the rotameric conformations of the active site residues favoring an active site geometry poised for catalysis. As the pH is lowered to ∼pH 4.5, electrostatic interactions holding the 3-strand leaf of the prodomain β-sandwich weaken and peel away from the interface. This is likely to result in repositioning of the linker loop bringing cleavage site 1 close to the active site. At progressively lower pH, interactions involving the 4-strand leaf of the propiece β-sandwich are disrupted, resulting in a more open conformation. This allows sufficient kinking in the linker conformation so that site 2 can be brought close to the active site and targeted for endopeptidase action. At pH <3.0, the interdomain binding interface is completely disrupted, allowing access to site 3.
We propose a model of activation that takes into the account the pH-induced change in binding together with the pH-specific cleavage site preference (Fig. 6D). In the previously determined prokumamolisin structure, the cleavage site His171-Phe172 is positioned close to the active site Ser (Ser466) (supplemental Fig. S4). This corresponds to cleavage site 1 (Ser181-Leu182) in pro-TPP1, which is relatively close to the active site Ser475, with the carbonyl carbon of the scissile peptide being 12.0 Å away. In the absence of the lattice interactions that alter the position of the linker in our crystals grown at pH 5.0, this site is likely to approach Ser475. However, under the conditions that we use in our in vitro activation reactions, autocatalytic cleavage at pH 4.5 and above is extremely slow, with the intact pro-TPP1 zymogen persisting for days (data not shown). It is likely that tight binding between the prodomain and catalytic domain at this pH holds the zymogen in a conformation that favors the nonoptimal active site geometry, and that the small amount of cleavage observed might arise from local breathing motions that allow acquisition of a conformationally active catalytic site. However, given that the prodomain retains high affinity for the catalytic domain and remains associated even after cleavage (68), the equilibrium would favor the inactive conformation. At lower pH, the prodomain-catalytic domain interface could become destabilized, allowing assumption of a more conformationally optimal active site in the catalytic domain, as well as reorientation of the prodomain and linker. After endoproteolytic cleavage, the liberated propeptide as well as portions of the linker region still attached to the catalytic domain could then be further digested by the free catalytic domain (see below).
Consistent with this model, rates of pro-TPP1 activation are greatly accelerated in the presence of denaturants such as urea (supplemental Fig. S7) and guanidine hydrochloride (data not shown). In addition, the environment of late endosomes and lysosomes may be conducive to destabilization of the prodomain-catalytic domain complex, allowing rapid activation at the physiological pH of these organelles (pH 6 to 4.5) (76, 77). For instance, glycosaminoglycans have been shown to accelerate autoactivation of pro-TPP1 at mildly acidic pH (16), as do liposomes containing bis(monoacylglycero)phosphate (also called lysobisphosphatidic acid),6 an anionic lipid enriched in late endosomes (78). Nonetheless, decreasing the pH to optimize the protonation states of residues in the catalytic center, combined with reorientation of the catalytic serine and glutamic acid that would be favored by destabilization of the prodomain-catalytic domain interaction, are likely to be key elements in acquisition of activity.
Pathway for Pro-TPP1 Processing in Vitro—Analysis of hexahistidine-tagged glycosylated recombinant TPP1 using SDS-PAGE (Fig. 7A), chemical sequencing (Fig. 7C), and MALDI-TOF MS (data not shown) corroborates the MS analysis of the Endo H-deglycosylated untagged TPP1 (Fig. 6A), and it also reveals that autocatalytic processing involves a series of pH-dependent endoproteolytic cleavages and tripeptidyl aminopeptidase cleavages. At pH 4.0, the predominant initial cleavage products correspond to an intermediate starting at Val190 (Fig. 7C) and residues 20-189 of the propeptide (Fig. 7A, band marked F2 and MALDI-TOF MS; data not shown). Over time, the intermediate undergoes two tripeptidyl-peptidase cleavages, yielding an intermediate starting at Thr193 and the mature protein starting at Leu196 (Fig. 7C), whereas the liberated propeptide is degraded (Fig. 7A). At pH 2.5, the predominant initial cleavage products correspond to an intermediate starting at Phe169 (Fig. 7, A, band marked *, and C) and residues 20-168 of the propeptide (Fig. 7A, band marked F3 and MALDI-TOF MS; data not shown). With increasing time, the intermediate undergoes a second endoproteolytic cleavage to liberate the intermediate form starting at Val190 with subsequent TPP1 cleavage as above (Fig. 7C). Initial endoproteolytic cleavages of peptide bonds Asp168-Phe169 and Gln189-Val190 occur both at pH 3.0 and 2.5 (Fig. 7, A and C). In addition to the tripeptidylpeptidase cleavages of the form with an N-terminal Val190 observed at pH 4.0, there is also processing of the form starting at Phe169, yielding intermediates with N-terminal Gly172 and Arg175.
FIGURE 7.

Proteolytic processing and autoactivation of pro-TPP1 zymogen. A, autoproteolytic processing. Activation reactions were conducted using 1-1.5 μm pro-TPP1, and aliquots (typically 0.5-1 μg) were analyzed by SDS-PAGE as described under “Experimental Procedures.” Z, position of ∼66-kDa glycosylated pro-TPP1 zymogen; asterisk, ∼49-kDa intermediate; M, ∼46-kDa glycosylated mature TPP1; F1, F2, and F3, fragments cleaved at sites 1, 2, and 3, respectively, with assignment of F1 being based on MALDI-TOF analysis of deglycosylated TPP1 (Fig. 6A). B, comparison of proteolytic processing and acquisition of TPP1 activity. Two to four gels for each pH were used for the analysis and background-corrected intensity data normalized for each gel. The green upward pointing triangles indicate disappearance of zymogen at each time point, normalized to the intensity at t = 0 (e.g. 1 - Zt/Zo). The red downward pointing triangles indicate levels of liberated propeptide (F1 + F2 + F3), normalized to intensity of the highest value obtained for the reaction time course. Open circles indicate TPP1 activity measurements on activation reactions initiated using 1.5 μm zymogen. For comparison, data from Fig. 1 conducted using 15 nm protein are presented using shaded circles. Although the time course was similar at both concentrations for pH 4 and 3.5, there were noticeable differences at pH 2.5 and 3.0. For the latter reactions, the initial phase for acquisition of activity was similar, but the second phase was noticeably slower for the high concentration reactions. The reason for this is not entirely obvious, but it may reflect experimental considerations such as some reassociation of the prodomain after the activation reactions were arrested by dilution into pH 5.0 buffer. Similarly, the faster time course and coincidence of the acquisition of enzymatic activity and endoproteolysis at pH 4.0 that we observed previously (14) may reflect differences in experimental design, where the reactions were terminated by dilution into pH 4.0 buffer and then assayed immediately for TPP1 activity. Under these conditions, the propeptide would be expected to be largely dissociated from the mature protein. C, N-terminal sequence analysis of autocatalytically processed TPP1. Activation reactions were conducted using 1.5 μm pro-TPP1. Protein (17 pmol/time point) was resolved by SDS-PAGE and transferred to membranes. Bands of molecular mass ranging between 46 and 49 kDa were analyzed by chemical sequencing. The table lists the amount of each form in picomoles as estimated from the sequencing data (see “Experimental Procedures” for details). D, model for pro-TPP1 processing. Symbols are as in C and Fig. 6 (see text for details).
The observed cleavage pattern can be summarized by the pathway shown in Fig. 7D. This is entirely consistent with the known exopeptidase specificity of TPP1, where peptides with prolines at either P1 or P1′ are extremely poor substrates (20), thus preventing further exopeptidyl digestion past Arg175. The Leu182 N terminus produced by cleavage at pH 4.5 (Fig. 6A) (13) could not be processed by progressive tripeptidyl peptidase cleavages to yield mature TPP1 because of both frame and specificity considerations, and would be predicted to undergo a second endoproteolytic cleavage at Gln189-Val190 to yield the form with an N-terminal Leu196. The liberated propeptide has an N terminus SYSP that is unlikely to be efficiently digested by TPP1 exopeptidase activity. Thus, in the absence of any additional proteases, its hydrolysis probably involves TPP1 endoproteolytic cleavage. Activation at pH 4.0 can be accelerated by other proteases (data not shown), and given the protease-rich environment of the lysosome, it is likely that downstream hydrolysis of the initial propeptide cleavage product is not rate-limiting in vivo.
It has been recently reported that the propeptide can inhibit the activity of mature TPP1 at neutral and mildly acidic pH, with an IC50 in the micromolar range at pH 4.0 (68). Our results on comparing zymogen cleavage and acquisition of TPP1 activity are consistent with this. There was a marked lag between the two events at pH 4.0 (Fig. 7B, t½ cleavage ∼20 min; t½ activation ∼8 h). In contrast, the appearance and disappearance of the free propeptide could be fit with a two-step consecutive irreversible reaction model (36), with the second phase having a t½ of ∼7 h, paralleling that of acquisition of TPP1 activity. Thus, the lag is likely to reflect the time required for proteolysis of the inhibitory prodomain. The lag at lower pH values was much less pronounced, with the difference in the half-times between the first phase of the activation reaction and zymogen cleavage differing by at most a factor of 2 and both processes occurring rapidly (Fig. 7B, pH 2.5, 3.0, and 3.5; t½ cleavage 3, 6, and 20 s; and t½ activation 5, 12, and 20 s, respectively). This is consistent with rapid dissociation of the cleaved propeptide at more acidic pH values.
Biomedical Relevance—The structure of pro-TPP1 provides some insights into disease-causing mutations in LINCL (discussed in the accompanying paper by Pal et al. (81) (supplemental Fig. S8). There are currently 57 mutations listed in the NCL mutation data base. Roughly half (29 mutations) are missense mutations, some of which could respond to small molecule chaperone therapy directed at stabilizing the aberrant protein and thus rescuing function. For these, the structure of pro-TPP1 may help in identifying such small molecule therapeutics. However, most individuals with LINCL would not benefit from this approach as two mutations that encode totally nonfunctional proteins represent the majority of disease alleles (79). One is an invariant 3′ splice acceptor mutation in intron five that introduces a frameshift upstream of the Phe169 codon; the other is a nonsense mutation at codon Arg208 that does not appear to efficiently respond to potential therapeutics that promote translation readthrough (80).
Based on these and other considerations, any widely applicable therapy will need to target sufficient amounts of TPP1 to the brain to digest both newly arriving substrates and pre-existing lysosomal storage material. For enzyme replacement therapy using peripherally administered protein, it will be necessary to modify pro-TPP1 so that it can cross the blood-brain barrier (e.g. creation of a chimeric protein containing elements that allow transcytosis). In addition, for both enzyme and gene therapy approaches, it would be extremely useful to engineer functional TPP1 derivatives that have a longer half-life in the lysosome, thus increasing the effectiveness of properly targeted protein. Knowledge of the structure of TPP1 should be invaluable for such efforts.
Supplementary Material
Acknowledgments
We thank the Steinfeld and Sheldrick laboratories for sharing their manuscript and coordinates prior to submission; Bill Canfield for suggesting the use of and providing kifunensin to produce Endo H-sensitive pro-TPP1; John Taylor for advice and synthesis of fluorogenic peptide substrates; and Henry Lackland, Yanhong Wang, Caifeng Zhao, and Haiyan Zheng for invaluable assistance in protein production and analysis. We also thank the beamline staff at X4C and Seetharaman Jayaraman at the National Synchrotron Light Source at Brookhaven National Laboratories for technical help during data collection, Matthew Miller for assistance with data processing, and David Sleat and Vikas Nanda for helpful discussions.
The atomic coordinates and structure factors (code 3EDY) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant NS037918 (to P. L.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains Figs. S1-S8, Tables S1-S4, and additional references.
Author's Choice—Final version full access.
Footnotes
The abbreviations used are: LINCL, late infantile ceroid lipofuscinosis; TPP1, tripeptidyl-peptidase 1; CHO, Chinese hamster ovary; MOPS, 3-(N-morpholino)propanesulfonic acid; PDB, Protein Data Bank; TLS, translation/libration/screw; GlcNAc, N-acetyl-d-glucosamine; r.m.s.d., root mean square deviation; PB, Poisson-Boltzmann; APBS, adaptive PB solver; MS, mass spectrometry; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight; BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)-propane-1,3-diol; Endo H, endoglycosidase H; SAD, single wavelength anomalous dispersion.
R. Steinfeld and G. M. Sheldrick laboratories, personal communication.
J. Clardy, personal communication.
I. Sohar and P. Lobel, unpublished data.
References
- 1.Sleat, D. E., Donnelly, R. J., Lackland, H., Liu, C. G., Sohar, I., Pullarkat, R. K., and Lobel, P. (1997) Science 277 1802-1805 [DOI] [PubMed] [Google Scholar]
- 2.Rawlings, N. D., and Barrett, A. J. (1999) Biochim. Biophys. Acta 1429 496-500 [DOI] [PubMed] [Google Scholar]
- 3.Vines, D. J., and Warburton, M. J. (1999) FEBS Lett. 443 131-135 [DOI] [PubMed] [Google Scholar]
- 4.Cabrera-Salazar, M. A., Roskelley, E. M., Bu, J., Hodges, B. L., Yew, N., Dodge, J. C., Shihabuddin, L. S., Sohar, I., Sleat, D. E., Scheule, R. K., Davidson, B. L., Cheng, S. H., Lobel, P., and Passini, M. A. (2007) Mol. Ther. 15 1782-1788 [DOI] [PubMed] [Google Scholar]
- 5.Passini, M. A., Dodge, J. C., Bu, J., Yang, W., Zhao, Q., Sondhi, D., Hackett, N. R., Kaminsky, S. M., Mao, Q., Shihabuddin, L. S., Cheng, S. H., Sleat, D. E., Stewart, G. R., Davidson, B. L., Lobel, P., and Crystal, R. G. (2006) J. Neurosci. 26 1334-1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sondhi, D., Hackett, N. R., Peterson, D. A., Stratton, J., Baad, M., Travis, K. M., Wilson, J. M., and Crystal, R. G. (2007) Mol. Ther. 15 481-491 [DOI] [PubMed] [Google Scholar]
- 7.Sondhi, D., Peterson, D. A., Edelstein, A. M., del Fierro, K., Hackett, N. R., and Crystal, R. G. (2008) Exp. Neurol. 213 18-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Worgall, S., Sondhi, D., Hackett, N. R., Kosofsky, B., Kekatpure, M. V., Neyzi, N., Dyke, J. P., Ballon, D., Heier, L., Greenwald, B. M., Christos, P., Mazumdar, M., Souweidane, M. M., Kaplitt, M. G., and Crystal, R. G. (2008) Hum. Gene Ther. 19 463-474 [DOI] [PubMed] [Google Scholar]
- 9.Lin, L., and Lobel, P. (2001) Biochem. J. 357 49-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang, M., Cooper, J. D., Sleat, D. E., Cheng, S. H., Dodge, J. C., Passini, M. A., Lobel, P., and Davidson, B. L. (2008) Mol. Ther. 16 649-656 [DOI] [PubMed] [Google Scholar]
- 11.Golabek, A. A., and Kida, E. (2006) Biol. Chem. 387 1091-1099 [DOI] [PubMed] [Google Scholar]
- 12.Sohar, I., Sleat, D. E., and Lobel, P. (2004) in Handbook of Proteolytic Enzymes (Barrett, A. J., Rawlings, N. D., and Woessner, J. F., eds) 2nd Ed., pp. 1893-1896 Elsevier/Academic Press, San Diego
- 13.Golabek, A. A., Wujek, P., Walus, M., Bieler, S., Soto, C., Wisniewski, K. E., and Kida, E. (2004) J. Biol. Chem. 279 31058-31067 [DOI] [PubMed] [Google Scholar]
- 14.Lin, L., Sohar, I., Lackland, H., and Lobel, P. (2001) J. Biol. Chem. 276 2249-2255 [DOI] [PubMed] [Google Scholar]
- 15.Golabek, A. A., Kida, E., Walus, M., Wujek, P., Mehta, P., and Wisniewski, K. E. (2003) J. Biol. Chem. 278 7135-7145 [DOI] [PubMed] [Google Scholar]
- 16.Golabek, A. A., Walus, M., Wisniewski, K. E., and Kida, E. (2005) J. Biol. Chem. 280 7550-7561 [DOI] [PubMed] [Google Scholar]
- 17.Tsiakas, K., Steinfeld, R., Storch, S., Ezaki, J., Lukacs, Z., Kominami, E., Kohlschutter, A., Ullrich, K., and Braulke, T. (2004) Glycobiology 14 C1-C5 [DOI] [PubMed] [Google Scholar]
- 18.Page, A. E., Fuller, K., Chambers, T. J., and Warburton, M. J. (1993) Arch. Biochem. Biophys. 306 354-359 [DOI] [PubMed] [Google Scholar]
- 19.Ezaki, J., Takeda-Ezaki, M., Oda, K., and Kominami, E. (2000) Biochem. Biophys. Res. Commun. 268 904-908 [DOI] [PubMed] [Google Scholar]
- 20.Tian, Y., Sohar, I., Taylor, J. W., and Lobel, P. (2006) J. Biol. Chem. 281 6559-6572 [DOI] [PubMed] [Google Scholar]
- 21.Wlodawer, A., Li, M., Gustchina, A., Oyama, H., Dunn, B. M., and Oda, K. (2003) Acta Biochim. Pol. 50 81-102 [PubMed] [Google Scholar]
- 22.Rawlings, N. D., Tolle, D. P., and Barrett, A. J. (2004) Nucleic Acids Res. 32 D160-D164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dauter, Z., Li, M., and Wlodawer, A. (2001) Acta Crystallogr. Sect. D Biol. Crystallogr. 57 239-249 [DOI] [PubMed] [Google Scholar]
- 24.Wlodawer, A., Li, M., Dauter, Z., Gustchina, A., Uchida, K., Oyama, H., Dunn, B. M., and Oda, K. (2001) Nat. Struct. Biol. 8 442-446 [DOI] [PubMed] [Google Scholar]
- 25.Wlodawer, A., Li, M., Gustchina, A., Dauter, Z., Uchida, K., Oyama, H., Goldfarb, N. E., Dunn, B. M., and Oda, K. (2001) Biochemistry 40 15602-15611 [DOI] [PubMed] [Google Scholar]
- 26.Wlodawer, A., Li, M., Gustchina, A., Oyama, H., Oda, K., Beyer, B. B., Clemente, J., and Dunn, B. M. (2004) Biochem. Biophys. Res. Commun. 314 638-645 [DOI] [PubMed] [Google Scholar]
- 27.Comellas-Bigler, M., Fuentes-Prior, P., Maskos, K., Huber, R., Oyama, H., Uchida, K., Dunn, B. M., Oda, K., and Bode, W. (2002) Structure (Lond.) 10 865-876 [DOI] [PubMed] [Google Scholar]
- 28.Comellas-Bigler, M., Maskos, K., Huber, R., Oyama, H., Oda, K., and Bode, W. (2004) Structure (Lond.) 12 1313-1323 [DOI] [PubMed] [Google Scholar]
- 29.Okubo, A., Li, M., Ashida, M., Oyama, H., Gustchina, A., Oda, K., Dunn, B. M., Wlodawer, A., and Nakayama, T. (2006) FEBS J. 273 2563-2576 [DOI] [PubMed] [Google Scholar]
- 30.Wlodawer, A., Li, M., Gustchina, A., Tsuruoka, N., Ashida, M., Minakata, H., Oyama, H., Oda, K., Nishino, T., and Nakayama, T. (2004) J. Biol. Chem. 279 21500-21510 [DOI] [PubMed] [Google Scholar]
- 31.Dodson, G., and Wlodawer, A. (1998) Trends Biochem. Sci. 23 347-352 [DOI] [PubMed] [Google Scholar]
- 32.Kraut, J. (1977) Annu. Rev. Biochem. 46 331-358 [DOI] [PubMed] [Google Scholar]
- 33.Wlodawer, A., Durell, S. R., Li, M., Oyama, H., Oda, K., and Dunn, B. M. (2003) BMC Struct. Biol. 3 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walus, M., Kida, E., Wisniewski, K. E., and Golabek, A. A. (2005) FEBS Lett. 579 1383-1388 [DOI] [PubMed] [Google Scholar]
- 35.Sohar, I., Lin, L., and Lobel, P. (2000) Clin. Chem. 46 1005-1008 [PubMed] [Google Scholar]
- 36.Ferscht, A. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, p. 144, W. H. Freeman & Co., New York
- 37.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 38.Schneider, T. R., and Sheldrick, G. M. (2002) Acta Crystallogr. Sect. D Biol. Crystallogr. 58 1772-1779 [DOI] [PubMed] [Google Scholar]
- 39.Pape, T., and Schneider, T. R. (2004) J. Appl. Crystallogr. 37 843-844 [Google Scholar]
- 40.Terwilliger, T. C. (2000) Acta Crystallogr. Sect. D Biol. Crystallogr. 56 965-972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Terwilliger, T. C., and Berendzen, J. (1999) Acta Crystallogr. Sect. D Biol. Crystallogr. 55 849-861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams, P. D., Grosse-Kunstleve, R. W., Hung, L. W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K., and Terwilliger, T. C. (2002) Acta Crystallogr. Sect. D Biol. Crystallogr. 58 1948-1954 [DOI] [PubMed] [Google Scholar]
- 43.Cowtan, K. D., and Main, P. (1993) Acta Crystallogr. Sect. D Biol. Crystallogr. 49 148-157 [DOI] [PubMed] [Google Scholar]
- 44.Project, C. C. (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-763 [DOI] [PubMed] [Google Scholar]
- 45.Lamzin, V. S., and Wilson, K. S. (1993) Acta Crystallogr. Sect. D Biol. Crystallogr. 49 129-147 [DOI] [PubMed] [Google Scholar]
- 46.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 47.Emsley, P., and Cowtan, K. D. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 48.Painter, J., and Merritt, E. A. (2005) Acta Crystallogr. Sect. D Biol. Crystallogr. 61 465-471 [DOI] [PubMed] [Google Scholar]
- 49.Painter, J., and Merritt, E. A. (2006) Acta Crystallogr. Sect. D Biol. Crystallogr. 62 439-450 [DOI] [PubMed] [Google Scholar]
- 50.Painter, J., and Merritt, E. A. (2006) J. Appl. Crystallogr. 39 109-111 [Google Scholar]
- 51.Kleywegt, G. J., and Jones, T. A. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 1119-1131 [DOI] [PubMed] [Google Scholar]
- 52.Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B., III, Snoeyink, J., Richardson, J. S., and Richardson, D. C. (2007) Nucleic Acids Res. 35 W375-W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 54.Kabsch, W. (1976) Acta Crystallogr. Sect. A 32 922-923 [Google Scholar]
- 55.Krissinel, E., and Henrick, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2256-2268 [DOI] [PubMed] [Google Scholar]
- 56.Jones, S., and Thornton, J. M. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 13-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reynolds, C., Damerell, D., and Jones, S. (2008) Bioinformatics (Oxf.) [DOI] [PubMed]
- 58.Diemand, A. V., and Scheib, H. (2004) Nucleic Acids Res. 32 W512-W516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vriend, G. (1990) J. Mol. Graphics 8 52-56, 29 [DOI] [PubMed] [Google Scholar]
- 60.DeLano, W. L. (2002) The Pymol Molecular Graphics System, DeLano Scientific, San Carlos, CA
- 61.Baker, N. A., Sept, D., Joseph, S., Holst, M. J., and McCammon, J. A. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 10037-10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dolinsky, T. J., Czodrowski, P., Li, H., Nielsen, J. E., Jensen, J. H., Klebe, G., and Baker, N. A. (2007) Nucleic Acids Res. 35 W522-W525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dolinsky, T. J., Nielsen, J. E., McCammon, J. A., and Baker, N. A. (2004) Nucleic Acids Res. 32 W665-W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li, H., Robertson, A. D., and Jensen, J. H. (2005) Proteins 61 704-721 [DOI] [PubMed] [Google Scholar]
- 65.Junaid, M. A., Wu, G., and Pullarkat, R. K. (2000) J. Neurochem. 74 287-294 [DOI] [PubMed] [Google Scholar]
- 66.Chang, V. T., Crispin, M., Aricescu, A. R., Harvey, D. J., Nettleship, J. E., Fennelly, J. A., Yu, C., Boles, K. S., Evans, E. J., Stuart, D. I., Dwek, R. A., Jones, E. Y., Owens, R. J., and Davis, S. J. (2007) Structure (Lond.) 15 267-273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wujek, P., Kida, E., Walus, M., Wisniewski, K. E., and Golabek, A. A. (2004) J. Biol. Chem. 279 12827-12839 [DOI] [PubMed] [Google Scholar]
- 68.Golabek, A. A., Dolzhanskaya, N., Walus, M., Wisniewski, K. E., and Kida, E. (2008) J. Biol. Chem. 283 16497-16504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Diederichs, K., and Karplus, P. A. (1997) Nat. Struct. Biol. 4 269-275 [DOI] [PubMed] [Google Scholar]
- 70.Weiss, M. S. (2001) J. Appl. Crystallogr. 34 130-135 [Google Scholar]
- 71.Weiss, M. S., and Hilgenfeld, R. (1997) J. Appl. Crystallogr. 30 203-205 [Google Scholar]
- 72.Menard, R., and Storer, A. C. (1992) Biol. Chem. Hoppe-Seyler 373 393-400 [DOI] [PubMed] [Google Scholar]
- 73.Guo, H., Wlodawer, A., Nakayama, T., and Xu, Q. (2006) Biochemistry 45 9129-9137 [DOI] [PubMed] [Google Scholar]
- 74.Xu, Q., Guo, H. B., Wlodawer, A., Nakayama, T., and Guo, H. (2007) Biochemistry 46 3784-3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khan, A. R., and James, M. N. (1998) Protein Sci. 7 815-836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Majumdar, A., Cruz, D., Asamoah, N., Buxbaum, A., Sohar, I., Lobel, P., and Maxfield, F. R. (2007) Mol. Biol. Cell 18 1490-1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maxfield, F. R., and McGraw, T. E. (2004) Nat. Rev. Mol. Cell Biol. 5 121-132 [DOI] [PubMed] [Google Scholar]
- 78.Kolter, T., and Sandhoff, K. (2005) Annu. Rev. Cell Dev. Biol. 21 81-103 [DOI] [PubMed] [Google Scholar]
- 79.Sleat, D. E., Gin, R. M., Sohar, I., Wisniewski, K., Sklower-Brooks, S., Pullarkat, R. K., Palmer, D. N., Lerner, T. J., Boustany, R. M., Uldall, P., Siakotos, A. N., Donnelly, R. J., and Lobel, P. (1999) Am. J. Hum. Genet. 64 1511-1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sleat, D. E., Sohar, I., Gin, R. M., and Lobel, P. (2001) Eur. J. Paediatr. Neurol. 5 Suppl. A, 57-62 [DOI] [PubMed] [Google Scholar]
- 81.Pal, A., Kraetzner, R., Gruene, T., Grapp, M., Schreiber, K., Gronborg, M., Urlaub, H., Becker, S., Asif, A. R., Gärtner, J., Sheldrick, G. M., and Steinfeld, R. (2009) J. Biol. Chem. 264 in press [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.