

Abstract
To study the oncogenic role of the NRAS oncogene (NRASG12V) in the context of acute myeloid leukemia (AML), we used a Vav promoter–tetracycline transactivator (Vav-tTA)–driven repressible TRE-NRASG12V transgene system in Mll-AF9 knock-in mice developing AML. Conditional repression of NRASG12V expression greatly reduced peripheral white blood cell (WBC) counts in leukemia recipient mice and induced apoptosis in the transplanted AML cells correlated with reduced Ras/Erk signaling. After marked decrease of AML blast cells, myeloproliferative disease (MPD)–like AML relapsed characterized by cells that did not express NRASG12V. In comparison with primary AML, the MPD-like AML showed significantly reduced aggressiveness, reduced myelosuppression, and a more differentiated phenotype. We conclude that, in AML induced by an Mll-AF9 transgene, NRASG12V expression contributes to acute leukemia maintenance by suppressing apoptosis and reducing differentiation of leukemia cells. Moreover, NRASG12V oncogene has a cell nonautonomous role in suppressing erythropoiesis that results in the MPD-like AML show significantly reduced ability to induce anemia. Our results imply that targeting NRAS or RAS oncogene-activated pathways is a good therapeutic strategy for AML and attenuating aggressiveness of relapsed AML.
Introduction
Kelly and Gilliand have proposed that acute myeloid leukemia (AML) is induced by the cooperation of 2 general classes of mutations.1 Class I mutations confer cell survival and proliferation advantages and generally result from mutations in genes encoding cell-signaling molecules like NRAS and FLT3. Class II mutations impair differentiation and subsequent apoptosis and, in general, result from mutations in genes encoding transcription factors or chromatin-modifying proteins such as AML1 or mixed lineage leukemia (MLL). Most studies to develop new anticancer drugs have focused on finding efficient inhibitors against the mutant products of class I oncogenes such as ABL, c-KIT, and FLT3.2–8 Although previous studies showed that FLT3 inhibition effectively suppresses the cell growth of leukemia induced by the cooperation of an FLT3-activating mutation and an MLL translocation,9,10 the appropriateness of class I oncogene inhibitors for AML therapy has not been well studied, particularly in leukemia induced in cooperation with a class II oncogene. Conditionally expressed transgenes in mice provide an ideal setting in which to address this issue. Studies have shown that continued expression of oncogenes such as c-Myc or Bcr/Abl is required for maintaining leukemia in transgenic mouse models.11–13 Cancer cells in these models are said to have “oncogene addiction,” because they die, differentiate, or become quiescent upon shutting off oncogene expression.13 To study the functions of a class I mutation in AML, we investigated the role of an NRAS-activating mutation, NRASG12V, in murine AML induced in combination with the Mll-AF9 oncogene
RAS mutants, which abnormally stimulate RAF/MEK/ERK, PI3K/AKT, and other pathways, are frequent in human cancers.14,15 RAS mutations are commonly single amino acid substitutions such as G12V and G13R that reduce guanine triphosphate (GTP) activating protein sensitivity of RAS, thus causing maintenance of high levels of RAS-GTP.14,16 Among the RAS family mutations, NRAS activating mutations are found in around 30% of hematopoietic malignances including AML, myelodysplastic syndrome, and myeloproliferative diseases (MPD).17,18 To study the function of oncogenic NRAS in hematopoietic malignancies, we previously developed a Vav promoter–tetracycline transactivator (Vav-tTA)–driven repressible system for tetracycline response element (TRE)–NRASG12V expression in mice, and found that NRASG12V expression induces a mast cell disease similar to human aggressive systemic mastocytosis (ASM).19 Our data are consistent with other results showing that an activated NRAS mutant can induce mastocytosis as well as AML or a chronic myeloid leukemia (CML)–like leukemia when delivered to mice using a retroviral vector.15,19,20
The MLL gene encodes a trithorax-homologous transcription factor and is a positive regulator of HOX gene expression in development. MLL translocations resulting in fusions with other genes are common in AML and acute lymphoblastic leukemia (ALL), and more than 40 fusion partners are known.21–23 The MLL-AF9 translocation is the most common MLL gene fusion found in AML and is associated with a poor prognosis.24,25 Mll-AF9 knockin mice provide a model of human t(9;11)(p22;q23) translocation.26 These mice significantly expand nonmalignant myeloid progenitor cells during fetal development and most of them develop AML after 5 to 6 months.27,28 It has been suggested that the MLL-AF9 oncogene disrupts the normal proliferation and/or survival of myeloid progenitors and that secondary oncogenic mutations are required for full hematopoietic malignancy.27–29 The MLL-AF9 translocation occurs predominantly in monocytic AML (AML-M5; French-American-British classification).30 Previous studies found that mutations of FLT3, RAS, or BRAF genes are associated with the MLL-AF9–mediated leukemogenesis in 140 therapy-related MDS and AML (tMDS/tAML) patients.21,31
Here we report a model of AML development using transgenic mice coexpressing NRASG12V and Mll-AF9 and a requirement of continuous NRASG12V expression for maintaining the AML. NRASG12V-independent MPD-like AML occurring upon subsequent relapse is less myelosuppressive and more differentiated than AML before the suppression of NRASG12V.
Methods
Mice and treatment
To generate Vav-tTA; TRE-NRASG12V; Mll-AF9 triply transgenic (TRM-transgenic) and control mice under the same strain (FVB/n × C57BL/6J) F1 background, we crossed the Vav-tTA; TRE-NRASG12V doubly transgenic (TR-transgenic) FVB/n line previously developed in our laboratory and the Mll-AF9 knockin C57BL/6J strain kindly provided by Dr Terence H. Rabbitts (MRC Laboratory of Molecular Biology, Cambridge, United Kingdom; Figure 1A).19,26,27 Severe combined immunodeficiency (SCID)/NCr-Balb/c (SCID) mice were purchased from National Cancer Institute (Frederick, MD) for transplantation of leukemia cells. To repress Vav-tTA–mediated TRE-NRASG12V transgene expression, we added doxycycline hyclate (Dox; Sigma-Aldrich, St Louis, MO) to sterilized drinking water with 0.1% sodium saccharin (Alfa Aesar, Ward Hill, MA) to a final Dox concentration of 5 mg/mL.20 To repress the NRASG12V expression in TR-transgenic new litters, we treated parental mice with Dox during the pregnancy (approximately 21 days) and maternal feeding periods (21 days) before weaning. For continuous treatment, the Dox solution was changed every 5 to 6 days and protected from light. For temporary treatment, normal drinking water bottles replaced the Dox-treated water bottle after 6 hours. Because some SCID mice were moribund with fulminant AML, we also washed mice's mouths with 250 μL Dox-treated water to ensure the uptake of the Dox in drinking water. The transgenic mice were housed under specific pathogen–free conditions and cared for by protocols approved by the Institutional Animal Care and Use Committee at the University of Minnesota. The SCID mice were housed under the aseptic conditions with autoclaved cages, beds, water, bottles, and irradiated foods. When mice were moribund they were humanely killed in a CO2 chamber for harvesting the required tissues.
Figure 1.

AML is induced by the combination of NRASG12V and Mll-AF9. (A) Breeding scheme for generating experimental animals. Boxes indicate transgenes present. (B) Kaplan-Meier survival curve. Vav-tTA; TRE-NRASG12V; Mll-AF9 triply transgenic (TRM-transgenic) and Vav-tTA; Mll-AF9 doubly transgenic (TM-transgenic) mice developed AML and died significantly earlier than Vav-tTA; TRE-NRASG12V doubly transgenic (TR-transgenic) mice (P < .05). The TRM-transgenic mice showed a trend of decreased latency for AML compared with those carrying the TM transgene (P = .072). Bidirectional arrow indicates the prenatal period of Dox treatment. (C) Immunophenotypes of BM and spleen cells in AML mice. Mac1 and Gr1 double-positive cells are greater than those of FBV/n × C57BL/6J F1 mice. (D) NRASG12V transcription is repressed by Dox treatment. Vav-tTA-driven TRE-NRASG12V expression is found in all Vav-tTA; TRE-NRASG12V cotransgenic BM cells with or without Mll-AF9 transgene and mastocytosis tumor cells (lanes 2, 5, 6, and 7). The NRASG12V expression is completely repressed by Dox treatment (lane 1). GAPDH is used as a loading control. No RT-PCR products were found in the same procedures without reverse transcriptase (data not shown). The genotypes and cell types along with numbers indicate the transgenic cells used for mRNA preparations. BM indicates bone marrow; MT, mastocytosis tumor; and W, water control.
Cell preparation and transplantation
Bone marrow (BM) cells were flushed from the femurs and tibias with 1× Dulbecco modified Eagle medium (DMEM; Cellgro, Herndon, VA). The single-cell suspensions from spleen, thymus, lymph node, and mastocytosis tumors were prepared by grinding tissues with glass caps and screening with 70-μm cell strainers (BD Falcon, Bedford, MA). As described in our previous paper,20 nucleated cells were isolated from the prepared cells and were viably saved in liquid nitrogen for further experiments.
BM cells from 4 independent TRM-transgenic mice and BM cells from 2 independent Vav-tTA; Mll-AF9 cotransgenic (TM-transgenic) FVB/n × BL6 F1 mice with AML were transplanted into 16 primary recipient SCID mice by intravenous injections at 1.6 to 2.6 × 106 cells per mouse and into 8 primary recipient SCID mice at 2.0 to 2.8 × 106 cells per mice, respectively (Figure 2A). Furthermore, BM cells originating from 2 independent TRM-transgenic mice with AML and from 1 TM-transgenic mouse with AML that were previously engrafted on the primary recipient SCID mice without Dox treatment (Figure 2A) were intravenously injected into secondary recipient SCID mice at 0.9 to 1.3 × 106 cells per mouse (7 and 8 mice used for the repeated transplantation experiments of AML2, and 8 mice used for a transplantation of AML4) and at 1.2 to 1.4 × 106 cells per mouse (12 mice), respectively (Figure 3A). At last, the BM cells from the secondary recipient SCID mice that showed NRASG12V-independent relapsed MPD-like AML after temporary (6-hour) or chronic Dox treatment (Figure 3A) were transplanted into 8 or 5 tertiary SCID mice, respectively, at 0.8 to 1.2 × 106 cells per mouse (see Figure 6A). As a control, TM-transgenic AML BM cells from previously xenografted SCID mice were also transplanted into 8 other SCID mice at 1.1 × 106 cells per mouse.
Figure 2.

NRASG12V expression is required for AML persistence. (A) Transplantation of AML BM cells via intravenous injection. Half of the transplanted SCID mice were continuously treated with Dox. (B) Kaplan-Meier survival curve in mice with or without Dox treatment. Chronic Dox treatment significantly delays or suppresses the establishment of AML in SCID mice after transplantation of the BM cells from triply transgenic mice with AML (P = .012). The numbers in circles represent each of the 4 independent AML BM cells (AML1, 2, 3, and 4) transplanted into SCID mice.
Figure 3.

Conditional repression of NRASG12V expression after the establishment of AML results in remission, prolong survival, and relapse. (A) Serial transplantation of previously engrafted AML BM cells into secondary recipient SCID mice. The secondary recipient SCID mice were treated with Dox either chronically or for 6 hours at a time after the establishment of full-blown AML. (B) Kaplan-Meier survival curve of all AML2 and AML4 transplantations shows that both chronic and temporary Dox treatments significantly increase the survival time compared with those transplanted mice not treated with Dox after the establishment of AML (P < .001). The arrow indicates the time that chronic or 6-hour Dox treatment started. (C) WBC concentration in the peripheral blood of the secondary recipient SCID mice that were transplanted with AML2 BM cells from TRM-transgenic mice. The first Dox in the arrowed square means the start of chronic and temporary (6-hour) Dox treatments. Other Dox in the arrowed squares indicated the repeated 6-hour Dox treatment. Chronic Dox in the top panel means the continuous Dox treatment instead of the 6-hour Dox treatment was started at this time point. In the top panel, n = 4 were treated with chronic and n = 3 with 6-hour Dox. In the bottom panel, n = 2 were treated with chronic, n = 2 with 6-hour, n = 4 with no, and n = 3 (Mll-AF9 only) with chronic Dox. Narrow bars show standard deviations for WBC concentration at each measurement. (D) Flow cytometry of spleen cells from the engraftment, remission, and relapse in secondary recipient SCID mice. The spleen of engrafted mice contains greater than 50% of myeloid (Mac1/Gr1+) lineage. After 8 days of chronic Dox treatment, most GFP-positive cells are eliminated. However, after relapse, the spleen contains more than 70% GFP/Mac1 or GFP/Gr1 double-positive cells. (E) Western blot analysis for transgenic NRASG12V and endogenous Nras proteins. Due to the EE tag at the N terminus of NRASG12V, the molecular weight of NRASG12V is approximately 1 kDa larger than endogenous Nras. Both NRASG12V and endogenous Nras proteins are decreased at day 4 and not detected at day 8 after chronic Dox treatment. GAPDH is used as a loading control.
Figure 6.

Relapsed MPD-like AML is transplantable but requires a longer latency for disease induction. (A) BM cells from secondary recipient mice with MPD-like AML (after chronic or temporary Dox treatment) were transplanted into tertiary recipient SCID mice. As a control, BM cells from previously transplanted AML (not treated with Dox) were intravenously injected into control SCID mice. (B) Kaplan-Meier survival curve shows that the SCID recipients of BM from relapsed MPD-like AML cases have significantly longer survival time in comparison with those mice transplanted with AML that were never treated with Dox (P < .001). (C) Western blot analysis of spleen cells shows that the NRASG12V transgene is expressed again in the engrafted AML after BM transplantation with the relapsed MPD-like AML that has been treated with Dox for 6 hours, but not with chronic Dox treatment. Circled numbers over each lane indicate each protein sample from tertiary recipient mice with (1) AML engraft without Dox treatment; (2) AML regression after 4-day Dox treatment; (3) AML relapse after 6-hour Dox treatment; (4) AML engraft without Dox treatment after relapsed AML (with 6-hour DOX) transplantation; (5) AML relapse with chronic Dox treatment; (6) AML engraft after relapsed AML (with chronic Dox) transplantation; and (7) TM-transgenic AML.
Peripheral blood analysis
We obtained peripheral blood by retro-orbital puncture every 4 weeks for the FVB/n × C57BL/6J F1 mice that developed AML and every 2 days for the SCID mice 2 to 3 weeks after AML BM transplantations. After anesthetizing eyes with 0.5% ophthalmic solution of proparacaine hydrochloride (Falcon Pharmaceuticals, Fort Worth, TX), 200 μL blood were collected with EDTA-coated capillary tubes (Drummond Scientific, Broomall, PA) and transferred to a blood collection tube (Ram Scientific, Needham, MA). The peripheral blood samples were analyzed by an automated veterinary blood analyzer (MASCOT; CDC Technology, Oxford, CT) to measure the concentrations of white blood cells (WBCs), red blood cells (RBCs), and other blood components. To analyze the nuclear shapes in leukemia blast cells and differentiated leukocytes under the microscopes, we stained peripheral blood smears on glass slides using modified Wright-Giemsa staining methods.19
Flow cytometry
To test the immunophenotypes of nucleated cells, 106 BM, 3 × 106 spleen, 106 thymus, or 106 lymph node cells from TRM-transgenic and control mice were used. The hematopoietic cells were suspended in 200 μL phosphate-buffered saline (PBS) buffer (2% fetal bovine serum, 0.1% sodium azide). We used allophycocyanin (APC)–anti-Mac1 and phycoerythrin (PE) or PE-Cy7–anti-Gr1 antibodies to sort myeloid cells, APC anti–T-cell receptor β (TCRβ) antibodies to sort T lymphoid cells, and PE anti-CD19 antibody to sort B lymphoid cells from the green fluorescent protein (GFP)–expressing TRE-NRASG12V–transgenic mice. Fluorescein isothiocyanate (FITC) anti-Mac1 or FITC anti-CD19 antibodies were used to sort myeloid and B cells, respectively, from wild-type mice. All fluorochrome-conjugated antibodies were purchased from BD Biosciences (San Jose, CA). The staining procedures were the same as described in our previous paper.20 The sorting procedures and conditions using the FACSAria Cell-Sorting System (BD Biosciences) were according to the manufacturer.
Reverse transcription polymerase chain reaction (RT-PCR) and cDNA cloning
Using a direct mRNA micro kit (QIAGEN, Valencia, CA), we prepared mRNA from BM cells or mastocytosis tumors in variously transgenic FVB/n × C57BL/6J F1 mice, as shown in Figure 1A. To avoid DNA contamination, we treated mRNA samples with DNaseI (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. To detect the expression of transgenic NRASG12V, we used a pair of DNA primers, 5′-GTTATAGATGGTGAAACCTGTT-3′ and 5′-ATCTTGTTACATCACCACACAT-3′, and RT-PCR conditions previously applied in our paper.19 Endogenous glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcripts as a loading control were also amplified using another pair of DNA primers, 5′-AACGACCCCTTCATTGAC-3′ and 5′-TCCACGACATACTCAGCAC-3′. RT-PCR was performed using the Robust-RT kit (Finnzymes; MJ Research, Waltham, MA). We also performed RT-PCR without reverse transcriptase to ensure the RT-PCR products were not amplified from contaminated chromosomal DNA.
To clone the cDNA of endogenous Nras transcripts in BM cells of the secondary recipient SCID mice shown in Figure 3A, we prepared mRNA using the same method described above. To amplify the endogenous Nras, we designed a pair of DNA primers, 5′-GGAGTTTGAGGTTTTTGCTG-3′ and 5′-GTGTCTTACTACATCAGCAC-3′, to anneal the 5′ untranslated region (UTR) and the 3′ UTR expanded to coding region, respectively. Using the Robust-RT kit, RT-PCR, entailed incubation at 45°C for 45 minutes was followed by 5 minutes at 94°C, then 40 cycles at 94°C for 30 seconds, 45°C for 1 minute, and 72°C for 1 minute. These cycles were followed by a final extension at 72°C for 10 minutes. The cDNA was purified using the agarose gel extraction, and cloned into pCR-XL-TOPO vector (Invitrogen). Using the EcoRI restriction enzyme reaction, we confirmed that the plasmid prepared from 10 independent transformant Escherichia coli colonies contained the RT-PCR products. The cloned plasmids were sent to an automated sequencing core facility with universal sequencing primers, M13 reverse primer and M13 forward primer, to learn whether any mutations occurred in the endogenous Nras.
Western blot analysis
Spleen cells from engrafted or relapsed AML SCID mice were lysed in RIPA buffer (0.05 M Tris [pH 8.0], 0.15 M NaCl, 0.005% sodium deoxycholate, 0.01% Igepal CA-630, and 0.1% sodium dodecyl sulfate [SDS]) containing protease inhibitors (Complete Mini; Roche, Indianapolis, IN). Each protein extract (40-50 μg) was separated by the electrophoresis on 4%-12% gradient polyacrylamide gels (Invitrogen) and transferred to nitrocellulose membranes (Trans-Blot Transfer Medium; Bio-Rad Laboratories, Hercules, CA). The protein-blotted membrane was blocked with TBS-T (1 M Tris base, 18% NaCl, 0.05% Tween 20, pH 7.4) solution containing 1% skim milk (Difco, Sparks, MD) and 4% bovine serum albumin (BSA; Sigma-Aldrich). Then, a mouse monoclonal antibody anti-Nras (Santa Cruz Biotechnology, Santa Cruz, CA), a mouse monoclonal antibody anti-Erk1/2, a rabbit polyclonal antibody anti–phospho-Erk1/2, or a rabbit monoclonal antibody anti-GAPDH (Cell Signaling Technology, Danvers, MA) was used as a primary antibody to detect the target proteins. The horseradish peroxidase linked whole antibody anti–mouse immunoglobulin G (IgG; from sheep) or anti–rabbit IgG (from donkey) purchased from GE Healthcare BioScience (Piscataway, NJ) was used as a secondary antibody. The antibody-labeled proteins were illuminated with chemiluminescent substrates (Pierce Biotechnology, Rockford, IL) and exposed to imaging films (Kodak, Rochester, NY).
Histopathologic analysis
Tissues (spleen, liver, lymph node, thymus, lung, and sternum containing BM) collected from the mice were fixed in 10% neutral-buffered formalin for 24 hours and transferred into 70% ethanol for storage. Tissues were processed by standard methods and embedded in paraffin. Four-micrometer-thick sections were cut and stained with hematoxylin and eosin. Tissues were read by American College of Veterinary Pharmacists (ACVP) board-certified veterinary pathologist (I.M.) to determine whether hematopoietic neoplasia was present and to classify lesions further as lymphoid or nonlymphoid hematopoietic neoplasia when possible. Bethesda guidelines for classification of nonlymphoid hematopoietic malignancies in mice were followed.32
Apoptotic cell staining
For cleaved caspase-3 detection in the tissues, 4-μm formalin-fixed paraffin sections of spleen and BM were cut, deparaffinized, and rehydrated. Except for antigen retrieval and counterstaining, all procedures were carried out on a DAKO autostainer (DAKO, Carpinteria, CA) using a rabbit monoclonal antibody against human cleaved Caspase-3 (clone Asp175 5A1; Cell Signaling Technologies) diluted at 1:200. Immunocytochemical staining procedure for detection of cleaved caspase-3 in peripheral blood smears was similar except for following details: air-dried blood smears were fixed in 10% neutral-buffered formalin for 2 minutes, antigen retrieval was not performed, and endogenous peroxidase was blocked with 0.3% hydrogen peroxide solution at room temperature for 15 minutes.
Terminal deoxynucleotidyl transferase (TdT)–mediated dUTP-biotin nick end labeling (TUNEL) staining was performed on 4-μm formalin-fixed paraffin sections of BM in selected mice using the ApopTag Plus kit (Chemicon, Temecula, CA). The sections were deparaffinized, rehydrated, and incubated with protease K (20 μg/mL; R&D Systems, Minneapolis, MN) for 15 minutes at room temperature. The staining procedures were performed according to manufacturer protocols. Methyl Green was used as the counterstain. Equilibration buffer was substituted for the volume of TdT enzyme to serve as a negative control. For semiquantitative evaluation of the extent of apoptosis in the BM, the number of positive (dark brown cells) were counted in 3 representative fields at ×400 magnification, and the average per field was estimated in selected mice. No attempts were made to distinguish between neoplastic cell apoptosis and background tissue cell apoptosis when evaluating the extent of apoptosis. For estimation of apoptosis in the peripheral blood smears, the number of positive (brown) staining cells was counted in 40 to 50 nonoverlapping fields starting from the feathered edge and ending at the thickest portion of the smear.
Statistical analysis
All graphs were generated using Microsoft Excel. Statistical differences (P values) were determined by the 2-tailed Student t test assuming equal variances of the Microsoft Excel data analysis program. Kaplan-Meier survival curve generation and statistical analysis were performed using Prism 4 (GraphPad Software, San Diego, CA).
Results
NRASG12V and Mll-AF9 coexpression induces AML
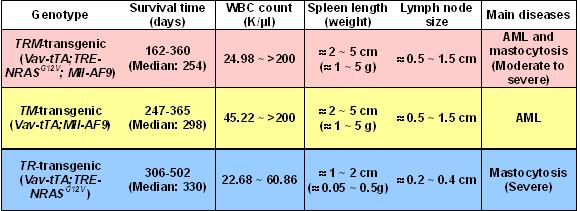
To test the hypothesis that 2 classes of mutations could cooperate to induce leukemia, we generated Vav-tTA; TRE-NRASG12V; Mll-AF9 triply transgenic (TRM-transgenic) mice by crossing Vav-tTA; TRE-NRASG12V doubly transgenic (TR-transgenic) FVB/n mice to Mll-AF9 knockin C57BL/6J mice (Figure 1A). The TRM-transgenic mice developed AML along with mastocytosis and showed a trend toward disease acceleration (P = .072) compared with Vav-tTA; Mll-AF9 doubly transgenic (TM-transgenic) control mice developing AML without mastocytosis (Figure 1B and Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). The TR-transgenic mice developed mastocytosis with no evidence of AML. Dox treatment before weaning as described in “Mice and treatment” did not significantly alter survival rates of TRM- and TM-transgenic mice (Figure S2).
The AML phenotype in TRM-transgenic mice was verified by flow cytometry. Mac1/Gr1 double-positive cells were enriched in BM (> 50% ≈ 70%) and spleen (> 20% ≈ 25%) of the TRM-transgenic and TM-transgenic mice, compared with only 40% and 1% in BM and spleen of wild-type mice, respectively (Figure 1C). The hematopoietic malignancies were also phenotyped by histopathologic analysis as described in Figure S3. Using RT-PCR, we determined that Dox treatment conditionally repressed the Vav-tTA–driven TRE-NRASG12V expression BM cells (Figure 1D). In conclusion, addition of an NRASG12V mutation to AML predisposed mice harboring an Mll-AF9 fusion knock-in causes a trend toward decreased latency for developing AML, although NRASG12V is not specifically required for AML development in cooperation with Mll-AF9.
Continuous NRASG12V expression is required for leukemia persistence and apoptosis suppression in AML induced in combination with Mll-AF9
To determine whether NRASG12V expression is essential for AML persistence, we transplanted BM cells from a single TRM-transgenic mouse into 4 SCID mice. Two of the transplanted mice were continuously treated with Dox, while the other 2 did not receive Dox (Figure 2A). We repeated this experiment 4 times with 4 different TRM-transgenic AML samples (Figure 2B, AML1, 2, 3, and 4). The transplantability of 3 of 4 established AMLs were significantly attenuated after Dox treatment (overall P = .012; Figure 2B). The survival rate of SCID mice transplanted with AML cells from TM-transgenic mice was not altered by Dox treatment (P = .508; data not shown). Mastocytosis was not transplantable as previously reported.19 Using flow cytometry and histopathologic analysis, we confirmed that the engrafted leukemia was equivalent to the AML seen in the donor mice (data not shown). Taken together, these data show that AML induced by the cooperation of NRASG12V and Mll-AF9 is transplantable and loss of NRASG12V expression significantly reduces the aggressiveness of the disease as indicated by survival time.
To test whether NRASG12V is required for persistence of established AML, we performed a serial BM transplantation experiment. We selected BM originating from 2 different TRM-transgenic mice (Figure 2B, AML2 and AML4). BM cells from “No Dox” primary recipients of AML2 were pooled and transplanted into secondary SCID recipients. This was repeated using AML4 BM cells (Figure 3A). By measuring the WBC concentration in peripheral blood every 2 days from 21 days after transplantation, we determined when AML had become established in recipient mice. Once the AML was established in the secondary recipients, we chronically or temporarily (6 hours) treated mice with Dox in drinking water. The conditional repression of NRASG12V expression after the establishment of full-blown AML (n = 17) significantly prolonged the survival time compared with recipients without Dox treatment (n = 6; P < .001; Figure 3B).
Four days after starting either chronic or 6-hour Dox treatment, WBC concentration in peripheral blood was dramatically reduced in all recipients of AML2 BM cells and completely normalized in 6 to 10 days (Figure 3C top panel). The regression of WBC was less dramatic in the secondary recipients of AML4 BM cells (Figure S4). We repeated the transplantation using the AML2 BM cells, but began Dox treatment earlier when the WBC concentration was in the range of 80 to 100 K/μL (Figure 3B bottom panel). Interestingly, however, the 6-hour Dox treatment could reduce WBC count twice in the first experiment, but only once in the second experiment before Dox resistance developed (Figure 3C).
To examine the phenotypes of the transplanted AML in the secondary recipient SCID mice, we used flow cytometry to measure Mac1 and Gr1 levels in GFP-positive cells. Both GFP/Mac1 and GFP/Gr1 double positive AML cells were greatly reduced by chronic Dox treatment for 8 days (Figure 3D). Since the transgenic NRASG12V cDNA is expressed with a glutamic acid–glutamic acid (EE) tag at the N terminus, the molecular weight of the NRASG12V protein is approximately 1 kDa greater than endogenous Nras. Due to the protein size difference, we could confirm the NRASG12V expression was repressed by the Dox treatment using Western blot analysis, and this correlated with the RT-PCR results shown in Figure 1D (Figure 3E).
To determine whether the regression of transplanted AML is correlated with the induction of apoptosis, BM tissues from transplanted mice were stained for double-strand DNA breaks (Figure 4A,B). Dox treatment for 4 to 8 days significantly increased the number of apoptotic cells in BM from TRM-transgenic AML recipients (P = .021), but not from TM-transgenic AML recipients (P = .259; data not shown). Moreover, the number of apoptotic cells stained for cleaved caspase-3 in peripheral blood was increased in TRM-transgenic AML recipients after 12 to 24 hours after Dox treatment (P = .045; Figure 4C). These data showed that repression of NRASG12V by Dox treatment induced apoptosis of TRM-transgenic AML cells in vivo, which are completely or partially addicted to NRASG12V oncogene in spite of the presence of other oncogenic mutations including the Mll-AF9 transgene.
Figure 4.

Conditional repression of NRASG12V expression induces apoptosis in the BM and circulating leukemia cells. (A) Apoptotic cell staining in the sternal BM with TUNEL method and Methyl Green as a counterstaining. Four days after Dox treatment, the number of apoptotic (brown) cells in the BM are increased in SCID mice transplanted with TRM-transgenic AML cells. Bars represent 50 μm. (B) Numerical comparison of apoptotic cells in BM. The average numbers of TUNEL-stained (TUNEL +) cells are shown with the standard deviation bars. The results indicate that conditional repression of NRASG12V expression significantly increases apoptotic death rates in the BM cells (P = .021). Apoptotic cell number is the average of stained cells in 3 fields. (C) Cleaved caspase-3 staining in peripheral WBCs. Apoptosis in peripheral bloods is observed 12 to 24 hours after Dox treatment. The number of apoptotic cells 12 to 24 hours is significantly increased in comparison with that of 0 to 6 hours (P = .045). The average number of apoptotic, cleaved caspase-3 positive cells was calculated by counting the stained cells in 41-45 fields at ×400 magnification. The narrow bars indicate the standard errors.
NRASG12V-independent relapsed MPD-like AML is more differentiated and less myelosuppressive than primary AML
The regression of AML blast cells after the conditional repression of NRASG12V expression was followed later (after approximately 21-25 days) by an increase of NRASG12V-independent WBC number in peripheral blood (Figure 3B). Using flow cytometry with spleen cells from secondary recipients, we found that the relapsed cells were GFP/Mac1 and GFP/Gr1 double-positive cells transplanted from the TRM-transgenic AML (Figure 3D). Morphologically, the blood smears from SCID mice with relapsed myeloid leukemia contained fewer large, undifferentiated blasts (≈20%-25%) than mice with primary AML before Dox treatment (≈85%-90%; Figure 5A). Mice with the NRASG12V-independent relapsed leukemia were histopathologically determined to have MPD-like AML and showed blast cells less than 20% in BM and spleen (Figure S5). The MLL protein was found in most (≈80%-90%) BM cells of engrafted AML and relapsed MPD-like AML in recipient SCID mice transplanted with TRM-transgenic AML cells. (Figure S6). Because the expression of both endogenous Mll and Mll-AF9 are regulated by the endogenous promoter of the mouse Mll gene, these results imply that Mll-AF9 is constantly expressed in TRM-transgenic AML and relapsed MPD-like AML cells independent of NRASG12V suppression. In addition, we analyzed the mRNA expression patterns of endogenous Mll, Mll-AF9, endogenous Nras, and NRASG12V in GFP-positive and GFP-negative BM cells of transplanted SCID mice. The NRASG12V transgene is expressed only in GFP-positive cells, but is completely repressed by Dox treatment. The Mll-AF9 transgene mRNA is only expressed in GFP-positive AML cells, but is not repressed by Dox treatment (Figure S7). Unlike the myelosuppressive effect of the TRM-transgenic primary AML evidenced by pronounced anemia, the numbers of RBCs in the relapsed MPD-like AML cases were significantly higher than that in mice with established AML expressing NRASG12V, in which the concentration of peripheral WBCs was more than 200 K/μL (Figure 5B). The activation of Ras/Erk signaling pathway was reduced after the conditional repression of NRASG12V expression, but up-regulated again in the NRASG12V-independent MPD-like AML as much as that of the engrafted AML before Dox treatment (Figure 5C). By analyzing 27 independent cDNA of Nras transcripts from the relapsed MPD-like AML and TM-transgenic AML cells, we found that the reactivation of Ras/Erk signaling pathway was not caused by an activating mutation (ie, 12, 13, or 61 amino acid substitution) of the endogenous mouse Nras gene (data not shown). Taken together, we concluded that the NRASG12V-independent relapsed MPD-like AML cells have reduced myelosuppression and are more differentiated than primary AML, although the Ras/Erk signaling pathway is reactivated without NRASG12V expression in relapsed disease.
Figure 5.

Relapsed NRASG12V-independent MPD-like AML has become less myelosuppressive and more differentiated. (A) Modified Wright-Giemsa staining of peripheral blood shows more differentiated myeloid lineage cells in the relapsed disease. In the blood smear from a mouse with established AML, more than 80% of WBCs have undifferentiated blast morphology. In the blood smear from the same mouse with relapsed MPD-like AML after Dox treatment, most cells are differentiated granulocytes (neutrophils, eosinophils) and less than 20% are undifferentiated blasts. Images taken at ×1000 magnification. Bars represent 25 μm. (B) For 0 through 6 days during which the WBC concentration reaches 200 K/μL or more, the RBC concentration in the relapsed MPD-like AML mice is significantly higher than in the mice harboring AML expressing NRASG12V (P < .001). Narrow bars indicate standard deviations. (C) Western blot analysis shows that the Ras/Erk pathway is inactivated with Dox treatment during AML regression, but is reactivated in the NRASG12V-independent relapsed MPD-like AML. The NRASG12V protein is not detected in Dox-resistant relapsed MPD-like AML. The Erk1/2 proteins acted as loading controls. Vertical lines have been inserted to indicate a repositioned gel line.
Relapsed MPD-like AML is transplantable, but shows greatly attenuated myelosuppression
To test the transplantability of the relapsed MPD-like AML, we transplanted 2 independent Dox-resistant MDP-like AML and Dox-sensitive primary AML BM cells from secondary SCID mice into tertiary recipient SCID mice (Figure 6A). The relapsed MPD-like AML, whether from mice given repeated 6-hour or chronic Dox treatment, were transplantable. The concentration of peripheral WBCs in all SCID recipients reached 150 to more than 200 K/μL3 weeks after transplantation in all tertiary recipients. However, SCID mice harboring the relapsed MPD-like AML lived significantly longer before becoming morbid than SCID mice harboring the primary AML (P < .001; Figure 6B), because anemia and/or thrombocytopenia in the MPD-like AML recipients appeared much later and was less severe than that in primary AML recipients. NRASG12V expression recurred in AML cells from the spleen of 6-hour Dox-treated MPD-like AML recipients, but not in the chronic Dox-treated MPD-like AML recipients in spite of reactivated Ras/Erk signaling in both cases (Figure 6C). Interestingly, the NRASG12V re-expression was correlated with the increase of undifferentiated cells in the peripheral blood in 6-hour Dox-treated MPD-like AML recipients, which looked like the established AML cells in the secondary recipient mice (Figure 5A).
To determine whether the Dox-sensitive AML and relapsed Dox-resistant MPD-like AML cells were clonally related, we used the comparative genome hybridization (CGH) technique with spleen cells from AML recipients. Compared with genomic DNA from a FVB/n × C57BL/6J F1 control mouse, we could find a unique gain on chromosome 12q in both samples (primary AML and relapsed MDP-like AML). Control SCID cell DNA did not show this chromosomal gain compared with FVB/n × C57BL/6J F1 DNA either (data not shown). Thus, the Dox-resistant, NRASG12V-independent disease is a subclone of the primary AML. From these results, we conclude that NRASG12V-independent MPD-like AML is a transplantable disease, but is less myelosuppressive than initial NRASG12V-dependent AML. These results provide evidences suggesting that NRASG12V expression suppresses the differentiation of AML cells maintaining a blastlike phenotype and induces a myelosuppressive phenotype in AML induced in cooperation with Mll-AF9.
Discussion
Based on the hypothesis that AML develops due to the cooperation of class I and II mutations,1 we sought to understand oncogenic roles of NRASG12V as a model class I mutation in the context of AML induced in combination with Mll-AF9 as a model class II mutation. Using a conditional repression system for NRASG12V expression, we found that continuous NRASG12V expression is required for AML persistence. Moreover, an initial apoptotic response is followed by some kind of signal pathway reprogramming and altered leukemic phenotypes upon relapse after the repression of NRASG12V expression.
Transgenic mice expressing both NRASG12V and Mll-AF9 developed AML (Figure 1). Although the difference in latency between NRASG12V/Mll-AF9 and Mll-AF9 alone was not statistically significant (Figure 1B), it is possible that by using more mice, we would detect a statistically significant acceleration of AML. Another possibility33,34 is that additional mutations may be required to induce oncogenic signaling pathways and/or inhibit tumor suppressing mechanisms despite the presence of NRASG12V and Mll-AF9. It is also possible that our Vav promoter-tTA–driven TRE-transgene system may not express the level of NRASG12V expression needed to shorten the latency of AML development in cooperation with Mll-AF9. We previously found that myeloid lineages and hematopoietic progenitor cells do not express very high levels of Vav-tTA–driven luciferase expression mice.20
Although the NRASG12V expression could not significantly shorten the latency of AML development induced in combination with Mll-AF9, most AML required ongoing NRASG12V expression for AML persistence (Figure 2B). The essential role of NRASG12V was revealed in the apoptosis of AML cells upon conditional repression of NRASG12V expression after the establishment of full-blown AML (Figures 3,4). This phenomenon has been observed and described as “oncogene addiction” or “oncogene shock.”5,6,13 The oncogene addiction hypothesis proposes that a single oncogene could be required for persistence of cancer cells, even though other mutations are present. Our data are consistent with this hypothesis and prove the situation exists for the NRAS oncogene even in AML cells cotransformed by a strong second oncogene. Our results show that the NRASG12V oncogene in AML induced in cooperation with Mll-AF9 could be a good molecular target for AML treatment. However, our data suggest that NRAS oncogene independence is likely to evolve, perhaps with an altered phenotype.
The pathogenic features of the NRASG12V-independent MPD-like AML were dramatically attenuated compared with NRASG12V-expressing primary AML. Our results suggest that NRAS oncogene may induce signals in AML cells that suppress the development of other myeloid lineages or allow AML cells to “outcompete” other lineages (Figure 5B). Others have shown that KRASG12D expression autonomously suppresses erythrocyte differentiation during embryonic hematopoiesis.35 Our results suggest that the NRASG12V oncogene has a cell nonautonomous role in suppressing erythropoiesis in SCID recipients with AML.
Previous studies found that NRAS, KRAS, cKIT, and FLT3 induced MPD in mice and concluded that the class I mutations mainly induce myeloid proliferation, but may not be associated with differentiation.15,36,37 However, other studies suggested that RAS oncogenes appear to promote the differentiation of monocytes and granulocytes in mouse and human hematopoiesis, but inhibit the differentiation of erythroid progenitors.38–43 Myeloid dysplasia, which was previously found in other mutant NRAS models, was not detected in our experiments for unknown reasons.44,45 Our results showed that NRASG12V expression is associated with not only inducing proliferation and survival, but also suppressing differentiation of myeloid lineage cells in the context of an AML cell (Figures 3–5 and Figure S5). Consequently, we conclude the oncogenic roles of NRASG12V expression in AML induced in cooperation with Mll-AF9 are (1) to induce proliferation of AML blast cells, (2) to induce cell nonautonomous myelosuppression, (3) to suppress apoptosis in AML blast cells, and (4) to inhibit differentiation of AML blast cells.
Since Mll-AF9 mutation expands the nonmalignant myeloid progenitor cells as in an MPD,27 we hypothesize that oncogenic NRASG12V induces proliferation and suppresses the differentiation of the expanded myeloid progenitors resulting in AML development, probably with other cooperating mutations. When NRASG12V expression is repressed, 2 processes are probably triggered in AML cells: (1) an apoptosis induction of most AML blast cells perhaps because of “oncogene shock”6 and (2) the return to MPD-like AML showing less myelosuppression and more differentiation after bypassing apoptosis that could be linked with other spontaneous mutations. The therapeutic implications of these results are that NRAS or RAS oncogene-activated pathways are good molecular targets and that a subset of cells escapes apoptosis and is responsible for relapse in a RAS oncogene-independent form. It will be important to determine what mechanisms are involved in substituting for the loss of the RAS oncogene. Our results suggest that this mechanism involves ERK activation. In addition, methods to prevent escape from this apoptotic response should be sought to achieve a better therapeutic outcome when RAS signal pathway inhibition is used for AML.
Supplementary Material
Acknowledgments
We thank Dr John H. Kersey (University of Minnesota, Minneapolis, MN) for kindly providing Mll-AF9 knockin mice with the permission of Dr Terence H. Rabbitts. We thank Dr Betsy Hirsch and LeAnn Oseth in the Cytogenetics Core Facility at the University of Minnesota Cancer Center for performing and analyzing the comparative genomic hybridization.
This work was supported by grants from the National Cancer Institute (U01 CA84221; Bethesda, MD), the Leukemia & Lymphoma Society of America (LLS 7019-04; New York, NY), and the Leukemia Research Fund (Minneapolis, MN; all to D.A.L.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: W.K. was responsible for writing, experiment planning, data collection, and data analysis; I.M. was responsible for histopathologic analysis and apoptotic cell staining; M.D.D. was responsible for assistance with transplantation experiments; and D.A.L. was responsible for writing, experiment design, and data analysis.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David A. Largaespada, University of Minnesota, Department of Genetics, Cell Biology and Development, 6-160 Jackson Hall, 321 Church Street SE, Minneapolis, MN 55455; e-mail: larga002@umn.edu.
References
- 1.Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. 2002;3:179–198. doi: 10.1146/annurev.genom.3.032802.115046. [DOI] [PubMed] [Google Scholar]
- 2.Weisberg E, Boulton C, Kelly LM, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1:433–443. doi: 10.1016/s1535-6108(02)00069-7. [DOI] [PubMed] [Google Scholar]
- 3.Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 4.Jonkers J, Berns A. Oncogene addiction: sometimes a temporary slavery. Cancer Cell. 2004;6:535–538. doi: 10.1016/j.ccr.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Hubner A, Jaeschke A, Davis RJ. Oncogene addiction: role of signal attenuation. Dev Cell. 2006;11:752–754. doi: 10.1016/j.devcel.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Sharma SV, Fischbach MA, Haber DA, Settleman J. “Oncogenic shock”: explaining oncogene addiction through differential signal attenuation. Clin Cancer Res. 2006;12:4392s–4395s. doi: 10.1158/1078-0432.CCR-06-0096. [DOI] [PubMed] [Google Scholar]
- 7.Sharma SV, Settleman J. Oncogenic shock: turning an activated kinase against the tumor cell. Cell Cycle. 2006;5:2878–2880. doi: 10.4161/cc.5.24.3598. [DOI] [PubMed] [Google Scholar]
- 8.Konig H, Holyoake TL, Bhatia R. Effective and selective inhibition of chronic myeloid leukemia primitive hematopoietic progenitors by the dual Src/Abl kinase inhibitor SKI-606. Blood. 2008;111:2329–2338. doi: 10.1182/blood-2007-05-092056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown P, Levis M, McIntyre E, Griesemer M, Small D. Combinations of the FLT3 inhibitor CEP-701 and chemotherapy synergistically kill infant and childhood MLL-rearranged ALL cells in a sequence-dependent manner. Leukemia. 2006;20:1368–1376. doi: 10.1038/sj.leu.2404277. [DOI] [PubMed] [Google Scholar]
- 10.Stubbs MC, Kim YM, Krivtsov AV, et al. MLL-AF9 and FLT3 cooperation in acute myelogenous leukemia: development of a model for rapid therapeutic assessment. Leukemia. 2008;22:66–77. doi: 10.1038/sj.leu.2404951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 12.Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet. 2000;24:57–60. doi: 10.1038/71691. [DOI] [PubMed] [Google Scholar]
- 13.Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction–a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006;3:448–457. doi: 10.1038/ncponc0558. [DOI] [PubMed] [Google Scholar]
- 14.Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. 2007;6:541–555. doi: 10.1038/nrd2221. [DOI] [PubMed] [Google Scholar]
- 15.Parikh C, Subrahmanyam R, Ren R. Oncogenic NRAS rapidly and efficiently induces CMML- and AML-like diseases in mice. Blood. 2006;108:2349–2357. doi: 10.1182/blood-2004-08-009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 17.Ayllon V, Rebollo A. Ras-induced cellular events [review]. Mol Membr Biol. 2000;17:65–73. doi: 10.1080/09687680050117093. [DOI] [PubMed] [Google Scholar]
- 18.Scheele JS, Ripple D, Lubbert M. The role of ras and other low molecular weight guanine nucleotide (GTP)-binding proteins during hematopoietic cell differentiation. Cell Mol Life Sci. 2000;57:1950–1963. doi: 10.1007/PL00000675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiesner SM, Jones JM, Hasz DE, Largaespada DA. Repressible transgenic model of NRAS oncogene-driven mast cell disease in the mouse. Blood. 2005;106:1054–1062. doi: 10.1182/blood-2004-08-3306. [DOI] [PubMed] [Google Scholar]
- 20.Kim WI, Wiesner SM, Largaespada DA. Vav promoter-tTA conditional transgene expression system for hematopoietic cells drives high level expression in developing B and T cells. Exp Hematol. 2007;35:1231–1239. doi: 10.1016/j.exphem.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 21.Ayton PM, Cleary ML. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene. 2001;20:5695–5707. doi: 10.1038/sj.onc.1204639. [DOI] [PubMed] [Google Scholar]
- 22.Eklund EA. The role of HOX genes in malignant myeloid disease. Curr Opin Hematol. 2007;14:85–89. doi: 10.1097/MOH.0b013e32801684b6. [DOI] [PubMed] [Google Scholar]
- 23.Daser A, Rabbitts TH. The versatile mixed lineage leukaemia gene MLL and its many associations in leukaemogenesis. Semin Cancer Biol. 2005;15:175–188. doi: 10.1016/j.semcancer.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Eguchi M, Eguchi-Ishimae M, Greaves M. Molecular pathogenesis of MLL-associated leukemias. Int J Hematol. 2005;82:9–20. doi: 10.1532/IJH97.05042. [DOI] [PubMed] [Google Scholar]
- 25.Ernst P, Wang J, Korsmeyer SJ. The role of MLL in hematopoiesis and leukemia. Curr Opin Hematol. 2002;9:282–287. doi: 10.1097/00062752-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Corral J, Lavenir I, Impey H, et al. An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell. 1996;85:853–861. doi: 10.1016/s0092-8674(00)81269-6. [DOI] [PubMed] [Google Scholar]
- 27.Dobson CL, Warren AJ, Pannell R, et al. The mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J. 1999;18:3564–3574. doi: 10.1093/emboj/18.13.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson JJ, Chen W, Hudson W, et al. Prenatal and postnatal myeloid cells demonstrate stepwise progression in the pathogenesis of MLL fusion gene leukemia. Blood. 2003;101:3229–3235. doi: 10.1182/blood-2002-05-1515. [DOI] [PubMed] [Google Scholar]
- 29.Pession A, Martino V, Tonelli R, et al. MLL-AF9 oncogene expression affects cell growth but not terminal differentiation and is down-regulated during monocyte-macrophage maturation in AML-M5 THP-1 cells. Oncogene. 2003;22:8671–8676. doi: 10.1038/sj.onc.1207125. [DOI] [PubMed] [Google Scholar]
- 30.Swansbury GJ, Slater R, Bain BJ, Moorman AV, Secker-Walker LM. Hematological malignancies with t(9;11)(p21-22;q23)–a laboratory and clinical study of 125 cases. European 11q23 Workshop participants. Leukemia. 1998;12:792–800. doi: 10.1038/sj.leu.2401014. [DOI] [PubMed] [Google Scholar]
- 31.Christiansen DH, Andersen MK, Desta F, Pedersen-Bjergaard J. Mutations of genes in the receptor tyrosine kinase (RTK)/RAS-BRAF signal transduction pathway in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2005;19:2232–2240. doi: 10.1038/sj.leu.2404009. [DOI] [PubMed] [Google Scholar]
- 32.Kogan SC, Ward JM, Anver MR, et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood. 2002;100:238–245. doi: 10.1182/blood.v100.1.238. [DOI] [PubMed] [Google Scholar]
- 33.Ikeda A, Shankar DB, Watanabe M, Tamanoi F, Moore TB, Sakamoto KM. Molecular targets and the treatment of myeloid leukemia. Mol Genet Metab. 2006;88:216–224. doi: 10.1016/j.ymgme.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 34.Irons RD, Stillman WS. The process of leukemogenesis. Environ Health Perspect. 1996;104(suppl 6):1239–1246. doi: 10.1289/ehp.961041239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Liu Y, Beard C, et al. Expression of oncogenic K-ras from its endogenous promoter leads to a partial block of erythroid differentiation and hyperactivation of cytokine-dependent signaling pathways. Blood. 2007;109:5238–5241. doi: 10.1182/blood-2006-09-047050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacKenzie KL, Dolnikov A, Millington M, Shounan Y, Symonds G. Mutant N-ras induces myeloproliferative disorders and apoptosis in bone marrow repopulated mice. Blood. 1999;93:2043–2056. [PubMed] [Google Scholar]
- 37.Shen SW, Dolnikov A, Passioura T, et al. Mutant N-ras preferentially drives human CD34+ hematopoietic progenitor cells into myeloid differentiation and proliferation both in vitro and in the NOD/SCID mouse. Exp Hematol. 2004;32:852–860. doi: 10.1016/j.exphem.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Braun BS, Tuveson DA, Kong N, et al. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci U S A. 2004;101:597–602. doi: 10.1073/pnas.0307203101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dorrell C, Takenaka K, Minden MD, Hawley RG, Dick JE. Hematopoietic cell fate and the initiation of leukemic properties in primitive primary human cells are influenced by Ras activity and farnesyltransferase inhibition. Mol Cell Biol. 2004;24:6993–7002. doi: 10.1128/MCB.24.16.6993-7002.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Darley RL, Pearn L, Omidvar N, et al. Protein kinase C mediates mutant N-Ras-induced developmental abnormalities in normal human erythroid cells. Blood. 2002;100:4185–4192. doi: 10.1182/blood-2002-05-1358. [DOI] [PubMed] [Google Scholar]
- 41.White MA, Nicolette C, Minden A, et al. Multiple Ras functions can contribute to mammalian cell transformation. Cell. 1995;80:533–541. doi: 10.1016/0092-8674(95)90507-3. [DOI] [PubMed] [Google Scholar]
- 42.Chan IT, Kutok JL, Williams IR, et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest. 2004;113:528–538. doi: 10.1172/JCI20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan IT, Gilliland DG. Oncogenic K-ras in mouse models of myeloproliferative disease and acute myeloid leukemia. Cell Cycle. 2004;3:536–537. doi: 10.4161/cc.3.5.828. [DOI] [PubMed] [Google Scholar]
- 44.Kogan SC, Lagasse E, Atwater S, et al. The PEBP2βMYH11 fusion created by Inv(16)(p13;q22) in myeloid leukemia impairs neutrophil maturation and contributes to granulocytic dysplasia. Proc Natl Acad Sci U S A. 1998;95:11863–11868. doi: 10.1073/pnas.95.20.11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Omidvar N, Kogan S, Beurlet S, et al. BCL-2 and mutant NRAS interact physically and functionally in a mouse model of progressive myelodysplasia. Cancer Res. 2007;67:11657–11667. doi: 10.1158/0008-5472.CAN-07-0196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}